





Abstract
The Paecilomyces lilacinus is the most widely tested fungus for the control of root-knot and cyst nematodes. The fungus has also been implicated in a number of human and animal infections, difficulties in diagnosis often result in misdiagnosis or delays in identification leading to a delay in treatment. Here, we report the development of species-specific primers for the identification of P. lilacinus based on sequence information from the ITS gene, and their use in identifying P. lilacinus isolates, including clinical isolates of the fungus. The primer set generated a single PCR fragment of 130 bp in length that was specific to P. lilacinus and was also used to detect the presence of P. lilacinus from soil, roots and nematode eggs. Real-time PCR primers and a TaqMan® probe were also developed and provided quantitative data on the population size of the fungus in two field sites. PCR, bait and culture methods were combined to investigate the presence and abundance of the fungus from two field sites in the United Kingdom where potato cyst nematode populations were naturally declining, and results demonstrated the importance of using a combination of methods to investigate population size and activity of fungi.
1 Introduction
Paecilomyces lilacinus is the most widely tested fungus for the control of root-knot and cyst nematodes. Many authors have investigated its use in the field to control nematode populations [1–4] and it has been routinely isolated from infected nematode eggs in soils suppressive to plant parasitic nematodes [5–9]. At present, it is the only commercially available fungal formulation to control nematode pests, and the commercial strain 251 is registered for sale in a number of countries [[1]; http://www.biocontrol.co.za/pl; http://www.prophyta.com].
The fungus has also been isolated from clinical samples [10–14], not only from patients who are immuno-compromised and, therefore, at greater risk from opportunistic pathogens, but also from otherwise healthy patients. The diverse incidences and epidemiology of the infections by the fungus are varied and were reviewed by Okhravi et al.[1]. Morphological characteristics of the fungus are often overlooked by histopathologists leading to a misidentification and a delay in the correct form of treatment[13].
PCR has provided a rapid and powerful tool to amplify specific regions of DNA to identify organisms. Specific primers exist for a number of fungal species including those under investigation as potential biological control agents of plant parasitic nematodes [15–17]. Relatively simple and rapid kit-based DNA extraction methods have allowed DNA sufficiently pure for PCR amplifications to be extracted from a range of soil, plant and clinical samples, making the process both routine and consistent. A selective medium has also been developed for the isolation and quantification of P. lilacinus from soil and plant samples[18], requiring the correct identification of conidia and are less sensitive than other methods. The use of culturable methods for identification and estimates of fungal abundance also requires a level of expertise, and between 3 and 5 days incubation before colonies can be identified and are prone to contamination. DNA based PCR methods, although not providing information on the viability of the fungus do provide a rapid, accurate and sensitive method of detection.
Conventional PCR provides a direct method for identification of the fungus. When it is combined with other methods such as serial dilution plating on a selective medium and/or baiting soil with a host such as nematode eggs the technique can provide a diagnostic method for the abundance and biological control capability of the fungus[17]. Conventional PCR is unable to provide quantitative data on fungal populations, but the development of competitive and real-time PCR has enabled this important advance in molecular diagnostics. Although competitive PCR[19] is quantitative, it requires the construction of a competitor molecule in the reaction, optimisation, and serial dilution of each sample to derive quantification and has been superseded by real-time PCR. TaqMan® based real-time PCR relies on the combination of a primer set and an additional dual-labelled fluorogenic probe to allow continuous monitoring of amplicon synthesis during thermocycling and requires no post-PCR handling for target quantification[20]. It has been used to successfully determine the abundance of a number of fungal species [17,20–25] and to investigate their ecology in soil [17,23,25].
To determine the efficacy of a biological control agent it is important to develop tools to monitor and identify the specific isolate released into the environment and to re-isolate it from soil and its pest target[26]. Several authors [27–29] have investigated the use of arbitrary primers to develop DNA profiles for specific isolates, and to monitor cultures following application in the field. Here, the development and testing of specific PCR primers and a real-time PCR probe and primer set based on the ITS region of the fungal genome are reported to identify, and detect P. lilacinus from nematode, root and soil samples, to investigate the incidence of the fungus in two test soils from the UK, and to determine if the primers can detect both these and medical isolates of the fungus.
2 Materials and methods
2.1 Isolates and media
Fungal cultures were taken from the Rothamsted culture collection or from other sources stated in Tables 1 and 2. Isolates were stored at 4 °C on corn meal agar (CMA-Oxoid, Basingstoke, UK) plates until needed, then sub-cultured onto fresh corn meal agar and incubated at 28 °C. In the pot trials the P. lilacinus isolate used was determined by the nematode host. Where potato cyst nematodes (PCN-Globodera pallida) were added, isolate RES Pl1 was used, whereas where root knot nematodes (Meloidogyne incognita) were added, isolate 251 was used. For clinical isolates, DNA was extracted from pure cultures by Dr. Chris Linton (Mycology Reference Laboratory, Bristol, UK) and supplied for testing.
