





Abstract
The irritable bowel syndrome (IBS) is a functional gastrointestinal disorder with a largely unknown aetiology and a wide range of symptoms. Most cross-sectional studies carried out so far suggest subtle alterations in the structure of the intestinal microbiota that are barely reproduced, partly because of the high inter-subject variation in the community composition and disorder-specific features. We performed a longitudinal study to explore the within-subject variation in the faecal microbiota in two patients with IBS classified into the diarrhoea subtype and the healthy spouse of one of them. Faecal communities were monitored over 6–8 weeks and analysed through metagenomic and metatranscriptomic approaches. We found a higher temporal instability in the fraction of active microbiota related to the IBS condition and fluctuating symptoms. Strong and quick shifts in the distribution of the active microbiota and changes in the global pattern of gene expression were detected in association with acute diarrhoea, whereas microbial composition and encoded functions were more stable. The specific alterations in the microbiota were barely reproduced within and between patients. Further research is needed to assess whether these changes are a consequence of the abnormal gut function in acute diarrhoeic episodes and the potential usefulness of tackling them.
Introduction
The irritable bowel syndrome (IBS) is the most common functional gastrointestinal disorder in Western countries. The main symptoms include abdominal pain or discomfort, bloating and alteration in bowel habits (Hungin et al., 2005; Longstreth et al., 2006). Over the past decade, there has been an accumulation of evidence suggesting a role of the gut microbiota in IBS, mainly coming from case–control studies based on molecular methods, although no consensus has been reached regarding the association of specific microorganisms with IBS (Salonen et al., 2010; Simrén et al., 2013). The detection of the subtle alterations that seem to characterise microbial dysbiosis in IBS is hampered by the relatively small size of the cohorts and the many sources of variability in the composition of the microbiota unrelated to the disorder. Moreover, the detection of patterns may have been further complicated by differences between patients regarding physiopathology. Longitudinal studies in which patients are followed over time may help to overcome this.
There are few longitudinal studies on IBS, apart from clinical trials targeting the GI microbiota that address the improvement in symptoms (Moayyedi et al., 2010; Basseri et al., 2011; Simrén et al., 2013). One study reported a higher long-term temporal variation in the predominant faecal bacteria of patients with IBS when compared to that of controls (Mättö et al., 2005). Later, the same cohorts were re-analysed excluding patients who had taken antibiotics (Maukonen et al., 2006). The authors found a greater instability in patients with IBS in the metabolically active clostridial populations and speculated that instability might be due to variation in symptomatology between sampling days. In support of this, stabilisation of the faecal microbiota has been observed in patients with IBS after a probiotic supplementation that alleviated the symptoms (Kajander et al., 2008).
Besides, the functions of the microbiota are an essential factor to consider in understanding the GI disorders. Tana et al., (2010) found higher levels of acetic and propionic acids in faecal samples of patients with IBS compared with those of controls, which correlate with worse GI symptoms and quality of life and higher amounts of Lactobacillus and Veillonella. Elevated levels of amino acids and phenolic compounds (Ponnusamy et al., 2011) or primary bile acids (Duboc et al., 2012) have also been found in faeces of patients with IBS. Le Gall et al., (2011) explored the metabolic activity of the faecal microbiota in patients with IBS, patients with ulcerative colitis (UC) and healthy controls. They found that specific metabolites were associated with each group, but the IBS condition could not be predicted from the metabolite profile, unlike the UC or control conditions, similar to what happens at the compositional level (Qin et al., 2010; Durbán et al., 2012). The functional impact of the gut microbiota on the IBS condition and on the associated symptoms warrants further investigation.
The objective of this study was to investigate the stability of the compositional and functional profiles of the microbiota of patients with IBS over time and the specific changes associated with acute diarrhoeic episodes. We followed two IBS patients with diarrhoea as predominant bowel habit and a matched healthy control of one of them over 6–8 weeks. Self-reported symptom diaries allowed relating microbiological attributes to the presence and severity of symptoms. The faecal microbiota were analysed for the first time in IBS using metagenomics and metatranscriptomics.
Materials and methods
The detailed study protocol is provided as Supplementary Material.
