





Abstract
Two methanogenic cultures were enriched from acidic peat soil using a growth medium buffered to c. pH 5. One culture, 6A, was obtained from peat after incubation with H2/CO2, whereas culture NTA was derived from a 10−4 dilution of untreated peat into a modified medium. 16S rRNA gene clone libraries from each culture contained one methanogen and two bacterial sequences. The methanogen 16S rRNA gene sequences were 99% identical with each other and belonged to the novel ‘R-10/Fen cluster’ family of the Methanomicrobiales, whereas their mcrA sequences were 96% identical. One bacterial 16S rRNA gene sequence from culture 6A belonged to the Bacteroidetes and showed 99% identity with sequences from methanogenic enrichments from German and Russian bogs. The other sequence belonged to the Firmicutes and was identical to a thick rod-shaped citrate-utilizing organism isolated from culture 6A, the numbers of which decreased when the Ti (III) chelator was switched from citrate to nitrilotriacetate. Bacterial clones from the NTA culture clustered in the Delta- and Betaproteobacteria. Both cultures contained thin rods, presumably the methanogens, as the predominant morphotype, and represent a significant advance in characterization of the novel acidiphilic R-10 family methanogens.
Introduction
The CH4 concentration in Earth's atmosphere has doubled in the past two centuries (Cicerone & Oremland, 1988), and CH4 is considered to be second only to CO2 as a greenhouse gas driving global climate change. Wetlands are considered to be the largest natural source of global atmospheric CH4 and are responsible for the release of c. 20% (115 Tg) of the total annual CH4 emission (Cicerone & Oremland, 1988). Peat-forming wetlands are responsible for c. 60% of the total wetland emission (Matthews & Fung, 1987). Although peatlands are overall net carbon sinks, as evidenced by the accumulation of an estimated 30% of terrestrial soil carbon (Gorham, 1991), they occupy a scant 3% of global land area (Yavitt, 1995). Whether peat-forming wetlands will continue to act as carbon sinks or become overall net sources, and whether peatland methanogenesis will increase or decrease in the face of global climate change are important and unsolved questions.
Molecular biological studies indicate that northern acidic (pH<5) peatlands harbor a large diversity of methanogenic Archaea. In a study of two peatlands in New York State Basiliko et al., (2003) found methanogenic sequences from three orders of methanogens with over half of the 86 clones belonging to a family-level clade in the H2/CO2-utilizing order Methanomicrobiales, the R10 family (Hales, 1996), which often dominates 16S rRNA gene or methyl-coenzyme M methylreductase subunit A (mcrA) gene clone libraries from acidic peatlands in diverse geographical locations. In a blanket peat bog in northern England, Hales et al., (1996) found methanogenic 16S rRNA gene sequences clustering in both the Methanosarcinales and Methanomicrobiales, with the R10 clone dominating the sequences. Similarly, in analyzing an oligotrophic fen in Finland, Galand et al., (2002) found mcrA gene sequences that clustered among the Methanosarcinales and Methanomicrobiales, with roughly half of the 21 sequences presumably belonging to the R10 family; they called this the ‘Fen cluster.’ In another study of the same site by Galand et al., (2003), 16S rRNA gene sequences revealed a diversity of novel methanogens, including at least eight clone sequences in the R10 family, even though these were not the most dominant clones.
We know relatively little about the methanogens adapted to low pH and oligotrophic environments in peat, as indigenous acidiphilic methanogens have previously resisted cultivation (Williams & Crawford, 1985). At present, the most acidiphilic hydrogenotrophic methanogen described is Methanobacterium espanolae (Patel, 1990), with an optimum pH between 5.5 and 6.0, and a pH minimum near 4.7. Some strains of Methanosarcina spp. can grow near pH 4.5 on methanol or H2/CO2, although their optimum is near neutrality (Maestrojuan & Boone, 1991). Recent progress has been made in obtaining acid-tolerant enrichment cultures. Horn et al., (2003) described enrichments from a German bog using an undefined H2/CO2 medium supplemented with 0.5 g L−1 tryptone and yeast extract that contained predominantly members of the Methanobacteriales. They also detected methanogenesis in pH 4.5 Trypticase-soy broth dilutions with no added methanogenic substrate (H2 was presumably produced from fermentation of the organic substrates). These cultures contained 16S rRNA gene sequence AMC 1 belonging to the R10 family. Sizova et al., (2003) described methanogenic cultures enriched from a large bog in Siberia, one called ‘26’ that contained members of the Methanobacteriales, and the other ‘K’, dominated by members of ‘Rice Cluster 1’ and also containing sequence K-4a2, a member of the R10 family.
Methanogenic Archaea carry out terminal reactions in the anaerobic conversion of organic matter to CH4 and rely on other organisms, usually Bacteria, to convert the organics to methanogenic precursors such as H2/CO2, acetate, formate or methanol. Not surprisingly, methanogens often grow in association with other microorganisms, and interdependencies, such as interspecies hydrogen transfer, are common (Zinder, 1993; Schink, 2002). Thus it can be difficult to separate methanogens from bacteria in culture, and such cultural studies may yield some insight into the associations between bacteria and methanogens in peatland ecosystems. Methanogenic enrichment cultures from bogs in Germany (Horn, 2003) and in Russia (Sizova, 2003) contained several novel bacterial 16S rRNA gene sequences.
