





Abstract
A primer pair was designed to selectively amplify a fragment of the Acinetobacter 16S rRNA gene from environmental samples by PCR. 16S rRNA gene products were only obtained in PCRs with DNA from members of the genus Acinetobacter and not with DNA from other bacterial species. Denaturing gradient gel electrophoresis (DGGE) of the Acinetobacter 16S rRNA gene amplicons enabled discrimination between different Acinetobacter species. PCR using the Acinetobacter primer pair allowed detection of Acinetobacter in soil with a detection limit of 104 cells g−1 soil, but attachment of the GC-clamp to the forward primer resulted in a 100-fold decrease in sensitivity. Using a nested PCR approach, the detection limit could be lowered to at least 10 cells g−1 of soil. The method was applied to assess Acinetobacter diversity in soil samples originating from different historically hydrocarbon-contaminated sites. In addition, for one oil-contaminated soil, the dynamics of the Acinetobacter community in response to different treatments was monitored over time in a laboratory biostimulation experimental set-up. In all cases, bands in the DGGE fingerprints were cloned and sequenced. Environmental samples taken from a mineral oil-contaminated site and from a kerosene-contaminated site demonstrated relatively simple Acinetobacter 16S rRNA gene fingerprints with A. lwoffii and A. johnsonii as dominant members. In contrast, soils derived from MTBE- and BTEX-contaminated sites did not harbor detectable Acinetobacter populations. Although Acinetobacter was detected in the soil employed for the biostimulation experiment prior to treatment, substantial changes in its populations were observed depending on the treatment.
1 Introduction
Bacteria belonging to the genus Acinetobacter have gained increasing attention in recent years not only due to their potential to cause severe nosocomial infections [1] but also because of their potential role and application in processes of major environmental importance. Acinetobacter is considered to play an important role in enhanced biological phosphorus removal in wastewater treatment [2]. Otherwise, many environmental isolates of Acinetobacter show hydrocarbon degrading capacities, which are of interest for soil bioremediation. Acinetobacter venetianus RAG-1 [3], Acinetobacter sp. M-1 [4], Acinetobacter sp. ODDK71 [5], and Acinetobacter venetianus spp. C3, C4 and B1/II [6] are able to degrade n-alkanes with carbon chain lengths of C20–C44 while Acinetobacter radioresistens strain S13 is able to degrade aromatic pollutants [7]. Besides their ability to utilize a variety of organic pollutants as carbon source, many Acinetobacter species produce extracellular bioemulsifiers [8]. Bioemulsifiers can be used to enhance oil recovery, are of interest for the detergent industry, and might be used for formation of stable oil-in-water emulsions in the food and cosmetic industries [9]. In addition, bioemulsifiers play a role in the growth of the microorganisms on hydrocarbons [10]. The best studied bioemulsifiers in this context are emulsan produced by Acinetobacter venetianus RAG-1 and alasan produced by Acinetobacter radioresistens KA53. Enhanced aqueous solubility and biodegradation rates of polyaromatic hydrocarbons (PAHs) were observed by Barkay et al. [10] in the presence of alasan. These characteristics make Acinetobacter spp. potential candidates for bioaugmentation of oil-contaminated soil [11]. Maximum biodegradation of petroleum hydrocarbons in an oily-sludge-contaminated soil was observed by Mishra et al. [12] after addition of an hydrocarbon-degrading bacterial inoculum including two A. baumannii strains. In addition, Acinetobacter has been isolated from environments contaminated with petroleum fuels [13,14]. For example Gallego et al. [13] found the prevalence of Acinetobacter spp. while studying the degradation of hydrocarbons in diesel-contaminated soils after supplementing inorganic nutrients or after bioaugmentation with sewage sludge.
Detection of Acinetobacter spp. in environmental samples mostly relies on cultivation-dependent plating techniques [13,14]. In order to provide better insight into the diversity of the Acinetobacter community in environmental habitats and its dynamics in response to changing conditions, there is a need for cultivation-independent techniques enabling a direct and rapid picture of these populations to be established. These techniques comprise molecular methods targeting specifically the Acinetobacter community such as fluorescent in situ hybridization (FISH) and specific PCR. Wagner et al. [2] monitored the in situ presence of members of the genus Acinetobacter in activated sludge based on FISH using a genus-specific rRNA-targeted oligonucleotide probe. This method provides quantitative information on the presence of Acinetobacter cells but failed to monitor the Acinetobacter diversity, as no probe is available specifically targeting different species of Acinetobacter.