Fungal isolates and their sources
| Name | Isolate | Substrate | Source | Detection using P. lilacinus specific PCR primers |
| Pochonia chlamydosporia var. chlamydosporia | 10 | Meloidogyne incognita eggs | 1 | – |
| Pochonia chlamydosporia var. catenulate | 392 | Meloidogyne incognita eggs | 2 | – |
| Verticilium dahliae | 327 | Strawberry | 3 | – |
| V. lecanii | 17.3 | Barley | 4 | – |
| Penicillium chrysogenum | Soil | 5 | – | |
| Plectosphaerella cucumerina | Globodera pallida eggs | 7 | – | |
| Pythium sp. | 31.5 | Wheat | 4 | – |
| Aspergillus niger | Soil | 5 | – | |
| A. niger | 8 | – | ||
| A. versicolor | Soil | 5 | – | |
| A. versicolor | 8 | – | ||
| Botrytis sp. | C1650 | Soil | 6 | – |
| Fusarium poae | 24.1 | Wheat | 4 | – |
| Paecilomyces variotii | CNCPF 2825 | Brain abscess | 8 | – |
| Gliocladium roseum | 10.1 | Wheat | 4 | – |
| A. fumigatus | 1 | 8 | – | |
| A. fumigatus | 2 | 8 | – | |
| A. fumigatus | 3 | 8 | – | |
| Non pigmented A. fumagatus | 1 | 8 | – | |
| A. nidulans | 1 | 8 | – | |
| A. flavus | 1 | 8 | – | |
| A. flavus | 2 | 8 | – | |
| A. flavus | 3 | 8 | – | |
| Penicillium sp. | 1 | 8 | – | |
| Penicillium sp. | 2 | 8 | – | |
| Penicillium sp. | 3 | 8 | – | |
| Penicillium sp. | 4 | 8 | – |
| Name | Isolate | Substrate | Source | Detection using P. lilacinus specific PCR primers |
| Pochonia chlamydosporia var. chlamydosporia | 10 | Meloidogyne incognita eggs | 1 | – |
| Pochonia chlamydosporia var. catenulate | 392 | Meloidogyne incognita eggs | 2 | – |
| Verticilium dahliae | 327 | Strawberry | 3 | – |
| V. lecanii | 17.3 | Barley | 4 | – |
| Penicillium chrysogenum | Soil | 5 | – | |
| Plectosphaerella cucumerina | Globodera pallida eggs | 7 | – | |
| Pythium sp. | 31.5 | Wheat | 4 | – |
| Aspergillus niger | Soil | 5 | – | |
| A. niger | 8 | – | ||
| A. versicolor | Soil | 5 | – | |
| A. versicolor | 8 | – | ||
| Botrytis sp. | C1650 | Soil | 6 | – |
| Fusarium poae | 24.1 | Wheat | 4 | – |
| Paecilomyces variotii | CNCPF 2825 | Brain abscess | 8 | – |
| Gliocladium roseum | 10.1 | Wheat | 4 | – |
| A. fumigatus | 1 | 8 | – | |
| A. fumigatus | 2 | 8 | – | |
| A. fumigatus | 3 | 8 | – | |
| Non pigmented A. fumagatus | 1 | 8 | – | |
| A. nidulans | 1 | 8 | – | |
| A. flavus | 1 | 8 | – | |
| A. flavus | 2 | 8 | – | |
| A. flavus | 3 | 8 | – | |
| Penicillium sp. | 1 | 8 | – | |
| Penicillium sp. | 2 | 8 | – | |
| Penicillium sp. | 3 | 8 | – | |
| Penicillium sp. | 4 | 8 | – |
(1) Pochonia chlamydosporia culture collection with the isolate number indicated: Rothamsted Research; (2) Dr. L. Hidalgo, CENSA, Cuba; (3) Dr. D.J. Barbara, HRI Wellesbourne, UK; (4) Dr. W. Dawson, Rothamsted Research, UK; (5) Prof. D.K. Arora, Rothamsted Research, UK; (6) Dr. J. Pell, Rothamsted Research, UK; (7) Dr. S.D. Atkins, Rothamsted Research, UK; (8) Dr. Chris Linton, CNCPF National collection of pathogenic fungi.
