





Abstract
Lichen symbioses were recently shown to include diverse bacterial communities. Although the biogeography of lichen species is fairly well known, the patterns of their bacterial associates are relatively poorly understood. Here we analyse the composition of Alphaproteobacteria in Cetraria aculeata, a common lichen species that occurs at high latitudes and various habitats. Using clone libraries we show that most of the associated Alphaproteobacteria belong to Acetobacteraceae, which have also been found previously in other lichen species of acidic soils and rocks in alpine habitats. The majority of alphaproteobacterial sequences from C. aculeata are very similar to each other and form a single clade. Data from C. aculeata reveal that alphaproteobacterial communities of high latitudes are depauperate and more closely related to each other than to those of extrapolar habitats. This agrees with previous findings for the fungal and algal symbiont in this lichen. Similar to the algal partner, the composition of lichen alphaproteobacterial communities is affected by environmental parameters.
Introduction
Lichens are usually characterized as symbioses between a fungus (mycobiont) and photoautotrophic organisms that belong to the green algae or cyanobacteria (photobionts). The photobionts provide the fixed carbon for the association and allow the fungus to develop a unique structure, the lichen thallus. The photobiont is enclosed by fungal textures in a kind of biological growth chamber for optimized symbiosis. In the symbiotic stage the lichen symbionts tolerate hostile and dynamic environmental conditions, especially in cold habitats. Therefore, they are abundant and often the dominant life form in terrestrial ecosystems of the Antarctic (Øvstedal & Smith, 2001), the Arctic (Longton, 1988; Printzen, 2008) and high alpine regions (Türk & Gärtner, 2001). Ecophysiological and experimental studies have revealed that lichens can survive long periods of cold and drought, and can even survive conditions of outer space in a cryptobiotic stage (Kappen, 1993; Barták et al., 2007; Raggio et al., 2011; Schroeter et al., 2011). Recent results from the continental Antarctic indicate that lichens are active for < 10% of the year (Schroeter et al., 2011). In these habitats, they can maintain active photosynthesis below 0 °C, and they have the capacity to become hydrated without contact with liquid water (Schroeter & Scheidegger, 1995; Pannewitz et al., 2006).
The patterns of partnerships between lichen symbionts have recently received much attention. Molecular studies have verified that lichen mycobionts are able to associate with different photobionts: green-algal lichens usually associate with genetically different photobiont species or lineages (e.g. Kroken & Taylor, 2000; Opanowicz & Grube, 2004; Piercey-Normore, 2004; Blaha et al., 2006). In fact, photobiont switching seems to be the rule rather than the exception (Piercey-Normore & DePriest, 2001; O'Brien et al., 2005; Nelsen & Gargas, 2008; Wornik & Grube, 2010). Evidence for an ecological influence on symbiotic interactions in lichens is accumulating (Blaha et al., 2006; Casano et al., 2011; Fernández-Mendoza et al., 2011; Peksa & Skaloud, 2011). The observed dynamics of many symbiont systems has led to the hypothesis that a symbiotic lifestyle increases the adaptive and evolutionary potential of symbiotic holobionts. A symbiotic host may adapt to changing environmental conditions by shifting its microbial community, thereby ‘outsourcing’ (Gilbert et al., 2010) parts of its stress response to the symbiotic partners (Rodriguez et al., 2008). Shifting of symbiotic partners has been demonstrated for coral–Symbiodinium associations and has led to the coral probiotic hypothesis (Reshef et al., 2006) and the hologenome theory of evolution (Rosenberg et al., 2007; Gilbert et al., 2010). Switching symbionts allows a much faster and individualized reaction to environmental changes than the slow evolutionary processes of mutation and selection.
Owing to their ecological success and symbiotic strategies, many polar and alpine lichens have extremely wide geographical distributions. About 55% of the lichens in the maritime Antarctic are cosmopolitan (Hertel, 1988; Sancho et al., 1999), which is also true for a similar fraction of arctic lichens (Printzen, 2008). Some of these widely distributed species (such as Cetraria aculeata, Physcia caesia, Physcia dubia and Tephromela atra) are not only known from both polar regions and high mountain ranges elsewhere, but also occur in warm and dry habitats such as steppes and semi-deserts.