Sampling
Two female IBS patients with diarrhoea subtype, according to the Rome III criteria (Longstreth et al., 2006), were included in the study (Patients 1 and 2). The husband of Patient 1 was included as an age-matched control who shared her environment (Control 1). Participants gave informed written consent to the study protocol, which was approved by the Ethics Committee of La Fe University Hospital (Valencia, Spain). Relevant volunteers' details are summarised in Table 1997). Faecal samples were collected in the morning every 2 days the first week and once a week thereafter. Additional samples were collected when patients reported acute symptoms. A summary of the symptom diaries is shown in Supporting Information, Table S1. Faeces were collected in tubes containing phosphate-buffered saline and kept at 4 °C for 1–2 h before being stored at −80 °C. Volunteers kept their routine lifestyle habits throughout.
Characteristics of Control 1 (C1), Patient 1 (P1) and Patient 2 (P2)
| Subject | C1 | P1 | P2 |
| Group | Control | IBS–diarrhoea | IBS–diarrhoea |
| Age | 66 | 62 | 21 |
| Sex | Male | Female | Female |
| Body mass index | 31.7 | 27.5 | 30.4 |
| Nationality | Spanish | Spanish | Spanish |
| Educational level | University/college | University/college | High school |
| Physical activity | Moderate | Moderate | Moderate |
| High fibre food intake | Daily | Weekly | Daily |
| Alcohol consumption | Monthly | Never | Never |
| Smoking | Ex-smoker | Ex-smoker | Smoker |
| Antibiotics (last 3 months) | No | No | No |
| Medication intake at sampling time | Hypertension, hyperlipidemia, urinary tract disorders | Hypertension, arthrosis, osteoporosis | Bowel spasms, contraceptives |
| Subject | C1 | P1 | P2 |
| Group | Control | IBS–diarrhoea | IBS–diarrhoea |
| Age | 66 | 62 | 21 |
| Sex | Male | Female | Female |
| Body mass index | 31.7 | 27.5 | 30.4 |
| Nationality | Spanish | Spanish | Spanish |
| Educational level | University/college | University/college | High school |
| Physical activity | Moderate | Moderate | Moderate |
| High fibre food intake | Daily | Weekly | Daily |
| Alcohol consumption | Monthly | Never | Never |
| Smoking | Ex-smoker | Ex-smoker | Smoker |
| Antibiotics (last 3 months) | No | No | No |
| Medication intake at sampling time | Hypertension, hyperlipidemia, urinary tract disorders | Hypertension, arthrosis, osteoporosis | Bowel spasms, contraceptives |
Characteristics of Control 1 (C1), Patient 1 (P1) and Patient 2 (P2)
| Subject | C1 | P1 | P2 |
| Group | Control | IBS–diarrhoea | IBS–diarrhoea |
| Age | 66 | 62 | 21 |
| Sex | Male | Female | Female |
| Body mass index | 31.7 | 27.5 | 30.4 |
| Nationality | Spanish | Spanish | Spanish |
| Educational level | University/college | University/college | High school |
| Physical activity | Moderate | Moderate | Moderate |
| High fibre food intake | Daily | Weekly | Daily |
| Alcohol consumption | Monthly | Never | Never |
| Smoking | Ex-smoker | Ex-smoker | Smoker |
| Antibiotics (last 3 months) | No | No | No |
| Medication intake at sampling time | Hypertension, hyperlipidemia, urinary tract disorders | Hypertension, arthrosis, osteoporosis | Bowel spasms, contraceptives |
| Subject | C1 | P1 | P2 |
| Group | Control | IBS–diarrhoea | IBS–diarrhoea |
| Age | 66 | 62 | 21 |
| Sex | Male | Female | Female |
| Body mass index | 31.7 | 27.5 | 30.4 |
| Nationality | Spanish | Spanish | Spanish |
| Educational level | University/college | University/college | High school |
| Physical activity | Moderate | Moderate | Moderate |
| High fibre food intake | Daily | Weekly | Daily |
| Alcohol consumption | Monthly | Never | Never |
| Smoking | Ex-smoker | Ex-smoker | Smoker |
| Antibiotics (last 3 months) | No | No | No |
| Medication intake at sampling time | Hypertension, hyperlipidemia, urinary tract disorders | Hypertension, arthrosis, osteoporosis | Bowel spasms, contraceptives |
Sequencing of metagenomes and metatranscriptomes
Faecal suspensions were centrifuged at 3200 g to remove big particles. Supernatants were centrifuged at 16100 g to pellet cells. Nucleic acids were extracted using the AllPrep DNA/RNA Mini Kit (QIAGEN). Total RNA was incubated with DNase I (Ambion) and then linearly amplified using the MessageAmp II-Bacteria Kit (Ambion). The resulting antisense RNA was converted to double-strand complementary DNA (cDNA) using random hexamers. This cDNA and total DNA were sent for pyrosequencing on a Genome Sequencer FLX system using the GS FLX Titanium chemistry (454 Life Sciences, Roche). The metatranscriptomes of all samples and the metagenomes of some samples were analysed (1st and 3rd samples from Patient 1, 1st–5th samples from Patient 2). Table S2 provides characteristics of the obtained libraries. The entire data set has been deposited in the Sequence Read Archive of the European Bioinformatics Institute under the accession number ERP001739.