Fungal isolates and their sources
| Name | Isolate | Substrate | Source | Detection using P. lilacinus specific PCR primers |
| Pochonia chlamydosporia var. chlamydosporia | 10 | Meloidogyne incognita eggs | 1 | – |
| Pochonia chlamydosporia var. catenulate | 392 | Meloidogyne incognita eggs | 2 | – |
| Verticilium dahliae | 327 | Strawberry | 3 | – |
| V. lecanii | 17.3 | Barley | 4 | – |
| Penicillium chrysogenum | Soil | 5 | – | |
| Plectosphaerella cucumerina | Globodera pallida eggs | 7 | – | |
| Pythium sp. | 31.5 | Wheat | 4 | – |
| Aspergillus niger | Soil | 5 | – | |
| A. niger | 8 | – | ||
| A. versicolor | Soil | 5 | – | |
| A. versicolor | 8 | – | ||
| Botrytis sp. | C1650 | Soil | 6 | – |
| Fusarium poae | 24.1 | Wheat | 4 | – |
| Paecilomyces variotii | CNCPF 2825 | Brain abscess | 8 | – |
| Gliocladium roseum | 10.1 | Wheat | 4 | – |
| A. fumigatus | 1 | 8 | – | |
| A. fumigatus | 2 | 8 | – | |
| A. fumigatus | 3 | 8 | – | |
| Non pigmented A. fumagatus | 1 | 8 | – | |
| A. nidulans | 1 | 8 | – | |
| A. flavus | 1 | 8 | – | |
| A. flavus | 2 | 8 | – | |
| A. flavus | 3 | 8 | – | |
| Penicillium sp. | 1 | 8 | – | |
| Penicillium sp. | 2 | 8 | – | |
| Penicillium sp. | 3 | 8 | – | |
| Penicillium sp. | 4 | 8 | – |
| Name | Isolate | Substrate | Source | Detection using P. lilacinus specific PCR primers |
| Pochonia chlamydosporia var. chlamydosporia | 10 | Meloidogyne incognita eggs | 1 | – |
| Pochonia chlamydosporia var. catenulate | 392 | Meloidogyne incognita eggs | 2 | – |
| Verticilium dahliae | 327 | Strawberry | 3 | – |
| V. lecanii | 17.3 | Barley | 4 | – |
| Penicillium chrysogenum | Soil | 5 | – | |
| Plectosphaerella cucumerina | Globodera pallida eggs | 7 | – | |
| Pythium sp. | 31.5 | Wheat | 4 | – |
| Aspergillus niger | Soil | 5 | – | |
| A. niger | 8 | – | ||
| A. versicolor | Soil | 5 | – | |
| A. versicolor | 8 | – | ||
| Botrytis sp. | C1650 | Soil | 6 | – |
| Fusarium poae | 24.1 | Wheat | 4 | – |
| Paecilomyces variotii | CNCPF 2825 | Brain abscess | 8 | – |
| Gliocladium roseum | 10.1 | Wheat | 4 | – |
| A. fumigatus | 1 | 8 | – | |
| A. fumigatus | 2 | 8 | – | |
| A. fumigatus | 3 | 8 | – | |
| Non pigmented A. fumagatus | 1 | 8 | – | |
| A. nidulans | 1 | 8 | – | |
| A. flavus | 1 | 8 | – | |
| A. flavus | 2 | 8 | – | |
| A. flavus | 3 | 8 | – | |
| Penicillium sp. | 1 | 8 | – | |
| Penicillium sp. | 2 | 8 | – | |
| Penicillium sp. | 3 | 8 | – | |
| Penicillium sp. | 4 | 8 | – |
(1) Pochonia chlamydosporia culture collection with the isolate number indicated: Rothamsted Research; (2) Dr. L. Hidalgo, CENSA, Cuba; (3) Dr. D.J. Barbara, HRI Wellesbourne, UK; (4) Dr. W. Dawson, Rothamsted Research, UK; (5) Prof. D.K. Arora, Rothamsted Research, UK; (6) Dr. J. Pell, Rothamsted Research, UK; (7) Dr. S.D. Atkins, Rothamsted Research, UK; (8) Dr. Chris Linton, CNCPF National collection of pathogenic fungi.
P. lilacinus isolates screened with specific primers
| P. lilacinus isolate | Country of origin/source | Source | Detection using P. lilacinus specific PCR primers |
| 8 | Malaysia | 1 | + |
| 10 | Malaysia | 1 | + |
| 11 | Malaysia | 1 | + |
| 12 | Malaysia | 1 | + |
| 13 | Malaysia | 1 | + |
| 18 | Malaysia | 1 | + |
| 21 | Malaysia | 1 | + |
| 23 | Malaysia | 1 | + |
| 27 | Malaysia | 1 | + |
| 30 | Malaysia | 1 | + |
| 33 | Malaysia | 1 | + |
| 34 | Malaysia | 1 | + |
| 35 | Malaysia | 1 | + |
| 251 | Philippines | 1 | + |
| Pl “Mor” | Russia | 2 | + |
| Pl(Ks) | Poland | 2 | + |
| RES Pl1 | PCN, Jersey, UK | 3 | + |
| CNCPF 2665 | UK/ corneal scraping | 4 | + |
| CNCPF 7507 | UK | 4 | + |
| 1 | 4 | + | |
| 2 | 4 | + | |
| Pl001 | South Africa | 5 | + |
| P. lilacinus isolate | Country of origin/source | Source | Detection using P. lilacinus specific PCR primers |
| 8 | Malaysia | 1 | + |
| 10 | Malaysia | 1 | + |
| 11 | Malaysia | 1 | + |
| 12 | Malaysia | 1 | + |
| 13 | Malaysia | 1 | + |
| 18 | Malaysia | 1 | + |
| 21 | Malaysia | 1 | + |
| 23 | Malaysia | 1 | + |
| 27 | Malaysia | 1 | + |
| 30 | Malaysia | 1 | + |
| 33 | Malaysia | 1 | + |
| 34 | Malaysia | 1 | + |
| 35 | Malaysia | 1 | + |
| 251 | Philippines | 1 | + |
| Pl “Mor” | Russia | 2 | + |
| Pl(Ks) | Poland | 2 | + |
| RES Pl1 | PCN, Jersey, UK | 3 | + |
| CNCPF 2665 | UK/ corneal scraping | 4 | + |
| CNCPF 7507 | UK | 4 | + |
| 1 | 4 | + | |
| 2 | 4 | + | |
| Pl001 | South Africa | 5 | + |
Sources (1) S. Pande, Reading University; (2) Prof. D. Sosnowska, Department of Biocontrol and Quarantine, Institute of Plant Protection, Poznan, Poland; (3) Dr. S.D. Atkins, Rothamsted Research, UK; (4) Dr. Chris Linton CNCPF; (5) Biological Control Products Ltd., South Africa.