The lichen symbiosis often involves more organisms than the two classical functional partners. Tripartite lichens (representing c. 2% of all lichenized fungal species) regularly internalize nitrogen-fixing cyanobacteria in their thalli, whereas the function of further partners of the lichen symbiosis is diverse. So-called lichenicolous fungi can live as commensals or parasites on lichens (Lawrey & Diederich, 2003), and more recent work has shown the diversity and abundance of lichen-associated bacterial communities (reviewed by Grube & Berg, 2009). Confocal laser-scanning microscopy with group-specific probes showed that Alphaproteobacteria form the major bacterial component in lichens (Cardinale et al., 2008). Their usual dominance was confirmed by subsequent studies, but other bacterial groups may also be fairly abundant (Bates et al., 2011; Hodkinson et al., 2012; Cardinale et al., 2012). These bacterial groups are poorly represented in the culturable fraction (Cardinale et al., 2006), suggesting their growth is dependent on conditions intrinsic to the lichen symbiosis. Several examples of highly specific associations between Eukaryota and Alphaproteobacteria strains are known, including members of Acetobacteraceae (Favia et al., 2007), which seem to be particularly abundant in samples of alpine lichens on acidic substrates (Grube et al., 2009).
Grube et al. (2009) revealed that bacterial communities are host-specific in lichens, which was then confirmed by subsequent studies (Bates et al., 2011). Hodkinson et al. (2012) inferred photobiont type and geography as the most important factors shaping the microbiome composition. As these authors investigated whole lichen communities, it is not clear how individual species vary in their bacterial composition throughout their geographical range. Some variation within and among thalli of the same species exists in different habitats, depending on thallus age, substrate and exposure to sun (Cardinale et al., 2012). In the present study, we surveyed Alphaproteobacteria communities associated with a single euryoecious lichen species, C. aculeata, and examined how the structure of these communities varied over large geographical distances, from polar regions on opposite sides of the equator as well as more temperate sites of continental Europe. For this, we focus here on Alphaproteobacteria. We analyse the microbiome of the lichen C. aculeata in relation to environmental and geographical differences. The range of distribution, including European Alps, the US Rocky Mountains, Andean and Afroalpine highlands, as well as temperate grasslands and woodlands from Euasia to Central Asia, makes this erect shrubby species ideal for biogeographical studies.
Material and methods
Sampling
We sampled five individual thalli from each of four populations on King George Island (South Shetland Islands, Antarctic), Iceland, Germany and Spain (Table 1). Specimens were transferred to sterile plastic bags and kept frozen at −80 or −20 °C between sampling and isolation of DNA. Alphaproteobacterial communities were characterized by sequencing a c. 250-bp segment of the 16S rRNA gene from clone libraries.
Sampling localities for specimens of Cetraria aculeata used in this study
| Population | Latitude/longitude | Locality, year and collector | Sample codes | |
| Antarctica | 62°14′21.56″S, 58°39′58.33″W | South Shetland Islands, King George Island, Potter Peninsula, 2010, S. Domaschke & F. Fernández Mendoza | 2735 | B9 |
| 2736 | B10 | |||
| 2737 | B11 | |||
| 2738 | B12 | |||
| 2739 | B13 | |||
| Spain | 40°53′56.77″N, 03°36′06.22″W | Spain, Madrid, El Berrueco, 2010, F. Fernández Mendoza | 2740 | B15 |
| 2741 | B18 | |||
| 2742 | B21 | |||
| 2743 | B23 | |||
| 2744 | B27 | |||
| Germany | 50°06′42″N, 08°57′58″W | Germany, Hessen, Hannau, 2011, S. Becker, M. Fibian, S. Domaschke & C. Printzen | 2745 | H1 |