Annotation of sequences
Genes and cDNAs of the 16S and 23S rRNA genes were searched in metagenomes and metatranscriptomes as described previously (Gosalbes et al., 2011). The taxonomic affiliation of 16S rRNA genes and cDNAs was determined using the Classifier tool of the Ribosomal Database Project (RDP) II (Wang et al., 2007). Species-level phylotypes were defined at 97% of sequence identity for 16S rRNA reads using the cluster tool of the usearch package, version 5.0 (Edgar, 2010). Metagenomic reads were assembled using the runAssembly tool of the newbler package, version 2.6 (454 Life Sciences). Metatranscriptomic reads not labelled as rRNA were aligned to the concatenated metagenomic assembly of the corresponding volunteer using the runMapping tool of the newbler package, version 2.6. Putative coding regions were identified in the metagenomic assemblies from the coordinates of best hits in a blastx search against the NCBI nonredundant protein sequence database (Altschul et al., 1990). Additional open reading frames were searched using glimmer, version 3.02 (Salzberg et al., 1998). Putative coding genes were compared with the KEGG GENES and the TIGRFAM databases. The taxonomic assignment of the putative coding regions identified from the blastx search was assessed using the blast2lca tool, version 0.02 (http://github.com/emepyc/Blast2lca). Metatranscriptomic reads that aligned to a genomic region adopted its annotations, while reads not labelled as putative mRNAs were aligned against the Rfam database. Table S2 shows the number of annotated sequences per sample, which were the input for descriptive and statistical analysis.
Statistical analyses
The similarity between samples according to their bacterial composition was assessed with correspondence analysis (CA) or detrended correspondence analysis (DCA) and with analysis of similarities (ANOSIM). The variation in symptom presence/absence and intensity was analysed with principal components analysis (PCA). Chi-squared tests were applied to assess the homogeneity in the relative abundance of each taxon between days with severe and milder symptoms within each patient. The LEfSe algorithm was applied to identify intermediate functional categories in the KEGG pathway and the TIGRFAM hierarchies characterising the differences between patients and between days with severe and mild/moderate symptoms within each patient (Segata et al., 2011). Samples with < 200 sequences with an assigned functional role were discarded for analysis due to the great uncertainty in the estimation of the functional profile.
Results
Classification of samples based on symptoms
Patients 1 and 2 differed in their symptomatology: Patient 1 complained about abdominal pain, abdominal distension and defaecatory urgency, whereas Patient 2 had a high number of depositions with diarrhoea (Table S1). In addition, Patient 1 remained rather stable over the follow-up, while Patient 2 went through several phases of acute diarrhoea. Sampling days were classified based on symptoms (Supporting Information, Fig. S1). Days with milder symptoms were considered those with no pain–distension–urgency (day 21 in Patient 1; days 1, 7 and 42 in Patient 2) and those with less stools–diarrhoea (days 1, 3 and 7 in Patient 1; day 35 in Patient 2). Days with severe symptoms were considered those in which the number of diarrhoeal stools was higher (days 14, 28, 37 and 42 in Patient 1; days 3 and 28 in Patient 2). Day 56 in Patient 2 was also classified as one with severe symptoms because the numbers of diarrhoeal stools in the surrounding days were among the highest.
Dynamics of the microbial taxonomic profile
DCA of the distribution of microbial families estimated from 16S rRNA genes separated samples of Patient 2 from samples of Patient 1 and Control 1, which were mixed (Fig. 0001). However, the last samples of Patient 1 (taken on days with more severe symptoms) were slightly separated from the rest and closer to samples of Patient 2. In Patient 2, the active microbiota on most days with severe symptoms differed markedly from each other and from those taken on days with mild/moderate symptoms. Within each patient, samples taken on days with milder symptoms were quite similar. These patterns were similar to the ones observed from the taxonomic affiliation of mRNAs, although the bacterial distributions obtained with both procedures differed significantly (Figs S2 and S3).