P. lilacinus isolates screened with specific primers
| P. lilacinus isolate | Country of origin/source | Source | Detection using P. lilacinus specific PCR primers |
| 8 | Malaysia | 1 | + |
| 10 | Malaysia | 1 | + |
| 11 | Malaysia | 1 | + |
| 12 | Malaysia | 1 | + |
| 13 | Malaysia | 1 | + |
| 18 | Malaysia | 1 | + |
| 21 | Malaysia | 1 | + |
| 23 | Malaysia | 1 | + |
| 27 | Malaysia | 1 | + |
| 30 | Malaysia | 1 | + |
| 33 | Malaysia | 1 | + |
| 34 | Malaysia | 1 | + |
| 35 | Malaysia | 1 | + |
| 251 | Philippines | 1 | + |
| Pl “Mor” | Russia | 2 | + |
| Pl(Ks) | Poland | 2 | + |
| RES Pl1 | PCN, Jersey, UK | 3 | + |
| CNCPF 2665 | UK/ corneal scraping | 4 | + |
| CNCPF 7507 | UK | 4 | + |
| 1 | 4 | + | |
| 2 | 4 | + | |
| Pl001 | South Africa | 5 | + |
| P. lilacinus isolate | Country of origin/source | Source | Detection using P. lilacinus specific PCR primers |
| 8 | Malaysia | 1 | + |
| 10 | Malaysia | 1 | + |
| 11 | Malaysia | 1 | + |
| 12 | Malaysia | 1 | + |
| 13 | Malaysia | 1 | + |
| 18 | Malaysia | 1 | + |
| 21 | Malaysia | 1 | + |
| 23 | Malaysia | 1 | + |
| 27 | Malaysia | 1 | + |
| 30 | Malaysia | 1 | + |
| 33 | Malaysia | 1 | + |
| 34 | Malaysia | 1 | + |
| 35 | Malaysia | 1 | + |
| 251 | Philippines | 1 | + |
| Pl “Mor” | Russia | 2 | + |
| Pl(Ks) | Poland | 2 | + |
| RES Pl1 | PCN, Jersey, UK | 3 | + |
| CNCPF 2665 | UK/ corneal scraping | 4 | + |
| CNCPF 7507 | UK | 4 | + |
| 1 | 4 | + | |
| 2 | 4 | + | |
| Pl001 | South Africa | 5 | + |
Sources (1) S. Pande, Reading University; (2) Prof. D. Sosnowska, Department of Biocontrol and Quarantine, Institute of Plant Protection, Poznan, Poland; (3) Dr. S.D. Atkins, Rothamsted Research, UK; (4) Dr. Chris Linton CNCPF; (5) Biological Control Products Ltd., South Africa.
2.2 Optimisation of species-specific primers
Primer set (PaeF: 5′ CTC AGT TGC CTC GGC GGG AA 3′; PaeR: 5′ GTG CAA CTC AGA GAA GAA ATT CCG 3′) was designed from an ITS sequence submitted to the GenBank/EMBL database (Accession Number AF368804). Primers were compared to other sequences in the database through BLAST and FASTA searches to confirm specificity, and their design was optimised using the NetPrimer software (Biosoft International, http://www.premierbiosoft.com/netprimer.html). PCR reactions of 20 μl contained 10 μl 2 × RedTaq mega mix (Sigma, UK), 2 pmol of each primer and 20 ng template DNA. Optimised PCR conditions were as follows: 95 °C followed by 35 cycles of 94 °C for 1 min, 61 °C for 1 min, 72 °C for 1 min, with a final incubation at 72 °C for 5 min. PCR products were separated on 2% agarose gels.
2.3 Confirmation of P. lilacinus primer specificity using fungal isolates
DNA was extracted from the fungal cultures listed (Tables 1 and 2) using the method described by Klimyuk et al.[30]. DNA from cultures was added to the PCR reagents and the conditions followed as described above.