| 2746 | H2 | |||
| 2747 | H3 | |||
| 2748 | H4 | |||
| 2749 | H5 | |||
| Iceland | 65°52′55″N, 18°03′03″W | Iceland, 2010, C. Printzen | 2750 | B1 |
| 2751 | B2 | |||
| 2752 | B3 | |||
| 2753 | B4 | |||
| 2754 | B5 | |||
| Population | Latitude/longitude | Locality, year and collector | Sample codes | |
| Antarctica | 62°14′21.56″S, 58°39′58.33″W | South Shetland Islands, King George Island, Potter Peninsula, 2010, S. Domaschke & F. Fernández Mendoza | 2735 | B9 |
| 2736 | B10 | |||
| 2737 | B11 | |||
| 2738 | B12 | |||
| 2739 | B13 | |||
| Spain | 40°53′56.77″N, 03°36′06.22″W | Spain, Madrid, El Berrueco, 2010, F. Fernández Mendoza | 2740 | B15 |
| 2741 | B18 | |||
| 2742 | B21 | |||
| 2743 | B23 | |||
| 2744 | B27 | |||
| Germany | 50°06′42″N, 08°57′58″W | Germany, Hessen, Hannau, 2011, S. Becker, M. Fibian, S. Domaschke & C. Printzen | 2745 | H1 |
| 2746 | H2 | |||
| 2747 | H3 | |||
| 2748 | H4 | |||
| 2749 | H5 | |||
| Iceland | 65°52′55″N, 18°03′03″W | Iceland, 2010, C. Printzen | 2750 | B1 |
| 2751 | B2 | |||
| 2752 | B3 | |||
| 2753 | B4 | |||
| 2754 | B5 | |||
Sampling localities for specimens of Cetraria aculeata used in this study
| Population | Latitude/longitude | Locality, year and collector | Sample codes | |
| Antarctica | 62°14′21.56″S, 58°39′58.33″W | South Shetland Islands, King George Island, Potter Peninsula, 2010, S. Domaschke & F. Fernández Mendoza | 2735 | B9 |
| 2736 | B10 | |||
| 2737 | B11 | |||
| 2738 | B12 | |||
| 2739 | B13 | |||
| Spain | 40°53′56.77″N, 03°36′06.22″W | Spain, Madrid, El Berrueco, 2010, F. Fernández Mendoza | 2740 | B15 |
| 2741 | B18 | |||
| 2742 | B21 | |||
| 2743 | B23 | |||
| 2744 | B27 | |||
| Germany | 50°06′42″N, 08°57′58″W | Germany, Hessen, Hannau, 2011, S. Becker, M. Fibian, S. Domaschke & C. Printzen | 2745 | H1 |
| 2746 | H2 | |||
| 2747 | H3 | |||
| 2748 | H4 | |||
| 2749 | H5 | |||
| Iceland | 65°52′55″N, 18°03′03″W | Iceland, 2010, C. Printzen | 2750 | B1 |
| 2751 | B2 | |||
| 2752 | B3 | |||
| 2753 | B4 | |||
| 2754 | B5 | |||
| Population | Latitude/longitude | Locality, year and collector | Sample codes | |
| Antarctica | 62°14′21.56″S, 58°39′58.33″W | South Shetland Islands, King George Island, Potter Peninsula, 2010, S. Domaschke & F. Fernández Mendoza | 2735 | B9 |
| 2736 | B10 | |||
| 2737 | B11 | |||
| 2738 | B12 | |||
| 2739 | B13 | |||
| Spain | 40°53′56.77″N, 03°36′06.22″W | Spain, Madrid, El Berrueco, 2010, F. Fernández Mendoza | 2740 | B15 |
| 2741 | B18 | |||
| 2742 | B21 | |||
| 2743 | B23 | |||
| 2744 | B27 | |||
| Germany | 50°06′42″N, 08°57′58″W | Germany, Hessen, Hannau, 2011, S. Becker, M. Fibian, S. Domaschke & C. Printzen | 2745 | H1 |
| 2746 | H2 | |||
| 2747 | H3 | |||
| 2748 | H4 | |||
| 2749 | H5 | |||
| Iceland | 65°52′55″N, 18°03′03″W | Iceland, 2010, C. Printzen | 2750 | B1 |
| 2751 | B2 | |||
| 2752 | B3 | |||
| 2753 | B4 | |||
| 2754 | B5 | |||
DNA isolation and PCR
Young growing tips of lichen thalli were prepared under sterile conditions and rinsed for several minutes in double-distilled water. Genomic DNA was isolated from these fragments using the DNeasy Plant Mini Kit (Qiagen, Hilden, Germany) following the manufacturer's instructions. Five microlitres of isolated DNA was used in 25-μL PCR reactions containing 1 μL of primers ADF681F (Blackwood et al., 2005; specific for Alphaproteobacteria) and unibac927r (Lieber et al., 2003) and Illustra PureTaq PCR Beads (GE Healthcare, Chalfont St Giles, UK). Cycling conditions were as follows: initial denaturation at 94 °C (3 min), six cycles of 94 °C (30 s), 53 °C [−1 °C per cycle] (30 s) and 72 °C (1 min), followed by 30 cycles of 94 °C (30 s), 50 °C (30 s) and 72 °C (1 min), and a final extension at 72 °C (10 min). PCR products were gel-purified using the Qiaquick gel extraction kit (Qiagen) and the five PCR products from each geographical region were pooled before cloning.