In this work, we describe the development of a method based on denaturing gradient gel electrophoresis (DGGE) of PCR-amplified Acinetobacter 16S ribosomal RNA (rRNA) gene fragments to assess directly the diversity of the genus Acinetobacter in environmental samples. The PCR primers were chosen such that they amplify a variable 16S rRNA gene sequence allowing differentiation by DGGE between the different corresponding Acinetobacter species. The method was used to examine the diversity of Acinetobacter populations in hydrocarbon-contaminated soil samples and their dynamics in soil samples undergoing biostimulation.
2 Materials and methods
2.1 Chemicals
The chemicals used in this work were purchased from Merck (Germany). All enzymes were purchased from Sigma–Aldrich Chemie GmbH (Germany). All primers were synthesized by Westburg (The Netherlands) and PCR buffer, dNTPs and polymerase were purchased from TaKaRa (TaKaRa Shuzo Co., Ltd., Biomedical Group, Japan).
2.2 Bacterial strains
The bacterial strains used in this study (Table 1) were routinely grown at 28 °C on 869 rich medium [15]. Acinetobacter spp. were tested for growth on diesel fuel by supplying 50 μl of the hydrocarbon mixture on Tris minimal medium plates prior to inoculation [16].
Bacterial strains used in this study
| Strain | Characteristics | Origin/referencea |
| γ-Proteobacteria | ||
| Acinetobacter baumannii ATCC 19606T | – | ATCC |
| Acinetobacter baumannii DVL5014 | – | [39] |
| Acinetobacter calcoaceticus DSM30006T | Produces isoprene | DSM |
| Acinetobacter sp. LH168 | Grows on diesel fuel | [40] |
| Acinetobacter haemolyticus DSM6962T | – | DSM |
| Acinetobacter johnsonii DSM6963T | – | DSM |
| Acinetobacter junii DSM6964T | – | DSM |
| Acinetobacter lwoffii DSM2403T | – | DSM |
| Acinetobacter radioresistens DSM6976 T | Resistant to radiation, biosurfactant production | DSM |
| Acinetobacter sp. BD4 (DSM586) | Used for natural transformation, biosynthesis of polysaccharide capsule | DSM |
| Acinetobacter sp. PCI-3 (ATCC10153) | Produces sorbose | ATCC |
| Acinetobacter sp. R (ATCC27197) | Produces glutamine-asparaginase | ATCC |
| Acinetobacter sp. DSM11652 | Fixes CO2 | DSM |
| Acinetobacter venetianus RAG-1 (ATCC31012) | Grows on diesel fuel, produces emulsan | ATCC |
| Aeromonas enteropelogenes DSM6394T | – | DSM |
| Escherichia coli CM2529 | – | [41] |
| Moraxella sp. SK12 | Grows on biphenyl | [42] |
| Pseudomonas corrugata LH120 | Grows on diesel fuel and naphthalene | [40] |
| Pseudomonas fluorescens VM380 | Transforms phenanthrene | K. Vanbroekhoven, unpublished |
| Pseudomonas mendocina AS302 | Resistant to heavy metals | L. Diels, unpublished |
| Pseudomonas putida BN10 | Grows on naphthalene | [43] |
| Xanthomonas campestris DSM1350 | – | DSM |
| α-Proteobacteria | ||
| Sphingomonas sp. LH128 | Grows on phenanthrene | [40] |
| Sphingomonas sp. EPA505 (DSM7526) | Grows on fluoranthene | DSM |
| β-Proteobacteria | ||
| Burkholderia sp. JS150 (DSM8530) | Grows on BTEX | DSM |
| Ralstonia metallidurans CH34 | Resistant to cadmium, cobalt and zinc ions | DSM |
| δ-Proteobacteria | ||
| Desulfovibrio desulfuricans DSM642 | Recovery of elemental palladium | DSM |
| Desulfomicrobium norvegicum DSM1741 | Utilizes sulphur | DSM |
| Actinomycetes | ||
| Mycobacterium sp. LB501T | Grows on anthracene | [16] |
| Mycobacterium sp. VM552 | Grows on pyrene and phenantrene | K. Vanbroekhoven, unpublished |