2.4 Detection of P. lilacinus in soil
DNA was extracted from two soils (Ely, Cambridgeshire and Spalding, Lincolnshire, UK) where it was reported that potato cyst nematode populations were naturally declining[31]. Triplicate DNA samples were extracted from 10 soil cores from each site using a soil DNA extraction kit (Mo-Bio Laboratories Inc, California, USA). All soil DNA extractions were diluted 1:10 before using in PCR reactions. DNA (20 ng) was added to a PCR as above with minor modifications, namely MgCl2 concentration increased from 1.5 to 2.5 mM and the number of cycles increased to 45. As a control, DNA was also extracted from potting compost where P. lilacinus had not been detected. The abundance of the fungus was also estimated at each site by plating soil onto a selective medium as described by Atkins et al.[32].
2.5 Detection of P. lilacinus from plant roots
Moist Terra-green (Oil Dri Corporation of America, USA) was added to Magenta boxes (Sigma) and sterilized by autoclaving for 15 min at 1.01 × 105 N/m2 (15 psi). Potato chits (var. Maris Piper) were added aseptically just below the surface of the Terra-green, above five 5 mm agar plugs taken from the edge of a culture of P. lilacinus RES Pl1 on a CMA plate. The plants were incubated in a growth chamber at 15 °C with 12 h light and dark periods. After three weeks the plants were inoculated with 1000 G. pallida second-stage juveniles. Control non-inoculated potato plants, and plants infected only with PCN were included. After eight weeks, plants were removed from pots and the Terra-green washed gently from the roots with water and cut into 1 cm sections. Genomic DNA was extracted from triplicate 1 g pooled roots samples using the DNeasy™ Plant mini kit (Qiagen). The presence of P. lilacinus was detected by PCR as above using the conditions outlined for detection in soil.
2.6 Detection of P. lilacinus from infected nematode eggs
Sterile sandy loam soil, that was free of P. lilacinus, was inoculated with P. lilacinus isolate 251 at a rate of 5000 spores g−1 soil. The soil was baited with root-knot nematode, M. incognita, females according to the methods described by Atkins et al.[17]. Females were removed from the soil after one week incubation, the eggs separated and plated out onto water agar containing antibiotics (8 g technical agar (Oxoid), 50 mg streptomycin (Sigma), 50 mg chloramphenicol (Sigma), 50 mg chlortetracycline l−1 (Sigma)). After three days, 30 infected eggs were chosen at random to assess the proportion containing P. lilacinus using the PCR based diagnostic primers.
Soil from the two field sites (Ely and Spalding) was sieved through a 5-mm sieve, and 1 kg was put into a 12-cm diameter pot for each treatment. Treatments were duplicated. The soils were baited with 25 G. pallida cysts in accordance to the method outlined by Atkins et al.[17]. A sandy loam soil in which P. lilacinus had not been previously detected was similarly baited with cysts as a control. Baits were collected after two weeks, the cysts crushed to release the eggs, and the eggs collected using standard techniques[33]. Eggs were plated onto water agar containing antibiotics as above. The percentage level of infection was calculated by observing the plates with a dissecting microscope after 2–3 days incubation and recording the level of infected and uninfected eggs. Fifty infected eggs were picked arbitrarily after three days incubation at 21 °C. DNA was extracted from each individual egg using the method described by Klimyuk et al.[30], and the presence of P. lilacinus detected using the specific primer set PaeF and PaeR as described previously. The proportion of eggs infected from each soil was compared to the control and analysed by one way analysis of variance by using the Genstat® program[34].
2.7 Real-time PCR quantification of P. lilacinus from field sites
TaqMan® PCR primers and probes were designed from the same ITS sequence described above, using the Primer Express software (Perkin–Elmer Biosystems, Norwalk, CT, USA), in accordance with the criteria required for real-time PCR primer design. The fluorogenic probe (PLrtP) was labelled at the 5′ end with the fluorogenic reporter dye FAM (6-carboxy-fluorescein), and the 3′ end was modified with the quencher dye TAMRA (6-carboxy-tetramethylrhodamine). The primer and probe sequences were as follows: PLrtF 5′ GAC CCA AAA CTC TTT TTG CAT TAC G 3′; PLrtR 5′ AGA TCC GTT GTT GAA AGT TTT GAT TCA TTT GTT TTG 3′; PLrt P 5′ FAM CCG GCG GAA TTT CTT CTC TGA GTT GC TAMRA 3′. Primers and probe were compared to other sequences in the database through BLAST and FASTA searches to confirm specificity, and tested in soil where P. lilacinus had not been previously detected using PCR.
2.8 Inoculation of soil for standard curve analysis for real-time PCR
An aliquot of sandy loam soil (10 g) was inoculated with 106 conidiospores of P. lilacinus isolates 251. The fungus had not been previously detected in this soil by using selective media or PCR methods prior to inoculation. DNA was extracted from triplicate 0.25 g samples with a soil DNA extraction kit (Mo-Bio). Serial dilutions were prepared from each sample in ultrapure dH2O so that 1 μl represented 1 × 105, 1 × 104 to a final dilution of 10 spores g−1 soil. DNA prepared from the uninoculated soil was used as a negative control.