Cloning and sequencing
In total, 15 ng of DNA was cloned into Escherichia coli JM 109 using the pGEM-T Easy Vector System I (Promega, Madison, WI). For each population, 96 clones were picked and used in 25-μL PCR reactions as described above. PCR products were purified using the Nucleospin Extract kit (Macherey-Nagel, Düren, Germany). Then, 2–5 ng of purified DNA was labelled with the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Carlsbad, CA) and cycle sequenced with primer ADF681F at 94 °C (30 s), and 29 cycles of 95 °C (15 s), 45 °C (15 s) and 60 °C (4 min). Sequences were determined on an ABI PRISM 3730 DNA Analyzer (Applied Biosystems).
Preparation of datasets
Sequences were loaded into geneious pro v. 5.3.6 (Biomatters Ltd, Auckland, New Zealand) and manually edited. A blast search (Altschul et al., 2009) was performed to verify that all sequences belonged to the Alphaproteobacteria. Sequences were aligned using the Muscle algorithm with default settings as implemented in geneious pro 5.3.6. Ends were trimmed to exclude all terminal gaps and sequences having more than three positions with ambiguous signal were removed from the dataset. The resulting alignment was 200 bp long and comprised 69 sequences from the Antarctic, 57 from Iceland, 47 from Spain and 15 from Germany. This dataset was used in the diversity analyses (see below). For the phylogenetic and community composition analyses, we collapsed sequences into operational taxonomic units (OTUs) by splitting the dataset into clusters (OTUs) of sequences with more than 98% similarity. We used 1 minus the raw percentage distance between sequence pairs as a measure of similarity. Distances were calculated using the dist.dna function in R package APE (Paradis et al., 2004). Gaps due to serial nucleotide repeats (poly-A for example) and ambiguities were not counted as differences. For each cluster that comprised more than one sequence, a consensus sequence (75% identity) was calculated. We identified 46 clusters, five of which were composed of single incongruent sequences that were finally excluded from the community analysis. We checked the systematic assignments of OTUs using the RDP classifier tool (http://rdp.cme.msu.edu/classifier/; Cole et al., 2009).
For the phylogenetic analysis, 23 sequences from the GenBank database were added to this dataset representing the major lineages of Alphaproteobacteria, including four sequences from the lichen-associated lineage LAR1 in the Rhizobiales (Hodkinson & Lutzoni, 2009), Pseudomonas aeruginosa (Gammaproteobacteria) and Chromobacterium violaceum (Betaproteobacteria) as outgroup sequences for the phylogenetic analysis (Fig. 1). To test whether Alphaproteobacteria from C. aculeata are similar to those of other terricolous lichens from similar habitats we also included 31 unique OTUs from an unpublished single-strand conformation polymorphism study by L. Muggia et al. on the microbiomes of the terricolous lichens Arthrorhaphis citrinella (2), Baeomyces placophyllus (4), Dibaeis baeomyces (6), Placynthiella icmalea (8) and Trapeliopsis granulosa (11) sampled between 1850 and 2020 m a.s.l in the Austrian Alps.
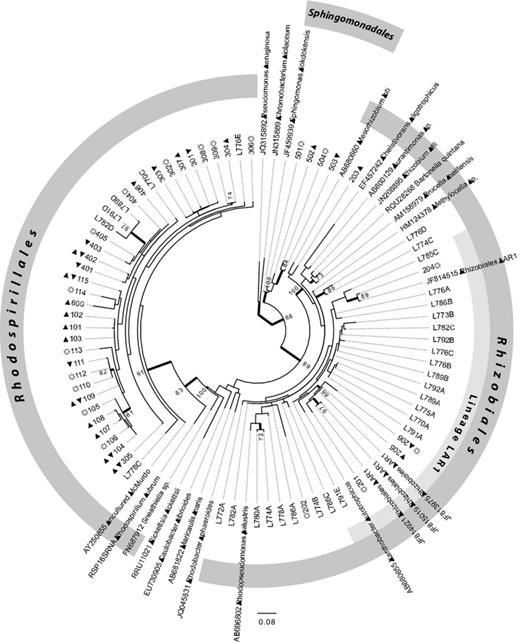
Maximum-likelihood bootstrap tree based on 16S rRNA gene sequences of 41 alphaprotebacterial OTUs from Cetraria aculeata, 31 sequences from other terricolous lichens and 23 GenBank sequences. Branches with > 60% bootstrap support are in bold. Geographical origin of OTUs is indicated by symbols next to the OTU number: ▲, north polar; ▼, south polar; ☼, temperate. Monophyly of the major orders of Alphaproteobacteria present in the microbiome of Ce. aculeata and other terricolous lichens (Rhizobiales, Rhodospirillales, Sphingomonadales) was enforced. The recently identified lichen-specifc clade LAR1 is also highlighted.