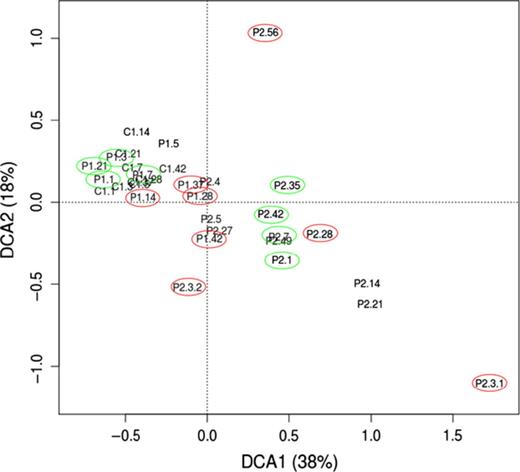
DCA of the distribution of microbial families in samples of Control 1, Patient 1 and Patient 2. Data were generated by taxonomic annotation of 16S rRNA genes in metatranscriptomes. Percentages correspond to the fraction of inertia explained by each axis. Samples are labelled with the code of the volunteer (C1, P1 and P2) and the sampling day over the follow-up. When there are two samples from a given individual in the same day, these are numbered after the day (e.g. P2.3.2 refers to the 2nd sample taken on day 3 from Patient 2). Days with milder and severe symptoms are circled in green and red, respectively.
The major groups of active bacteria were similar and remained quite constant in faeces of Control 1 and Patient 1 (Fig. 0002, Table S3a). The Clostridia and Bacteroidia classes accounted for the largest number of sequences all days in the communities of these subjects. Conversely, the faecal microbiota of Patient 2 was characterised by great temporal variation in the distribution of active bacteria (Fig. 0002, Table S3b). For example, the Firmicutes/Bacteroidetes ratio and the proportion of Proteobacteria varied widely over time (from 0.26 to 4.10 and from 3% to 88%, respectively). Also, the fraction of each bacterial family experienced greater variation than in Control 1 and Patient 1 (see, for example, the fluctuations in the amount of Clostridiaceae or Rikenellaceae over the follow-up). ANOSIM at the family level revealed that the median rank of distances between samples of Patient 1 was four times that of Control 1, and half of that found within Patient 2. Thus, temporal instability in the microbiota was associated with the IBS condition (greater in Patients than in Control 1) and with severe diarrhoea (greater in Patient 2 than in Patient 1). The discrepancies between the taxonomic composition estimated from metagenomes and from metatranscriptomes indicated a different contribution to the active fraction of the predominant bacteria (Table S4). Furthermore, when comparing the temporal variation in metagenomes and metatranscriptomes over the first 5 days of the follow-up of Patient 2, it could be appreciated that temporal dynamics was subtler at the metagenomic level (Fig. 0002).
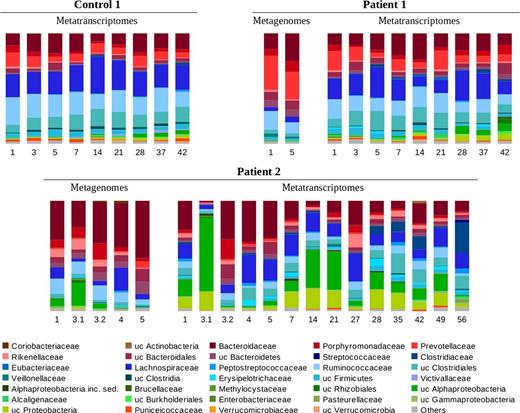
Relative abundance of microbial families in samples of Control 1, Patient 1 and Patient 2. Data were generated by taxonomic annotation of 16S rRNA genes and transcripts. The sampling day is indicated below each column. When there are two samples from a given individual in the same day, these are numbered after the day.
The alterations detected in association with worsening of symptoms were barely consistent between patients and within a single patient (Table S5). In Patient 2, the morning sample of day 3 had the highest level of activity of Alphaproteobacteria of all samples; the afternoon sample, the highest level of total Bacteroidia and Porphyromonadaceae; and sample of day 56, the highest level of Streptococcaceae,Clostridiaceae,Betaproteobacteria and Gammaproteobacteria. In Patient 1, sample of day 14 had an increase in Verrucomicrobia, while samples of days 28, 37 and 42, an increase in unknown members of Alphaproteobacteria and Proteobacteria, although this trend was shared with Control 1, which might reflect common environmental factors affecting the microbiota. The increases in the relative abundance of Proteobacteria were not due to a single or a few species, as demonstrated by the detection of multiple phylotypes defined at 97% of sequence identity mapped along reference 16S rRNA genes.