In a previous study (Bräuer, 2004) we determined some growth conditions for naturally occurring methanogens from our study site, McLean bog, located in central New York State, USA. We demonstrated that peat samples from McLean bog incubated with H2/CO2 produced acetic acid at levels that were toxic to methanogenesis and that the antibiotic rifampicin prevented this phenomenon, presumably by inhibiting acetogenic bacteria. We showed that indigenous methanogens in McLean bog produced CH4 from H2/CO2 optimally at pH values near 5 in samples buffered with Homopipes (pKa=4.7 at 28°C) and at temperatures between 33 and 37°C, and that methanogenesis was initially inhibited by NaCl and KCl concentrations as low as 2 mM. Here we extend these studies to the development of a culture medium with a pH near 5.0 into which methanogenic activity was successfully transferred, the improvement of the medium composition and growth conditions that allowed dilution of this culture to 10−6, and minimization of growth of bacterial contaminants. This culture, called 6A, was derived from enrichment of undiluted peat material. We describe the development of a second methanogenic enrichment culture, called NTA, derived from a most probable number (MPN) dilution of the peat material into our improved growth medium. We used 16S rRNA gene clone libraries to identify methanogenic and bacterial members of the cultures and mcrA sequences to characterize the methanogens. We also describe the isolation of one of the bacterial members of culture 6A.
Materials and methods
Growth of methanogenic culture 6A
We cultivated microorganisms in peat soil from McLean bog, an ombrotrophic (rain fed) kettle hole bog near Ithaca, NY (42°30′N, 76°30′W). Peat soil was collected in August 2003, using the same procedures described previously (Bräuer, 2004). For culture 6A, enrichments were obtained in anaerobic soil slurries (Bräuer, 2004) except that deionized water was substituted with Peat Medium 1 (PM1) mineral solution containing (in mg L−1) 1.5 KCl, 13.6 KH2PO4, 26.8 NH4Cl, 0.024 CoCl2·6H2O, 0.075 ZnCl2, 0.019 H3BO3, 0.024 NiCl2·6H2O, 0.024 Na2Mo4·2H2O, 1.344 FeCl2·4H2O, 0.026 MnSO4·4H2O, 1.556 MgSO4, 2.336 CaCl2·2H2O, 0.009 CuSO4·5H2O, 3.446 AlK(SO4)2·12H2O. The medium was dispensed inside an anaerobic chamber (Coy, Ann Arbor, MI) into 18 × 150 mm crimp-top tubes, which were sealed with thick blue butyl rubber stoppers (Bellco, Vineland, NJ), and the tubes were autoclaved. The headspaces of the tubes were flushed with sterile O2-scrubbed 70%N2/30%CO2, and sterile anaerobic additions were set to the following final concentrations: 0.2 mM titanium (III) citrate (pH 5.0), 5 mM Homopipes (homopiperazine-N, N′-bis-2-(ethanesulfonic acid); filter-sterilized 0.5 M stock solution adjusted to pH 5.5; Research Organics Inc., Cleveland, OH), 0.5 mM coenzyme-M (2-mercapthoethanesulfonic acid), 0.5 mM sodium acetate, 0.2 mmol L−1 H2S (added as a sterile gas), and 10 mg L−1 rifampicin. During the course of these studies, the medium was modified numerous times and additions may have included one or more of the following (final concentrations): 10% (v/v) filter-sterilized anaerobic digestor sludge supernatant (Maymo-Gatell, 1995), 1–10% (v/v) filter-sterilized anaerobic culture supernatant or culture extracts from less purified enrichments, from a pure culture of the citrate-utilizing thick rod or from a pure culture of Methanospirillum hungatei JF1, 5–10% (v/v) filter-sterilized anaerobic peat water, and/or a 100 μM fatty acid mixture containing 75 μM NaOH and 25 μM each of isobutyric, valeric, isovaleric and dl-2(α)-methylbutyric acid. For the final medium used to dilute the methanogenic enrichment cultures to 10−6, the following changes were made to PM1 medium: 0.2 mM sodium acetate (instead of 0.5 mM), 1.0 mM Ti (III) nitrilotriacetate (NTA, instead of citrate), 0.2 g L−1 yeast extract, 100 μM FAM, plus a 10-fold concentrated solution of vitamins (Balch, 1979) were added (Table 1). The final liquid volumes in the tubes were c. 5 mL, and 0.7 atm. H2/CO2 was added to the headspaces. The cultures were incubated on a gyratory shaker at 28°C and 200 r.p.m. The pH of the medium was 5.1. CH4 production by cultures was followed using a flame ionization gas detector as previously described (Bräuer, 2004).
Additions used in final culturing media for each of the two mixed cultures and for strain TR1 (final concentrations)
PM1 contains the following final concentrations of minerals (in mg L−1): 1.5 KCl, 13.6 KH2PO4, 26.8 NH4Cl, 0.024 CoCl2·6H2O, 0.075 ZnCl2, 0.019 H3BO3, 0.024 NiCl2× 6H2O, 0.024 Na2Mo4·2H2O, 1.344 FeCl2·4H2O, 0.026 MnSO4·4H2O, 1.556 MgSO4, 2.336 CaCl2·2H2O, 0.009 CuSO4·5H2O, 3.446 AlK(SO4)2·12H2O.
83 mM Ti (III) nitrilotriacetate (NTA) or citrate stock solutions were made by combining the following anaerobic solutions and filter sterilizing in an anaerobic glove box: 4.8 mL of 0.5 M sodium citrate or sodium nitrilotriacetate, 0.55 mL of Ti (III) chloride (15% solution in HCl; Riedel-de Haen, Seelze, Germany), and 7.2 mL of one of the following: 1 M Tris buffer (pH 8) for a pH 7 solution, 0.4 M solution for a pH 5 solution of Ti (III) nitrilotriacetate, or 0.12 M NaOH for a pH 5 solution of Ti (III) citrate.