Phylogenetic relations of bacterial sequences
To determine how the Alphaproteobacteria OTUs found in C. aculeata are related to the bacteria found in other terricolous lichens and whether they form separate clades, we constructed a maximum-likelihood (ML) tree with bootstrap support values using RAxML v 7.2.6 (Stamatakis, 2006) and RAxMLGui v 1.0 (Silvestro & Michalak, 2012). We used the ‘ML + thorough bootstrap’ option with 1000 bootstrap replicates and the GTRGAMMA substitution model. A preliminary analysis revealed that the backbone of the tree was poorly resolved and the Rhizobiales appeared as polyphyletic. Because all sequences from C. aculeata belonged to Rhizobiales, Rhodospirillales or Sphingomonadales, we repeated the analysis, this time enforcing these three orders to be monophyletic.
For the analysis of phylogenetic diversity, an ultrametric phylogeny of all 188 bacterial sequences from C. aculeata was calculated using beast (Drummond & Rambaut, 2007) on two separate runs of 10 × 106 generations sampling every 1000 trees. Convergence of the Markov chain was assessed in Tracer (http://beast.bio.ed.ac.uk/Tracer) and a Maximum Clade Credibility with median heights was summarized using Treeannotator (Drummond & Rambaut, 2007) after 10% burn-in trees were discarded. For the ultrametric phylogeny we implemented a strict molecular clock scaled to substitutions as we lack temporal calibration. The adequacy of using a strict molecular clock was tested under the ML test implemented in mega v.5 (Tamura et al., 2011). FigTree version 1.3.1 was used to draw the phylogenetic tree.
Association between OTUs and bacterial communities
We used contingency tables to assess the association between OTUs and community samples. Deviations from independence were assessed using Pearson's chi-squared test. Results were summarized by means of an extended association plot (Cohen, 1980; Friendly, 1992; Meyer et al., 2003) generated using the function assoc implemented in the R package vcd (Meyer et al., 2006).
Species richness and diversity
Patterns of species (OTU) richness and diversity are difficult to compare due to the unequal sample sizes. Rarefaction allows the comparison of richness and diversity values by standardizing all community samples to the smallest sample size. Species richness was rarefied by calculating the rarefaction function in R package vegan (Oksanen et al., 2011) using the mean value of 10 000 random re-samples of 14 individuals taken from each population (without repetition). Species diversities were also calculated in vegan using the rarefaction function.
Phylogenetic structure and diversity of bacterial communities
The condensed dataset was processed in R where metrics of phylogenetic structure of communities were calculated using package picante (Kembel et al., 2010). We described the phylogenetic structure of lichen-inhabiting bacterial communities using six descriptive metrics of phylogenetic diversity: phylogenetic species variability (PSV), clustering (PSC), richness (PSR) and evenness (PSE) (Helmus et al., 2007); as well as Faith's phylogenetic diversity (PD) (Faith, 1992) and Rao's quadratic entropy (Rao, 1982). Both PSV and PSC reflect relatedness of species in a sample. Their values range between zero and one, where zero indicates complete relatedness and one unrelatedness. PSR indicates species richness after discounting species relatedness. It decreases towards zero as relatedness of species in the sample increases. PSE is a modification of PSV that incorporates relative species abundances. The maximum attainable value of one occurs only if species abundances are equal and the sample follows a star phylogeny, i.e. has no phylogenetic structure. Rao's quadratic entropy is a measure of diversity that takes the species' phylogenetic dissimilarities into account.
Community classification
To infer the similarity of bacterial communities at the four lichen sampling sites we generated a full cophenetic distance matrix between OTUs based on the ultrametric phylogeny using picante (Kembel et al., 2010). We then calculated the average distance between individuals in each community sample, which we used in a hierarchical clustering algorithm (hclust, R package stats) to generate a community classification.