Dynamics of the microbial functional profile
Figure 0003 shows the distribution of broad categories in the KEGG pathway hierarchy. The encoded functions were highly conserved between subjects and over time (within a few days) within each subject. Temporal variation was higher at the gene expression level, but lower than variation in the distribution of active microorganisms. Given the different community assemblies found in each sample, it supports the concept of functional redundancy among faecal microorganisms. CA of the distribution of intermediate categories in the TIGRFAM and the KEGG pathway hierarchies separated the metagenomic samples of Patient 1 and Patient 2, while plots of metatranscriptomic samples gave a picture similar to that found with the distribution of active bacteria (Fig. S4). It should be noted that only a small number of sequences had a functional annotation in metatranscriptomes, so these results should be interpreted with caution.
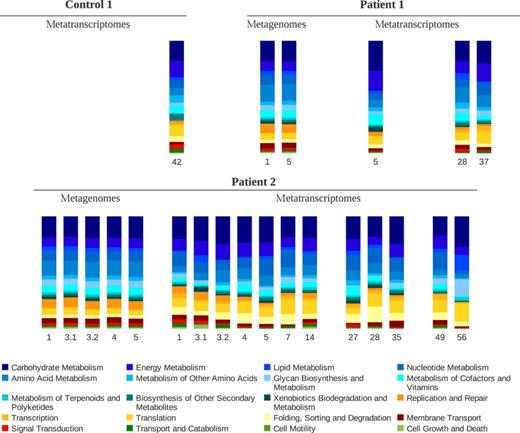
Relative abundance of functional categories in the KEGG pathway hierarchy in samples of Control 1, Patient 1 and Patient 2. Data were generated by functional annotation of protein coding genes and mRNAs. The sampling day is indicated below each column. When there are two samples from a given individual in the same day, these are numbered after the day. Samples with < 200 sequences with an assigned functional role are not shown.
Although statistically significant, the few differentially abundant functional features between and within patients detected with LEfSe analysis are difficult to interpret considering the limited number of samples per group and the rather low number of sequences per sample, which also complicate the detection of significant changes (Table S6). These limitations are more evident when we focus on specific taxa, because the number of sequences within each taxon was small.
Discussion
Temporal variation in the structure and function of the faecal microbiota was associated with the presence and intensity of IBS symptoms. Relapse and remission of acute diarrhoea occurred within short intervals and were associated with strong and quick changes in the microbiota of Patient 2. The increased instability in the patients with IBS could be reflecting the inability of the gut microbiota itself to maintain its structure, which might contribute to symptom development, or the microbial alterations produced by diarrhoea with the associated changes in motility and luminal contents. Moreover, a high degree of temporal variation is typical of re-establishing communities after disturbance, as seen in the faecal microbiota of subjects treated with antibiotics or dietary interventions or in patients with recurrent Clostridium difficile–associated diarrhoea (Chang et al., 2008; Dethlefsen & Relman, 2011; Wu et al., 2011).
We focused on the study of microbial activity at the level of gene expression because there are few functional studies on IBS and because we found greater variation between and within subjects at this level than at the genomic level. Compositional shifts in the active fraction of the microbiota characterised days with acute diarrhoea. Similarly, changes in the pattern of gene expression were associated with worsening of symptoms. Unfortunately, we had a relatively small number of sequences with a functional annotation in metatranscriptomes due to methodological limitations (the difficulty to enrich mRNAs prior to sequencing and the small length of non-rRNA sequences that makes difficult a confident assignment in homology searches). This leads to higher uncertainty in the estimated distributions of functional categories, so these results should be interpreted with caution.
Quantitatively, it is difficult to assess the influence of the difference in the number of sequences per sample on the analysis of similarities between samples. However, we expect that it has little effect on our analyses based on relative abundances. Although the number of sequences is important for the estimation of OTU richness, the estimation of relative abundances is in general less sensitive to it (although the associated standard errors are obviously larger for smaller samples). The smallest samples in metatranscriptomes (P1.3 in Patient 1 and P2.42 in Patient 2) were quite similar according to the distribution of microbial families to the other samples taken on days with similar symptoms. Therefore, undersampling did not appear to lead to a significant overestimation of dissimilarities.