The stock solution of vitamins contains the following (in mg L−1): 20 biotin, 20 folic acid, 100 pyridoxine hydrochloride, 50 thiamine hydrochloride, 50 nicotinic acid, 50 dl-calcium pantothenate, 1 vitamin B12, 50 p-aminobenzoic acid, 50 lipoic acid. Note: this is a 10-fold increase from that used by Balch et al., (1979).
The stock solution of FAM contains 7.5 mM NaOH and 2.5 mM each of isobutyric, valeric, isovaleric and dl-2(a)-methylbutyric acid.
Additions used in final culturing media for each of the two mixed cultures and for strain TR1 (final concentrations)
PM1 contains the following final concentrations of minerals (in mg L−1): 1.5 KCl, 13.6 KH2PO4, 26.8 NH4Cl, 0.024 CoCl2·6H2O, 0.075 ZnCl2, 0.019 H3BO3, 0.024 NiCl2× 6H2O, 0.024 Na2Mo4·2H2O, 1.344 FeCl2·4H2O, 0.026 MnSO4·4H2O, 1.556 MgSO4, 2.336 CaCl2·2H2O, 0.009 CuSO4·5H2O, 3.446 AlK(SO4)2·12H2O.
83 mM Ti (III) nitrilotriacetate (NTA) or citrate stock solutions were made by combining the following anaerobic solutions and filter sterilizing in an anaerobic glove box: 4.8 mL of 0.5 M sodium citrate or sodium nitrilotriacetate, 0.55 mL of Ti (III) chloride (15% solution in HCl; Riedel-de Haen, Seelze, Germany), and 7.2 mL of one of the following: 1 M Tris buffer (pH 8) for a pH 7 solution, 0.4 M solution for a pH 5 solution of Ti (III) nitrilotriacetate, or 0.12 M NaOH for a pH 5 solution of Ti (III) citrate.
The stock solution of vitamins contains the following (in mg L−1): 20 biotin, 20 folic acid, 100 pyridoxine hydrochloride, 50 thiamine hydrochloride, 50 nicotinic acid, 50 dl-calcium pantothenate, 1 vitamin B12, 50 p-aminobenzoic acid, 50 lipoic acid. Note: this is a 10-fold increase from that used by Balch et al., (1979).
The stock solution of FAM contains 7.5 mM NaOH and 2.5 mM each of isobutyric, valeric, isovaleric and dl-2(a)-methylbutyric acid.
Development of culture NTA
The number of H2/CO2-utilizing methanogens in the peat was estimated using the three-tube most probable number (MPN) dilution technique. Peat was collected from McLean bog in April 2004. An anaerobic soil slurry of 2 g peat with 18 mL PM1 mineral solution was homogenized using a tissue homogenizer (Tissuemiser, Fisher Scientific, Pittsburgh, PA) at 30 000 r.p.m. for 1.5 min inside an anaerobic glove box and used to make 10-fold serial dilutions into anaerobic crimp-top tubes containing sterile anaerobic PM1 medium. We used 5 mL final volumes instead of the standard 10 mL to allow maximum H2/CO2 gas diffusion for the methanogens. The medium was prepared as above for 6A, and the following sterile anaerobic additions were made (final concentrations): 0.5 mM titanium (III) nitrilotriacetate, 5 mM Homopipes, 0.5 mM coenzyme-M, 0.2 mM sodium acetate, 5% (v/v) filter-sterilized culture supernatant from the pure culture of the citrate-utilizing thick rod, 200 mg L−1 yeast extract, vitamin solution (Balch, 1979), 0.2 mmol L−1 H2S, and 10 mg L−1 rifampicin. H2/CO2 (0.7 atm.) was added to the headspaces, and the cultures were incubated on a gyratory shaker at 28°C and 200 r.p.m. MPN tubes with >1 mmol L−1 CH4 were scored positive for methanogenesis. The MPN was calculated from the dilution factor, the dry weight of the soil (by drying at 60°C for 48 h), and using a table for MPN standard error and confidence intervals (Cochran, 1950). Subsequent transfers and dilutions were made from the highest positive dilution, which eventually led to the stable methanogenic enrichment culture NTA. The additions and incubation conditions in the final growth medium for this culture were the same as in the final medium for culture 6A (Table 1).
Isolation from culture 6A and characterization of a citrate-utilizing member of the Firmicutes
The citrate-utilizing bacterium was enriched from the methanogenic enrichment culture 6A by transfer into PM1 mineral solution containing the following additions: 0.8 mM titanium (III) citrate (pH 5.0), 5 mM Homopipes (0.5 M stock solution adjusted to pH 4.5), 0.5 mM coenzyme-M (2-mercapthoethanesufonic acid), 0.5 mM sodium acetate, 0.2 mmol L−1 H2S, vitamin solution (Balch, 1979), 10% (v/v) filter-sterilized anaerobic digestor sludge supernatant (Maymo-Gatell, 1995), and 5% (v/v) filter-sterilized peat pore water. The bacterium was isolated after making serial dilutions to 10−9 three times sequentially. In the final medium used for growing the culture, 200 mg L−1 yeast extract and 100 mM sodium citrate (pH 4.5) were added and sewage sludge, peat pore water and H2S were omitted (Table 1). The culture was grown in either 5 or 10 mL volumes and incubated under static conditions at 28°C. For experiments on the effect of temperature, the final medium was used, and for those on pH, Homopipes was omitted and the pH of the medium was manipulated by addition of 100 mM citrate adjusted to various pH values. The pH of the cultures was assessed at the beginning of the incubations. For experiments on substrate utilization, citrate was omitted except in the positive control tubes. Temperature, pH, and substrate tests were performed in triplicate.