Results and discussion
Relationships between Alphaproteobacteria from Cetraria and other lichens
Figure 1 shows the ML tree of the reduced dataset comprising 95 OTUs, 41 from C. aculeata, 31 from other terricolous lichens and 23 GenBank sequences. The majority of bacterial sequences from C. aculeata belong to the Rhodospirillales and, together with five sequences from other terricolous lichens and an uncultured bacterial sequence from the Antarctic, form a well-supported clade (Fig. 1). All lichen-associated sequences within this clade were assigned to Acetobacteraceae with relatively high confidence. Three of the five sequences from other terricolous lichen species form a well-supported separate clade within the Acetobacteraceae. Six alphaproteobacterial OTUs from C. aculeata and all other lichen-associated bacteria belong to Rhizobiales, and four bacterial sequences from C. aculeata belong to Sphingomonadales. The relationships within the major orders Rhizobiales and Rhodospirillales are poorly resolved due to low overall bootstrap support. This reflects the short length and accordingly relatively low number of substitutions between the studied OTUs.
Acetobacteraceae have previously been identified as a major component of lichen microbiomes (Grube et al., 2009; Bates et al., 2011) as have Rhizobiales (Hodkinson & Lutzoni, 2009). The confidence values for an assignment to specific genera are mostly below 50% in our dataset (data not shown). However, a few OTUs can be assigned to families and genera with relatively high confidence. Among them, five species of the acidophilic genus Acidisoma are notable (OTUs 109, 110, 111, 304 and 307, Fig. 1, see Table 3 below). The two recognized species of the genus have been described from arctic and boreal Sphagnum bogs (Belova et al., 2009) and previously been identified in samples of the lichen Umbilicaria cylindrica (Grube et al., 2009), as have species of Acidiphilium (Bates et al., 2011). The fact that the five OTUs reported here appear in different positions on the phylogeny should not be overemphasized given the limited sequence lengths. The family Sphingomonadaceae, represented in our sample by three OTUs, has also been reported from lichens (Hodkinson et al., 2012). The microbiomes of the other terrestrial lichen species seem to be dominated by bacteria from the order Rhizobiales. Our findings support previous results indicating that lichens harbour specific bacterial communities (Grube et al., 2009; Bates et al., 2011), because no OTUs were shared among the other lichen species and C. aculeata. Because the species were not collected in the same localities, the differences could also reflect different environmental conditions or simply spatial effects. However, this is unlikely because the Cetraria microbiomes investigated here come from widely disjunct populations and still appear to be more closely related to each other than to bacteria from the other lichens.
Species richness, diversity and phylogenetic structure of communities
The unequal sample sizes make direct comparisons of the OTU numbers found in the different populations impossible (the Antarctic population has four times more sequences than the German population). Rarefied values of species richness are lowest in the Antarctic population, followed by Spain and Iceland. Although the highest number of OTUs was found in the sample from Iceland, the German population is probably more species rich, as indicated by rarefied richness values (Table 2). PD and Rao's quadratic entropy, two indices that also take relative abundance and phylogenetic distances into account, are also lowest in the Antarctic community.
Phylogenetic structure of bacterial communities in terms of species richness (SR), rarefied richness (RR), phylogenetic species variability (PSV), clustering (PSC), richness (PSR) and evenness (PSE), Faith's phylogenetic diversity (PD) and Rao's quadratic entropy (QE)
| n | SR (RR) | Phylogenetic species | Faith's PD | Rao's QE | ||||||
| Variability (PSV) | Clustering (PSC) | Richness (PSR) | Evenness (PSE) | |||||||
| Mean | Variance | Mean | Mean (rm) | Variance | Mean | |||||
| Antarctica | 69 | 12 (5.3) | 0.483 | 0.010 | 0.814 | 5.802 (2.56) | 1.470 | 0.192 | 0.574 | 0.026 |
| Iceland | 57 | 16 (7.3) | 0.485 | 0.006 | 0.853 | 7.768 (3.54) | 1.656 | 0.275 | 0.646 | 0.038 |
| Spain | 47 | 12 (6.1) | 0.571 | 0.010 | 0.807 | 6.856 (3.48) | 1.471 | 0.360 | 0.616 | 0.048 |
| Germany | 15 | 8 (8) | 0.704 | 0.018 | 0.686 | 5.632 (5.6) | 1.157 | 0.552 | 0.590 | 0.071 |
| n | SR (RR) | Phylogenetic species | Faith's PD | Rao's QE | ||||||
| Variability (PSV) | Clustering (PSC) | Richness (PSR) | Evenness (PSE) | |||||||
| Mean | Variance | Mean | Mean (rm) | Variance | Mean | |||||
| Antarctica | 69 | 12 (5.3) | 0.483 | 0.010 | 0.814 | 5.802 (2.56) | 1.470 | 0.192 | 0.574 | 0.026 |
| Iceland | 57 | 16 (7.3) | 0.485 | 0.006 | 0.853 | 7.768 (3.54) | 1.656 | 0.275 | 0.646 | 0.038 |
| Spain | 47 | 12 (6.1) | 0.571 | 0.010 | 0.807 | 6.856 (3.48) | 1.471 | 0.360 | 0.616 | 0.048 |
| Germany | 15 | 8 (8) | 0.704 | 0.018 | 0.686 | 5.632 (5.6) | 1.157 | 0.552 | 0.590 | 0.071 |
n, number of sequences. Phylogenetic distances between sequences are based on an ultrametric tree of 188 bacterial sequences from Cetraria aculeata (see text for more information).