Diarrhoea per se may result in the alterations detected in Patient 1 and Patient 2 on days with worse symptoms. The high rate of purging may disrupt the anaerobic environment in the gut, leading to the displacement of commensal anaerobes, and wash down more bacteria from the proximal regions of the gut, which have higher proportions of aerobes and facultative anaerobes than distal regions (Hayashi et al., 2005). Previous analyses of the faecal microbiota in sufferers from cholera and acute diarrhoea have shown reduced levels of obligate anaerobes and increases in facultative anaerobes (both commensal and pathogenic bacteria) (Albert et al., 1978; Balamurugan et al., 2008; Monira et al., 2013). Accordingly, we detected an overrepresentation of facultative anaerobes within Proteobacteria and Streptococcaceae on days with acute diarrhoea. However, it is important to note that we assessed the change in relative abundances of taxa comparing different samples, which does not necessarily translate into absolute changes. Diarrhoea may also increase the washout of mucosa-associated bacteria in the colon. The Proteobacteria,Streptococcaceae,Clostridiaceae and Verrucomicrobia detected in our surveys on days with worse symptoms could represent at least in part bacteria detached from the colonic mucosa, because the prevalence of these taxa seems to be higher in the colonic mucosa than in faeces (Durbán et al., 2011, 2012).
Besides, it has been repeatedly demonstrated an expansion of Enterobacteriaceae in unsubtyped IBS and diarrhoea-predominant IBS (Si et al., 2004; Krogius-Kurikka et al., 2009; Carroll et al., 2012). We did not detect this abnormality in our patients with IBS, but the levels of unknown members of Alphaproteobacteria and Proteobacteria were unusually high. Interestingly, an enrichment in Alphaproteobacteria in diarrhoea-predominant IBS sufferers compared with healthy controls has been reported previously (Krogius-Kurikka et al., 2009).
In this study, the global distribution of active bacteria discriminated Patient 2 from Control 1 and Patient 1. The last two were of similar age and shared environmental factors, which most probably contributed to their differentiation from Patient 2. However, the mild symptoms experienced by Patient 1 compared with Patient 2, together with the large alterations in the microbiota of Patient 2 concomitant with changes in symptom intensity, support that the singularities of Patient 2 were also related to the disorder. The inclusion of single samples from patients that were fairly asymptomatic at the time of sampling might partly explain the failure of most of the previous studies to discriminate IBS cases from controls on the basis of the global distribution of gut bacteria (Salonen et al., 2010; Durbán et al., 2012; Simrén et al., 2013). Besides, the analysis of the microbial activity instead of the microbial composition may have helped to differentiate our study subjects. Further studies should follow more patients and over longer periods to increase the chance of collecting samples that cover changes in bowel symptoms and relapse and remission of acute disease. Moreover, it would be helpful to have a finer classification of patients based on the pattern of symptoms, which can vary widely between patients and over time.
Cross-sectional studies on IBS to date have not found consistent alterations in the gut microbiota and are sometimes contradictory (Salonen et al., 2010; Simrén et al., 2013). Similarly, the alterations we found associated with acute symptoms are not systematic within a single patient. Although our results need to be interpreted with caution because of the limited sample size and they may also not be extrapolated to IBS patients with other symptom patterns, we consider it unlikely that dysbiosis is the underlying cause of the development of IBS symptoms. Other mechanisms may trigger the acute phases, for example stress. Animal studies suggest that psychological stress can change the composition of the microbiota via perturbation of the normal gastrointestinal habitat (Collins & Bercik, 2009). Besides, IBS sufferers tend to have a low threshold for coping with stressful situations and a high incidence of psychiatric comorbidity (Drossman, 1999; Hungin et al., 2005). Thus, stress may promote the alterations in the gut function observed in IBS and, subsequently, the alterations in the gut microbiota. We have proved that IBS is associated with a decrease in the stability (this study) and a decrease in the biodiversity of the gut microbiota (Durbán et al., 2012). Regardless of whether the community imbalances are a cause or a consequence of the development of symptoms (a topic that warrants further research), treatments that potentially counteract these attributes of the microbiota could be helpful in IBS.
Authors' contribution
A.L. and A.M. contributed equally to this work.
Acknowledgements
This work was funded by Grants BFU2009-04501-E, SAF2009-13032-C02-01 and SAF2012-31187 from Ministerio de Economía y Competitividad, Spain, and Prometeo/2009/092 from Generalitat Valenciana to A.M. A.D. is recipient of a fellowship from Instituto de Salud Carlos III, Spain.
References
Supporting Information
Additional Supporting Information may be found in the online version of the article:
Fig. S1. PCA based on the symptoms on sampling days.
Author notes
Editor: Julian Marchesi

{kind=link}
{kind=link}
{kind=link}