Molecular and phylogenetic analyses
A MoBio Ultraclean Microbial DNA extraction kit that includes a bead-beating step was used to extract and purify DNA from the culture (MoBio, Carlsbad, CA). Primers 1Af(-1) (5′ TCY GKT TGA TCC YGS CRG A) and 1100Ar (5′ TGG GTC TCG CTC GTT G) (Hales, 1996) were used to amplify archaeal 16S rRNA genes, ME1 (5′ GCM ATG CAR ATH GGW ATG TC) and ME2 (5′ TCA TKG CRT AGT TDG GRT AGT) (Hales, 1996) were used to amplify methanogenic mcrA gene fragments and 27f (AGA GTT TGA TCM TGG CTC AG) and 1492r (TAC GGY TAC CTT GTT ACG ACT T) (Lane, 1991) were used to amplify bacterial 16S rRNA genes. The PCR reaction contained 1.25 U Brinkmann Taq polymerase, 1 × Brinkmann Taq buffer, 0.18 μM primer, 200 μM dNTPs, and 4 μL of a 1 : 1 diluted DNA extract in a total volume of 55 μL. For both the bacterial and archaeal 16S rRNA gene amplifications as well as the methanogenic mcrA gene amplifications, the program consisted of 4 min at 95°C, 25 cycles of 95°C for 1 min, 50°C for 1 min, and 72°C for 1.5 min, then a final elongation step at 72°C for 10 min. The PCR products were screened on a 1% agarose gel in TBE buffer.
Ligations and transformations were performed using the Invitrogen TA Cloning kit (Invitrogen, Carlsbad, CA). Potential transformants were screened using the M13f(-20) and M13r(-27) primers using the following amplification program: 4 min at 95°C, 35 cycles of 95°C for 1 min, 46°C for 1 min, and 72°C for 1.5 min, then an elongation step of 72°C for 10 min. Clones with proper-sized inserts were digested overnight using restriction endonucleases HaeIII and HhaI (Moyer, 1996) (New England BioLabs, Ipswich, MA). Representatives of distinct restriction digestion types were sent to Cornell's Bioresources Center for sequencing.
Phylogenetic analyses of 16S rRNA gene sequences were performed on sequences aligned by clustalx using the PHYLIP 3.62 package (Felsenstein, 2004). Of the bases that would be amplified by primers 1Af and 1100Ar, 957 well-aligned bases were analyzed using SEQBOOT, DNADIST using the F84 distance model, and NEIGHBOR, or by DNAML using the global rearrangement option, and jumbling the sequences twice. For McrA-predicted amino acid sequences, the sequences were aligned using clustalx, and analyzed using PROTDIST and NEIGHBOR in the PHYLIP package.
Fluorescence microscopy
Acridine orange staining was performed by adding 10 μL of cell culture and 1 μL 0.01% weight in volume (w/v) Acridine orange for each wet mount. Agar slides were prepared by adding c. 1 mL of 2% molten agar to a clean slide held at a 45° angle. The slides were dried at room temperature overnight or longer. Approximately 5–7 μL of Acridine orange-stained cells were added to the slides, mounted with a coverslip, and allowed to settle c. 20 min prior to viewing. Slides were viewed using a Nikon Eclipse E600 epifluorescence microscope equipped with a Hamamatsu CCD digital camera.
Results
Development of methanogenic cultures 6A and NTA
From our previous results with rifampicin-treated peat (Bräuer, 2004), we decided to poise the pH of our growth medium near 5.0 with Homopipes buffer, maintain a low ionic strength in the medium, and incubate at 28°C. PM1 mineral solution was designed using peat pore water chemistry as a guide (see Materials and methods). Another important consideration in the design of anaerobic media is the reducing agent. Often a sulfur compound is used, which doubles as a sulfur source. We tested a number of potential compounds, including H2S and cysteine, and found nearly all were highly inhibitory with the exceptions of coenzyme M (mercaptoethane sulfonate) and dithiothreitol (Fig. 1). Ti (III) citrate was stimulatory and was chosen as the reducing agent, and a small amount (25 μM) of H2S was added as a potential sulfur source.
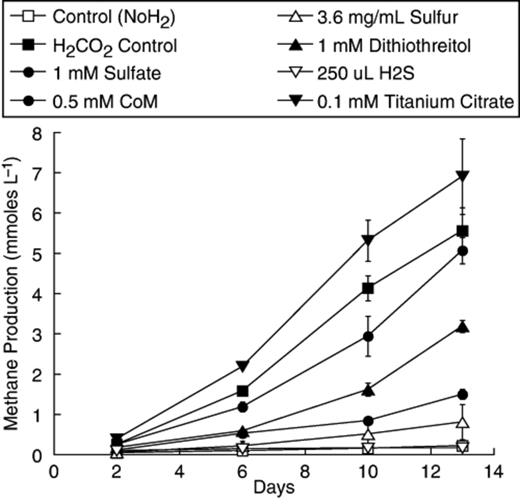
Methanogenesis from H2/CO2 by McLean bog peat slurries incubated with different potential reducing agents/sulfur sources. Besides the additions shown, less than 1 mmol L−1 CH4 was produced in 13 days by peat amended with 0.5 mM sulfite, 0.5 mM sodium thiosulfate, 1 mM cysteine, 1 mM sodium thioglycolate, or 2.9 mM mercaptoethanol. Points represent an average of duplicate tubes and the bars represent the range around those means.