Phylogenetic structure of bacterial communities in terms of species richness (SR), rarefied richness (RR), phylogenetic species variability (PSV), clustering (PSC), richness (PSR) and evenness (PSE), Faith's phylogenetic diversity (PD) and Rao's quadratic entropy (QE)
| n | SR (RR) | Phylogenetic species | Faith's PD | Rao's QE | ||||||
| Variability (PSV) | Clustering (PSC) | Richness (PSR) | Evenness (PSE) | |||||||
| Mean | Variance | Mean | Mean (rm) | Variance | Mean | |||||
| Antarctica | 69 | 12 (5.3) | 0.483 | 0.010 | 0.814 | 5.802 (2.56) | 1.470 | 0.192 | 0.574 | 0.026 |
| Iceland | 57 | 16 (7.3) | 0.485 | 0.006 | 0.853 | 7.768 (3.54) | 1.656 | 0.275 | 0.646 | 0.038 |
| Spain | 47 | 12 (6.1) | 0.571 | 0.010 | 0.807 | 6.856 (3.48) | 1.471 | 0.360 | 0.616 | 0.048 |
| Germany | 15 | 8 (8) | 0.704 | 0.018 | 0.686 | 5.632 (5.6) | 1.157 | 0.552 | 0.590 | 0.071 |
| n | SR (RR) | Phylogenetic species | Faith's PD | Rao's QE | ||||||
| Variability (PSV) | Clustering (PSC) | Richness (PSR) | Evenness (PSE) | |||||||
| Mean | Variance | Mean | Mean (rm) | Variance | Mean | |||||
| Antarctica | 69 | 12 (5.3) | 0.483 | 0.010 | 0.814 | 5.802 (2.56) | 1.470 | 0.192 | 0.574 | 0.026 |
| Iceland | 57 | 16 (7.3) | 0.485 | 0.006 | 0.853 | 7.768 (3.54) | 1.656 | 0.275 | 0.646 | 0.038 |
| Spain | 47 | 12 (6.1) | 0.571 | 0.010 | 0.807 | 6.856 (3.48) | 1.471 | 0.360 | 0.616 | 0.048 |
| Germany | 15 | 8 (8) | 0.704 | 0.018 | 0.686 | 5.632 (5.6) | 1.157 | 0.552 | 0.590 | 0.071 |
n, number of sequences. Phylogenetic distances between sequences are based on an ultrametric tree of 188 bacterial sequences from Cetraria aculeata (see text for more information).
The Antarctic sample is not only the most depauperate but also shows higher phylogenetic structure, while the German community seems to have a higher complexity than can be estimated from our data. Despite the small sample size, the species found in the German sample are more distantly related to each other than those in the other communities. Iceland and the Antarctic have the lowest PSV and PSE values, indicating that species are more closely related to each other, while Spain shows an intermediate value. All communities except that from Germany show similar PSC values, indicating that their samples are similarly clustered. Finally, PSR is highest in the sample from Iceland, but rarefied PSR values, which account for unequal sample sizes, again show that the Antarctic community is poorest and the German population more than twice as rich in OTUs. In summary, the Antarctic population is consistently identified as the one with lowest species richness and diversity and also shows relatively high phylogenetic clustering of taxa.
This low diversity of Alphaproteobacteria in the Antarctic population is in line with results from other lichen symbionts. González et al. (2005) found that culturable lichen-associated Actinobacteria were considerably less diverse in polar than in tropical lichens. Domaschke et al. (2012) also reported lower genetic variability of the mycobiont and photobiont of high-latitude samples of C. aculeata. Our results, however, differ from those of adjacent habitats: Chu et al. (2010) found no significant differences between high-latitude and other soil microbiomes. The fact that spatial diversity patterns of bacteria in C. aculeata appear to be similar to those of the mycobiont and photobiont, but are in conflict with general diversity patterns observed in bacteria, may be a further hint to a specific role of bacteria in the lichen symbiosis.