Real-time PCR was performed in Bioplastics (EU) 96 × 0.2 ml PCR plates capped with Bioplastics EU Optical thin wall 8-cap strip low background (Bioplastics Landgraaf, Netherlands) with the automated ABI Prism 7700 sequence detector (PE Applied Biosystems). Stratagene Brilliant® QPCR Master Mix TaqMan® Kit was used for real-time reactions (Stratagene, Amsterdam, Netherlands). Primer and probe concentrations were optimised according to the manufacture's guide. Primers PLrtF and PLrtR were included at a final concentration of 900 nM each and the TaqMan® probe (PLrtP) was used at 225 nM. Additional MgCl2 was added to increase the concentration to 3.5 mM. Reference dye was added in accordance to the manufacture's recommendations for use with the ABI 7700. The reaction was made up to 20 μl using ultrapure dH2O. The manufacturer's recommended universal thermal cycle protocol (Stratagene) was used for PCR amplification: stage 1; 50 °C for 2 min; stage 2; 95 °C for 10 min (activation of Taq DNA polymerase); and stage 3; 50 cycles of 95 °C for 15 s and 60 °C for 1 min. Reactions were performed in triplicate. An aliquot (1 μl) from each spore dilution was used to calculate the standard curve. Aliquots (20 ng) of DNA were used from each of the soil DNA preparations taken from the field sites at Spalding and Ely. The CT (cycle threshold) value (determined as the cycle number at which statistically significant increases in the reporter fluorescence can be detected) for each PCR was automatically calculated and analysed by the ABI Prism sequence detection software (version 1.5).
2.9 Calculation of ITS copy number in P. lilacinus spores
The ITS gene was cloned from P. lilacinus isolate RES Pl1 into TOPO vector (Invitrogen, Paisley, UK). The plasmid was extracted from a 16 h culture of Escherchia coli using a plasmid mini kit (Qiagen, Crawley, UK). The DNA was quantified and diluted so that 1 μl corresponded to 1 × 103 and 1 × 102 copies. The plasmid was added to a background of soil DNA (20 ng) where P. lilacinus had not been previously detected. Real-time PCR was performed as outlined above for detection of P. lilacinus in soil and the CT values for the ITS products were compared to a standard curve of P. lilacinus spores in soil.
3 Results
3.1 Specificity of P. lilacinus primers for conventional PCR
The specific ITS primers for the detection of P. lilacinus generated a single PCR product of 130 bp. The product was generated from DNA extracted from all the isolates of P. lilacinus (Table 2) but not samples taken from the other fungal cultures (Table 1). The primers generated a PCR fragment of the predicted length from the commercial strain 251 (Table 2), as well as isolates of P. lilacinus from different geographic areas (Table 2). The four human pathogenic isolates (CNCPF 2665; CNCPF 7507, 1 and 2 supplied by Dr. C. Linton) were also detected using these primers, whereas the closely related human pathogen P. variotii (CNCPF 2825) was not amplified (Tables 1 and 2). Isolates of other fungal species, including human pathogens supplied by Dr. C. Linton, were also not detected using the specific primer set, demonstrating its specificity.
3.2 Detection of P. lilacinus from soil and roots
Optimisation of the PCR conditions and reagents was necessary for detection of the fungus from soil and root samples. P. lilacinus was detected in all samples taken from Ely and Spalding by PCR (Fig. 1), whereas it was not detected from the control soil (potting compost). The fungus was detected at a population density of 3000 CFU g−1 soil (±456) at the Spalding site, whereas no colonies were detected on the selective medium from plates inoculated with soil from Ely, suggesting population densities lower than 102 CFU g−1 soil.
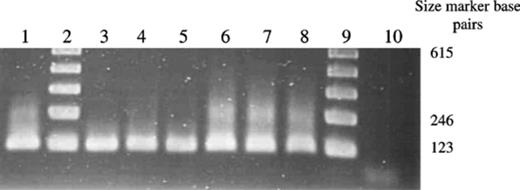
Detection of P. lilacinus in soil using P. lilacinus specific primers. Lane order: Lanes 2 and 9,123 bp marker; Lane 1, Positive control RES Pl1; Lanes 3–5, DNA extracted from soil taken at Ely; Lanes 6–8, DNA extracted from soil taken at Spalding; Lane 10, DNA extracted from control soil.
The proportion of fungal infected eggs in baited cysts incubated in the soil from Ely was 15% (±1.9 n= 8), and for Spalding was similar at 14% (±1.6 n= 8). Both these results were significantly greater than the 9% (±1.09 n= 6) infection of eggs observed in the control (the probability value corresponding to the variance ratio [Fpr] was 0.015 [standard errors of differences, 2.7] at Ely and 0.021 [standard errors of differences, 2.74] at Spalding). However, the fungus infecting the eggs was shown not to be P. lilacinus because the species-specific primer set for conventional PCR did not detect P. lilacinus in any of the G. pallida eggs taken from the baits from the two field sites Ely and Spalding. In contrast, in the control infections using soil inoculated with P. lilacinus isolate 251, 27% of the fungal infected M. incognita eggs taken arbitrarily from the baits gave a positive result for the presence of P. lilacinus. The presence of P. lilacinus RES Pl1 was detected in plant roots inoculated with the fungus with no detectable band resulting from DNA extracted from uninoculated roots, or roots infested with nematodes.