Methanogenic activity was successfully transferred from a 3 mL inoculum into 10 mL of PM1 medium, which after 20 days of incubation produced c. 12 mmol CH4 L−1, whereas previous attempts to transfer activity into growth media produced <1 mmol CH4 L−1 (data not shown). Transfers from this culture into new medium were inconsistent with the large inoculums needed, long lag periods, and poor agreement between duplicates. For example, a fifth-generation transfer receiving a 10% (0.5 mL/5 mL) inoculum produced over 40 mmol L−1 CH4 in 21 days with a doubling time for methanogenesis near 2.5 days, whereas a duplicate culture lagged considerably, and two tubes with 0.1 mL inoculum produced a small amount of CH4 and stopped (Fig. 2). Microscopic examination of positive enrichments revealed the dominant morphotype to be long thin rods (Figs 3a and b), although coccoid cells and thicker rods were also present. In some cases, aggregates consisting mainly of thin rods and coccoid cells were seen (Figs 3c and d).
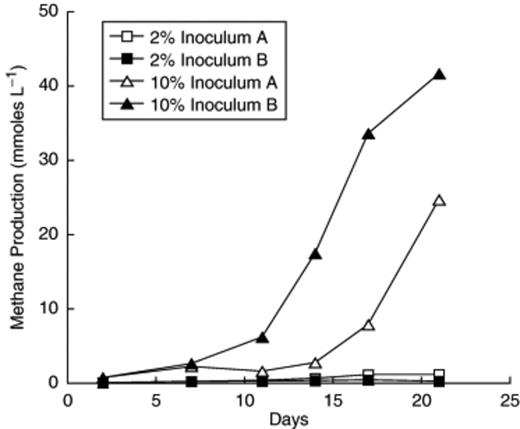
Methanogenesis by individual tubes of a 5th generation transfer of the H2/CO2 enrichment culture in pH 5.1 PM1 medium receiving either a 2% or 10% inoculum.
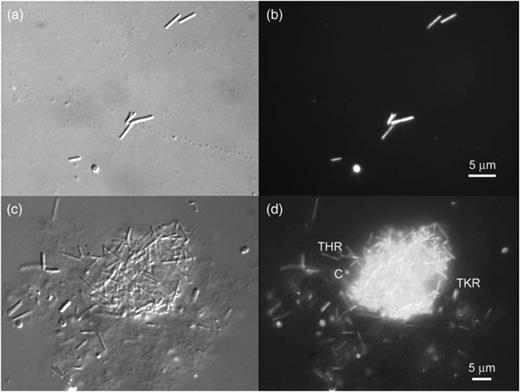
Destructive interference contrast (DIC) (a) and fluorescence (b) images of an acridine orange (AO)-stained preparation from a 4th generation transfer of the H2/CO2 enrichment culture from McLean bog, showing thin rods plus a coccoid cell. DIC (c) and fluorescence (d) images of a large aggregate containing thin rod (THR)-shaped and coccoid (c) cells as well as thick rod-shaped cells (TKR) outside the aggregate.
We endeavored to improve the reliability of transfer of the culture and included various nutrient supplements (see Materials and methods) with variable results. On the other hand, procedures that improved anaerobiosis improved reliability, including transferring cultures inside an anaerobic glovebox with syringes that had equilibrated at least 24 h with the glovebox headspace, and increasing the Ti (III) citrate concentration. Under these conditions, we were able to achieve growth of liquid dilutions up to 10−6, which was not sufficient for dilution to extinction. One of these dilutions was called 6A and was used for further studies.
Culture 6A contained thin rods, coccoid cells, and thick rods. Increasing Ti (III) citrate allowed more reliable transfer and also led to increased numbers of thick rods. We determined that an organism with this morphology utilized citrate as a growth substrate (see below), and its numbers were greatly decreased when we replaced citrate with nitrilotriacetate as the Ti (III) chelator (Moench & Zeikus, 1983). In subsequent transfers, the thin rods appeared shorter and tended not to clump with the cocci (Fig. 4a).

Fluorescence images of acridine orange stained preparations from a 20th generation transfer of the 6A enrichment culture after the switch to Ti (III) NTA as a reducing agent (a) and from a 7th generation transfer of the NTA culture (b). Note the similar morphologies and presence of both thin rods and coccoid organisms in each culture and spirals (s) in the NTA culture.
Culture 6A was developed from an enrichment using c. 0.1 g (dry wt) of peat as inoculum. Such enrichments often select for ‘weeds’, whereas dilution series directly from a habitat can enrich for more numerically important organisms (Ferris, 1996). Once we had developed procedures and a growth medium that succeeded in achieving reliable dilution of culture 6A, we used the Ti (III) nitrilotriacetate-based medium for MPN dilutions directly from a fresh peat sample. Serial dilutions of fresh peat yielded MPN counts of 1.9 × 106 cells per g (dry wt) (95% confidence limits=1.3 × 105–2.7 × 107). A tube from the highest positive dilution was transferred and called the NTA culture.
Like the 6A culture, the NTA culture also contained thin rods and coccoid cells, but also contained motile curved and spiral rods instead of thick rods (Fig. 4b). None of the cells in either culture showed visible autofluorescence found in many methanogens, attributable to high concentrations of cofactor F420. Both cultures produced significant amounts of CH4 and had estimated doubling times of 2–2.5 days at pH 5.1 and 28°C, the typical growth conditions.