Community patterns
The distribution of bacterial OTUs among the four lichen sampling sites is summarized in Table 3. Lichens at all four sites were dominated by a few common bacterial species. Two bacterial species were found in Germany and Spain. More interestingly, in spite of their strong geographical isolation, five OTUs were found in both polar communities. Although most OTUs were too rare to show any obvious trend, the more common ones are often strongly associated with certain sampling localities, for example OTU 109 with Iceland, 403 with the Antarctic and 306 with Spain. While bipolar distribution patterns have previously been reported from various microorganisms, e.g. marine bacterial and archaeal assemblages (Mergaert et al., 2001; Bano et al., 2004) and freshwater bacteria (Pearce et al., 2007), this is the first time a bipolar distribution pattern is observed in lichen-associated bacteria. Bipolar patterns are common among lichen mycobionts (Galloway & Aptroot, 1995) and were also observed in the photobionts of C. aculeata (Domaschke et al., 2012).

Distribution of OTUs in different community samples
The similarities between alphaproteobacterial communities are depicted by the neighbour-joining tree in Fig. 2 and support these observations. The two polar communities are more similar to each other than to the two temperate communities. Whether the isolated position of the German population is an artefact resulting from undersampling or reflects a real trend in our data, due to as yet unknown parameters, could not be determined. As with diversity levels, these results are concordant with the geographical patterns found in the green-algal photobiont of C. aculeata, Trebouxia jamesii (Fernández-Mendoza et al., 2011), which is best explained by climatic differences and codispersal with the mycobiont rather than geographical distances alone. Polar and temperate populations of C. aculeata are consistently associated with two different genetic groups of photobionts.
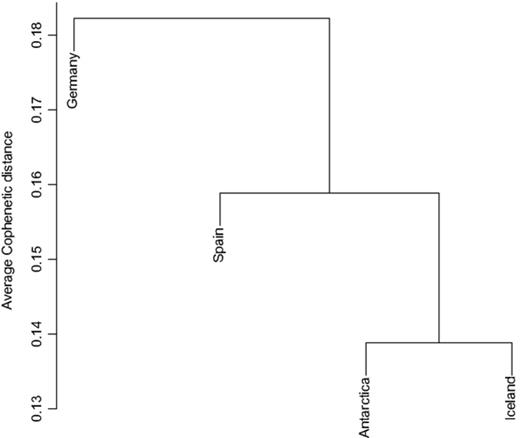
Community classification of the four populations investigated in this study based on the average cophenetic distances between individuals. The bar on the left indicates the value of the cophenetic distances between communities.
The fact that the photobionts of C. aculeata and its associated bacteria display similar patterns suggests that both may either respond similarly to environmental factors or are similarly transmitted, together with the mycobiont. A number of recent studies (Blaha et al., 2006; Yahr et al., 2006; Fernández-Mendoza et al., 2011) have shown a major influence of environmental factors on photobiont associations of lichens. These findings could agree with recent hypotheses that the symbiotic lifestyle may increase the evolutionary potential of the symbiotic holobiont by adaptation of symbiont constellations to ecological circumstances. A symbiotic host may ‘outsource’ (Gilbert et al., 2010) parts of its stress response to microbial partners and respond to changing environmental conditions by a habitat-adapted symbiont association (Rodriguez et al., 2008). Bacterial symbiotic communities are also known to vary in response to environmental factors, such as heat stress in corals or the nutritional diet of insects (Littman et al., 2010; Feldhaar, 2011). Our results provide first evidence for a similar response of lichen-associated bacterial communities. Whether microbial shifts contribute to the resilience of lichens to different environmental conditions throughout their wide geographical ranges, however, awaits further, perhaps experimental, studies.
Acknowledgements
We thank Heike Kappes (Grunelius-Möllgaard-Labor, Frankfurt) and Sigrun Kraker (Graz) for technical assistance. C.P. is supported financially by the German Research Foundation (DFG grant Pr 567/13-1) and the research funding programme ‘LOEWE – Landes-Offensive zur Entwicklung Wissenschaftlich-ökonomischer Exzellenz' of Hesse's Ministry of Higher Education, Research, and the Arts. M.G. is supported by FWF (I799).
References
Author notes
Editor: Max Häggblom

{kind=link}
{kind=link}
{kind=link}