3.3 Real-time quantification of P. lilacinus from field sites
The primers PLrtF and PLrtR generated a PCR product of 94 bp. No fluorescence was recorded in the DNA samples extracted from the negative controls, or from unseeded soil. The CT was 0.038, measuring amplification during the logarithmic phase of the PCR. A standard curve for P. lilacinus was generated by using data derived from the serial dilution of P. lilacinus spores in soil (Fig. 2). The linear correlation coefficient of the standard curve was r2= 0.973, demonstrating the accuracy of the PCR-based quantification. Based on three sample replications, the ABI Prism fluorescence detection system automatically calculated the starting concentrations of P. lilacinus DNA by comparison of the CT values from the unknown samples with those values on the standard curve. Using this method, it was possible to detect populations of 10 spores g−1 soil. The CT values from the Ely samples was significantly lower (Fpr < 0.001 SED 506) than those from Spalding, indicating that the fungal population was smaller at Ely (Fig. 2, Table 3), which corresponded to the data derived from the plating of soil onto the selective medium, but differences in the activity of the fungus as measured by egg parasitism were not found. To determine the relative population size of P. lilacinus in each soil sample, the CT value was compared to the standard curve (Fig. 2). Previous work[17] had demonstrated that PCR was not affected by differences in the soil type between these two sites. The copy number of ITS genes per spore was calculated as two by comparison of the ITS plasmid to the standard curve of spores in soil.
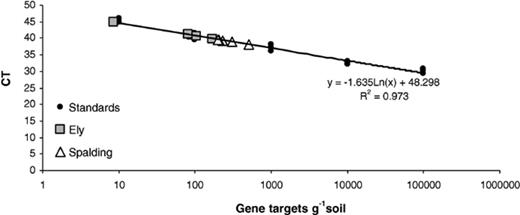
Real-time quantification of P. lilacinus from Ely and Spalding by plotting CT values against the P. lilacinus (RES Pl1) standard curve. CT set at 0.038.
CT values and corresponding gene target levels of P. lilacinus (g−1) soil from samples taken from Ely and Spalding
| Site | CT | Gene target g−1 soil | Mean population | Standard error |
| Ely | 40.9 | 920 | 880 | 162.7 |
| 40.9 | 920 | |||
| 40.7 | 1040 | |||
| 39.9 | 1700 | |||
| 44.8 | 80 | |||
| 41.1 | 810 | |||
| 40.7 | 1030 | |||
| 41.7 | 540 | |||
| Spalding | 38.1 | 5110 | 2981.3 | 478.8 |
| 39.4 | 2310 | |||
| 39.6 | 2040 | |||
| 38.1 | 5110 | |||
| 39.6 | 1990 | |||
| 38.5 | 3040 | |||
| 39.5 | 2120 | |||
| 39.5 | 2130 |
| Site | CT | Gene target g−1 soil | Mean population | Standard error |
| Ely | 40.9 | 920 | 880 | 162.7 |
| 40.9 | 920 | |||
| 40.7 | 1040 | |||
| 39.9 | 1700 | |||
| 44.8 | 80 | |||
| 41.1 | 810 | |||
| 40.7 | 1030 | |||
| 41.7 | 540 | |||
| Spalding | 38.1 | 5110 | 2981.3 | 478.8 |
| 39.4 | 2310 | |||
| 39.6 | 2040 | |||
| 38.1 | 5110 | |||
| 39.6 | 1990 | |||
| 38.5 | 3040 | |||
| 39.5 | 2120 | |||
| 39.5 | 2130 |
Values determined by plotting CT values against the P. lilacinus (RES Pl1) standard curve. CT set at 0.038.
CT values and corresponding gene target levels of P. lilacinus (g−1) soil from samples taken from Ely and Spalding
| Site | CT | Gene target g−1 soil | Mean population | Standard error |
| Ely | 40.9 | 920 | 880 | 162.7 |
| 40.9 | 920 | |||
| 40.7 | 1040 | |||
| 39.9 | 1700 | |||
| 44.8 | 80 | |||
| 41.1 | 810 | |||
| 40.7 | 1030 | |||
| 41.7 | 540 | |||
| Spalding | 38.1 | 5110 | 2981.3 | 478.8 |
| 39.4 | 2310 | |||
| 39.6 | 2040 | |||
| 38.1 | 5110 | |||
| 39.6 | 1990 | |||
| 38.5 | 3040 | |||
| 39.5 | 2120 | |||
| 39.5 | 2130 |
| Site | CT | Gene target g−1 soil | Mean population | Standard error |
| Ely | 40.9 | 920 | 880 | 162.7 |
| 40.9 | 920 | |||
| 40.7 | 1040 | |||
| 39.9 | 1700 | |||
| 44.8 | 80 | |||
| 41.1 | 810 | |||
| 40.7 | 1030 | |||
| 41.7 | 540 | |||
| Spalding | 38.1 | 5110 | 2981.3 | 478.8 |
| 39.4 | 2310 | |||
| 39.6 | 2040 | |||
| 38.1 | 5110 | |||
| 39.6 | 1990 | |||
| 38.5 | 3040 | |||
| 39.5 | 2120 | |||
| 39.5 | 2130 |
Values determined by plotting CT values against the P. lilacinus (RES Pl1) standard curve. CT set at 0.038.