Phylogenetic analyses of Archaea and Bacteria
In a clone library of culture 6A, all of 39 archaeal 16S rRNA gene clones amplified with primers 1Af and 1100Ar for methanogens (Hales, 1996; Basiliko, 2003) were of a single restriction type as were all of 29 successfully restricted clones in a clone library of culture NTA. These two sequences were 99% identical to each other and clustered with those in the R10 family, including sequences from McLean bog, although neither was identical to any of them (Fig. 5a, GenBank accession nos. DQ205187, and DQ205188). The closest sequence was that found in an anaerobic culture dechlorinating chlorinated ethenes (KB-1) (Duhamel, 2004). Both sequences also resembled sequences K4-a2 and AMC 1 (98% identity) from mixed methanogen cultures enriched from peat bogs in Germany and Russia (Sizova, 2003; Horn, 2003) and the R10 sequence from a British bog (Hales, 1996).
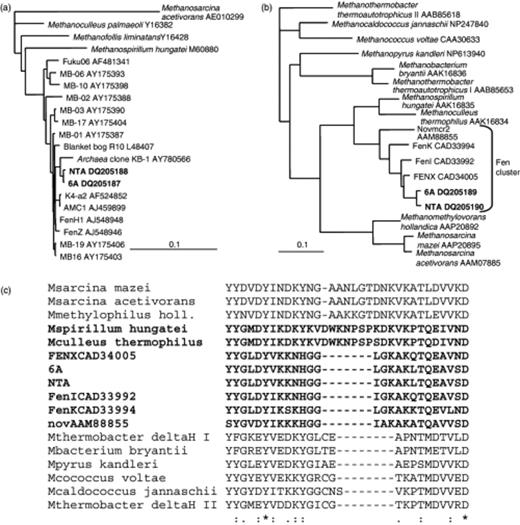
Dendrograms for the sequences from 6A and NTA cultures constructed for the 16S rRNA genes (a) and mcrA deduced amino acid sequences (b) using Methanosarcina acetivorans 16S rRNA gene sequence (a) and Methanothermobacter thermoautotrophicus McrA II (b) as outgroups. The dendrograms were constructed using neighbor joining methods as described under Molecular and phylogenetic analyses, and similar phylogenies were found using maximum likelihood methods. (c) Alignment of a section of mcrA-predicted amino acid sequences in (b) showing the insertion in the sequences in Methanospirillum, Methanoculleus, and the Methanosarcinales relative to the other methanogen sequences. The sequences in the Methanomicrobiales order are shown in bold.
Analysis of the PCR-amplified fragments of the mcrA gene, a faster-evolving gene often used in parallel with 16S rRNA gene for phylogenetic studies on methanogens (Hales, 1996; Lueders, 2001), revealed 96% nucleotide and 97% deduced amino acid identity between the mcrA sequences in cultures 6A and NTA, confirming that these strains are distinct. The 6A and NTA mcrA nucleotide sequences (GenBank accession nos. DQ205189 and DQ205190) were 92% and 91% identical over 666 bases (95% and 94% amino acid identity) with clone sequence FENX CAD34005 from an oligotrophic fen (Galand, 2003) (Fig. 5b). The closest sequence from a cultured organism was from Methanoculleus thermophilus, which was 87% identical over the 420 bases that BLAST aligned, and 76% identical at the AA level not counting a 7 AA insert in the M. thermophilus gene product relative to the ones from 6A (Fig. 5c). This insert was also present in the mcrA from Methanospirillum hungatei and other cultured members of the Methanomicrobiales (not shown) as well as members of the Methanosarcinales examined, but not in the Methanobacteriales or Methanococcales. Thus this previously ‘order-specific’ insert is not present in all members of the Methanomicrobiales.
Small clone libraries for each culture were generated from PCR amplifications using the bacterial 16S rRNA gene primers 27f and 1492r. Each library contained two bacterial sequences; MB6A-bac1 and MB6A-bac2 in culture 6A, and NTA-bac-1 and NTA-bac-2 in culture NTA (GenBank accession nos. DQ205191, DQ205192, DQ205193, and DQ205194). Culture 6A was examined after switching from citrate to nitrilotriacetate as the Ti (III) chelating agent. Clone MB6A-bac2 was the dominant bacterial clone in the 6A library representing 43 out of 47 clones. It clustered in the Bacteroidetes phylum in the Bacteroidaceae and had 99% sequence identity with clones BED7 and 26-4b2 from acidic methanogenic enrichment cultures of peat in Germany and Russia, respectively (Horn, 2003; Sizova, 2003) (Fig. 6). These sequences also clustered with sequences from anaerobic sludge digestors, including HA54 (Godon, 1997), and only showed 88% sequence identity with cultured organisms like Bacteroides fragilis. The remaining four (out of 47) clones had the same restriction pattern and the corresponding sequence, MB6A-bac1, belonged to Cluster IX of the Firmicutes (Fig. 6). Its closest relative was BED4, also from the methanogenic enrichment culture in Germany (Horn, 2003), although the clones shared only 96% sequence identity. MB6A-bac1 also displayed 95% sequence identity with clone SJA-143 from an anaerobic mixed culture that degraded trichlorobenzene (Wintzingerode, 1999).
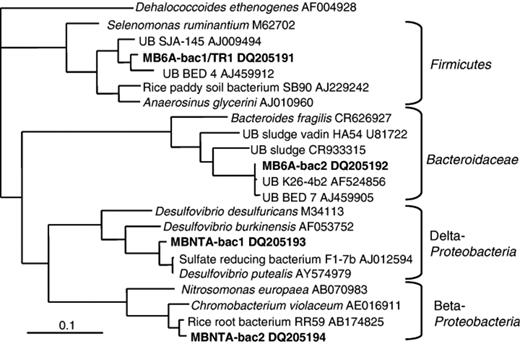
Dendrogram for the 16S rRNA gene sequence from the citrate-utilizing thick rod TR1 and for bacterial 16S rRNA gene sequences from 6A and NTA cultures. The sequences were analyzed as described under Molecular and phylogenetic analyses and the tree was produced using a maximum likelihood method, which gave the expected branching order for the phyla, but otherwise was similar to a neighbor-joining tree. Dehalococcoides ethenogenes, a member of the Chloroflexi, was used as the outgroup.
Clone MBNTA-bac2 was the most dominant clone in the NTA culture, representing 30 out of 38 clones. It exhibited 94% sequence identity with Chromobacterium violaceum, the nearest isolated relative (Fig. 6). MBNTA-bac2 showed a greater sequence identity of 97% with an unpublished sequence from Betaproteobacterium RR59, an acidogenic organism from rice roots. Clone MBNTA-bac1 represented eight of the 38 clones and clustered most closely (97% sequence identity) with a recently isolated species of Desulfovibrio, Desulfovibrio putealis (Basso, 2005) (Fig. 6). MBNTA-bac1 showed only 93% sequence identity to the next closest pure culture, Desulfovibrio burkinensis isolated from a rice paddy in Africa (Ouattara, 1999). MBNTA-bac1 and D. putealis both clustered with sequence F1-7b from a rice paddy.
Isolation of a citrate-utilizing member of the Firmicutes from culture 6A
Inoculation of culture 6A into medium containing 0.8 mM Ti (III) citrate and lacking H2/CO2 led to growth predominantly of organisms with the thick rod morphology. More copious growth was obtained when the citrate concentration was increased to 10 mM, and after three successive 10−9 dilutions in liquid medium, we considered it isolated and called it strain TR1. Its 16S rRNA gene sequence was identical to that of MB6A-bac1.
Figure 7 is a phase contrast micrograph of a culture of strain TR1.
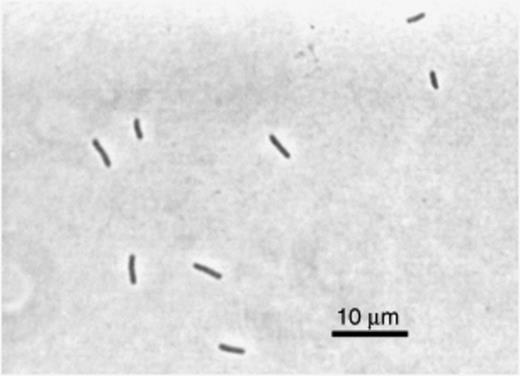
Phase contrast micrograph of the citrate-utilizing thick rod TR1 isolated from culture 6A.
Strain TR1 is moderately acidiphilic, growing best near pH 5.2, and growth was observed between pH 4.4 and 5.7 (Fig. 8). The culture exhibited a temperature optimum near 30°C (data not presented). In addition to citrate, the culture grew in anaerobic medium containing glucose, salicin, rhamnose, galacturonic acid, xylose, mannitol, arabinose, mannose, glucuronic acid, or cellibiose, but did not use peptone, glycerol, lactose, sucrose, meletizitose, polygalacturonic acid, cellulose, maltose, starch, pectin, ribose, vanillic acid, or syringic acid. Endospores or motility were not observed.
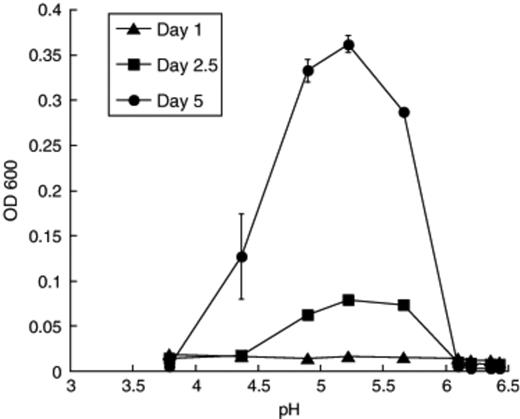
Effect of pH on growth (OD600) of the citrate-utilizing thick rod TR1.
Discussion
In this study, we obtained two well-defined methanogenic enrichment cultures each with one methanogen and two bacteria detected in clone libraries. All the organisms in these enrichments were acid-tolerant/mildly acidiphilic and capable of growth at pH 5, and we demonstrated that strain TR1 in the Firmicutes grows optimally near pH 5. Thus, these organisms are capable of growth at pH values near those in situ and represent indigenous peat bog species. The methanogens in both cultures represent a new genus in the novel R-10 family, from which there are currently no isolates described in the literature. Methanogens in this family of the Methanomicrobiales appear to be ubiquitous in peatlands throughout the Northern Hemisphere and sequences have been detected in peat or in enrichment cultures derived from peat in Germany (Horn, 2003), Russia (Sizova, 2003), England (Hales, 1996), Finland (Galand, 2002, 2003), and the United States (Basiliko, 2003).