4 Discussion
A rapid, diagnostic test to confirm the presence of P. lilacinus in samples is an essential step in tracking the fungus in the environment. Both for studies of the epidemiology of the fungus, and for registration purposes, it is important to develop tools for the monitoring of isolates. If the fungus fails as a biological control agent in trials it is important to be able to identify whether the inoculum is still present but is ineffective, or failed to establish. Just as important is the ability to demonstrate conclusively that the fungus is able to parasitise the target pest without any significant impact on non-target organisms in the environment.
The ability to detect the fungus in single infected eggs using PCR is an important step forward providing reliable estimates of the efficacy of a specific fungal isolate to control a pest population. The results reported in this paper demonstrate clearly the need for more than one diagnostic protocol when investigating the efficacy of a biological control agent, as P. lilacinus was only detected at Spalding using selective media and, although, P. lilacinus was detected at both the field sites using PCR diagnostics the fungus was shown not to have infected any of the eggs in the baits. Previous work,[17] had shown that the nematophagous fungus Plectosphaerella cucumerina was infecting a significant proportion of the PCN eggs at both the sites tested. Therefore, although P. lilacinus was present in the soil it was unlikely to be having any effects on the potato cyst nematode population at the two field sites investigated. However, in other cases, the fungus has been shown to significantly control nematode populations [1–9] and PCR demonstrated that 27% of the M. incognita eggs sampled in soil inoculated with P. lilacinus isolate 251 were infected with the fungus. This isolate is commercially available as a product to control root-knot nematodes, however, it appears that P. lilacinus isolates from the Spalding and Ely soils would not be efficacious for controlling PCN (although other isolates show different host nematode specificity).
The importance of combining molecular diagnostic tools with other more conventional diagnostic methods such as serial plate dilution, or baiting has already proved of value in demonstrating the activity of specific nematophagous fungi[17]. Linked with other DNA-based processes such as DNA profiling, specific P. lilacinus isolates can be tracked in the environment. The species-specific primers were able to detect all isolates of the fungus tested from different geographic areas and from clinical specimens, as well as the commercial strain 251. However, the primers did not give any amplification product with the closely-related species, Paecilomyces variotii, or other environmental or clinical fungal species, demonstrating potential for detecting the fungus in both clinical and agricultural samples.
The real-time PCR primer and probe set also demonstrated specificity for P. lilacinus. This technique provided a qualitative and quantitative assay for detection of the fungus from soil. Real-time PCR will provide a valuable tool in monitoring fungal spatio-temporal population changes in response to exogenous inputs, including changes in crops, farm practices, chemical inputs, and the presence of nematodes on roots and in soil. This will provide a more accurate assessment of the influence of the environment on the activity of this biological control agent. Adaptation of the real-time technique could provide rapid quantitative data on the incidences of P. lilacinus infecting humans and animals. The primer sets outlined in this paper have already demonstrated their ability to detect medically important isolates of P. lilacinus. Bretagne[10] argued the case for the use of real-time PCR in clinical parasitology and mycology, stating that the use of real-time PCR provides both qualitative and quantitative data rapidly whilst minimising the risk of contamination, and reviews the use of real-time PCR in these fields. At present identification relies on histology and sequencing infective agents. This is a time consuming process and relies on expert identification of the infective agent often leading to a delay in treatment or misidentification. Techniques such as real-time PCR could quickly identify and provide quantitative data on infective agents or confirm their absence, leading to quick and accurate identification and swift treatment.
P. lilacinus has been associated with human infections, and it is important to recognise that handling this fungus may have adverse health implications. The commercial strain 251 has undergone full toxicological and ecological testing for registration, yet other laboratory isolates may not have had this rigorous testing and, therefore, particular care needs to be taken when handling the fungus. The species-specific PCR primers and real-time primers and TaqMan® probe provide rapid diagnostic tests for the fungus which would greatly enhance identification and quantification, and should prove invaluable in screening programmes both for clinical and agricultural purposes.
Acknowledgements
Rothamsted Research receives grant-aided support from the Biotechnology and Biological Sciences Research Council. The work formed part of a DEFRA funded Project HPO1150. Ms. Sonal Pande was funded by a FELIX scholarship at the University of Reading, UK. The authors acknowledge the assistance of Dr. Chris Linton for providing the fungal DNA from clinical isolates, and Prof. D. Sosnowska and Dr. D. Neethling for supplying P. lilacinus isolates.
References

{kind=link}
{kind=link}