Here we have attempted to bridge the gap between methanogens naturally occurring in peat and those growing in culture by cultivating indigenous acidiphilic methanogens. Although we have succeeded in culturing methanogens from an important novel family (R-10), the 6A and NTA archaeal sequences are not identical with those obtained in clone libraries from McLean bog (Basiliko, 2003) suggesting that they represent a small proportion of the population there. Supporting this contention is a recent determination using quantitative PCR that numbers of methanogens from the R10 family are present in McLean bog peat on the order of 108 g−1 (dry weight) (H. Cadillo-Quiroz et al., unpublished), whereas the MPN counts presented here are nearly two orders of magnitude lower, although they are likely to be underestimates because of problems of clumping and attachment to particles.
We have succeeded in growing novel methanogens unrelated to other pure cultures. Yet, despite extensive studies to develop unique culturing conditions to support growth of prominent indigenous methanogens in McLean bog, the methanogens in our mixed cultures bear a striking resemblance to other methanogens recently grown in enrichment cultures. The methanogens in the 6A and NTA cultures were enriched by different strategies and are distinct organisms with clearly different mcrA sequences, although their 99% 16S rRNA gene sequence identity is near the threshold of differences that can be detected due to PCR amplification and sequencing artifacts. Their closest relative was archaeal sequence KB-1 (Duhamel, 2004), derived from a Canadian mixed culture reductively dechlorinating chlorinated ethenes. The KB-1 culture produced HCl from reductive dechlorination and was operated until dechlorination ceased and the pH was near 5.3, at which point the pH was neutralized, so that the culture endured significant periods of low pH (M. Duhamel and E. Edwards, pers. commun.).
The 6A and NTA 16S rRNA gene sequences also were closely related to those from enrichments from acidic peats. Sequence AMC 1 was detected in a 10−6 MPN dilution of peat from a German bog into pH 4.5 glucose-trypticase-soy medium (Horn, 2003). This was the highest dilution positive for CH4, and indicates that the organisms were numerically important in the peat. Although further characterization of these dilutions was not described, it is likely that fermentative heterotrophs using the rich organic matter supplements were the predominant organisms in the culture. Also related is the K-4a2 sequence from the ‘K’ methanogenic enrichment culture derived from Russian peat (Sizova, 2003). This H2/CO2 enrichment was developed in a pH 5.3 medium in which Ti (III) citrate was the primary organic constituent, similar to PM1. It is of interest that members of the ‘R10’ family appear in cultures lacking high concentrations of sulfide as the reducing agent, consistent with our findings that sulfide was inhibitory to methanogenesis from H2/CO2 in peat (Fig. 1). Culture K also contained sequence K-5a2 belonging to rice-cluster I (RC-I) methanoarchaeal group. According to fluorescence in situ hybridization analyses, most of the cells were thick straight rods that hybridized with a probe specific for K-5a2, whereas 1–5% of the cells were long thin filaments that hybridized with a Methanomicrobiales-specific probe, and presumably represent K-4a2. Thus, the 6A and NTA cultures are the first in which a member of this methanogenic group represents a majority of the organisms present. With these cultures we have made significant progress towards potential isolation of a novel acidiphilic methanogen from the R-10 clade of the Methanomicrobiales.
It is also interesting that the closest relatives of the two bacterial sequences detected in the 6A culture were from other methanogenic enrichment cultures from acidic peat soil. This is particularly striking for MB6A-bac2 in the Bacteroidetes, whose 16S rRNA gene sequence was essentially identical to sequences derived from methanogen enrichments from peat in Russia and Germany (Horn, 2003; Sizova, 2003). Although we did not attempt isolation of the organism bearing this sequence from culture 6A, we now have several isolates with nearly identical sequences, and will describe them elsewhere. Similarly, the closest relatives to sequence MB6A-bac1/TR1 were from enrichment cultures, although not as closely related.
The bacterial partners detected in the NTA culture were members of the Proteobacteria. MBNTA-bac2 was most closely related to a Betaproteobacterium isolated from rice roots and the genus Chromobacterium. The other, MBNTA-bac1, was most closely related to members of the genus Desulfovibrio, which is commonly found in methanogenic cultures using oxidized sulfur compounds and syntrophically utilizing fermentation products. There is at least one report in the literature suggesting that some sulfate-reducing bacteria may grow syntrophically with methanogens in peat (Watson & Nedwell, 1998); however, little is known about the roles these organisms play in carbon flow in peat.
These studies reveal limitations in current culturing techniques applied to acidic bogs. The 16S rRNA gene sequences of the methanogens in our enrichment did not match sequences in clone libraries from McLean bog, but rather were closest to those from enrichments from Canada, Germany, and Russia. Although different growth medium formulations and methods were used in each of these studies, only a small subset of diversity of the R10 methanogen family was cultured in these studies. Similarly, the four bacterial sequences in our cultures had sequences from cultures as their closest relatives despite differences in culturing conditions including, in our case, the addition of 10 mg L−1 rifampicin. Moreover, none of the four sequences matches those in a 77-clone bacterial 16S rRNA gene library we developed for McLean bog (E. Yashiro and S. Zinder, unpublished), although the bacterial diversity was high and the library was therefore far from saturation. It appears that some fundamental requirements of many of the organisms in acidic bogs are not being met by standard culture techniques applied to them. By developing media that more closely approximate the in situ conditions (low ionic strength, low sulfide), our lab and others (Horn, 2003; Sizova, 2003) have managed to culture organisms from a previously uncultured clade of methanogens; however, culture techniques will need to be developed further to culture more environmentally relevant species within this clade.
Acknowledgements
We thank Hinsby Cadillo-Quiroz for assistance in primer design and advice, Esther Angert and Bill Ghiorse for assistance with microscopy, and Lorenz Adrian for advice on culturing. This work was supported by the NSF Microbial Observatories program grant (0132049).
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}