| Rhodococcus globerulus P6 | Grows on biphenyl | [44] |
| Strain | Characteristics | Origin/referencea |
| γ-Proteobacteria | ||
| Acinetobacter baumannii ATCC 19606T | – | ATCC |
| Acinetobacter baumannii DVL5014 | – | [39] |
| Acinetobacter calcoaceticus DSM30006T | Produces isoprene | DSM |
| Acinetobacter sp. LH168 | Grows on diesel fuel | [40] |
| Acinetobacter haemolyticus DSM6962T | – | DSM |
| Acinetobacter johnsonii DSM6963T | – | DSM |
| Acinetobacter junii DSM6964T | – | DSM |
| Acinetobacter lwoffii DSM2403T | – | DSM |
| Acinetobacter radioresistens DSM6976 T | Resistant to radiation, biosurfactant production | DSM |
| Acinetobacter sp. BD4 (DSM586) | Used for natural transformation, biosynthesis of polysaccharide capsule | DSM |
| Acinetobacter sp. PCI-3 (ATCC10153) | Produces sorbose | ATCC |
| Acinetobacter sp. R (ATCC27197) | Produces glutamine-asparaginase | ATCC |
| Acinetobacter sp. DSM11652 | Fixes CO2 | DSM |
| Acinetobacter venetianus RAG-1 (ATCC31012) | Grows on diesel fuel, produces emulsan | ATCC |
| Aeromonas enteropelogenes DSM6394T | – | DSM |
| Escherichia coli CM2529 | – | [41] |
| Moraxella sp. SK12 | Grows on biphenyl | [42] |
| Pseudomonas corrugata LH120 | Grows on diesel fuel and naphthalene | [40] |
| Pseudomonas fluorescens VM380 | Transforms phenanthrene | K. Vanbroekhoven, unpublished |
| Pseudomonas mendocina AS302 | Resistant to heavy metals | L. Diels, unpublished |
| Pseudomonas putida BN10 | Grows on naphthalene | [43] |
| Xanthomonas campestris DSM1350 | – | DSM |
| α-Proteobacteria | ||
| Sphingomonas sp. LH128 | Grows on phenanthrene | [40] |
| Sphingomonas sp. EPA505 (DSM7526) | Grows on fluoranthene | DSM |
| β-Proteobacteria | ||
| Burkholderia sp. JS150 (DSM8530) | Grows on BTEX | DSM |
| Ralstonia metallidurans CH34 | Resistant to cadmium, cobalt and zinc ions | DSM |
| δ-Proteobacteria | ||
| Desulfovibrio desulfuricans DSM642 | Recovery of elemental palladium | DSM |
| Desulfomicrobium norvegicum DSM1741 | Utilizes sulphur | DSM |
| Actinomycetes | ||
| Mycobacterium sp. LB501T | Grows on anthracene | [16] |
| Mycobacterium sp. VM552 | Grows on pyrene and phenantrene | K. Vanbroekhoven, unpublished |
| Rhodococcus globerulus P6 | Grows on biphenyl | [44] |
aT refers to Type strain; ATCC: American Type Culture Collection, USA; DSM: Deutsche Sammlung von Mikroorganismen, Germany.
Bacterial strains used in this study
| Strain | Characteristics | Origin/referencea |
| γ-Proteobacteria | ||
| Acinetobacter baumannii ATCC 19606T | – | ATCC |
| Acinetobacter baumannii DVL5014 | – | [39] |
| Acinetobacter calcoaceticus DSM30006T | Produces isoprene | DSM |
| Acinetobacter sp. LH168 | Grows on diesel fuel | [40] |
| Acinetobacter haemolyticus DSM6962T | – | DSM |
| Acinetobacter johnsonii DSM6963T | – | DSM |
| Acinetobacter junii DSM6964T | – | DSM |
| Acinetobacter lwoffii DSM2403T | – | DSM |
| Acinetobacter radioresistens DSM6976 T | Resistant to radiation, biosurfactant production | DSM |
| Acinetobacter sp. BD4 (DSM586) | Used for natural transformation, biosynthesis of polysaccharide capsule | DSM |
| Acinetobacter sp. PCI-3 (ATCC10153) | Produces sorbose | ATCC |
| Acinetobacter sp. R (ATCC27197) | Produces glutamine-asparaginase | ATCC |
| Acinetobacter sp. DSM11652 | Fixes CO2 | DSM |
| Acinetobacter venetianus RAG-1 (ATCC31012) | Grows on diesel fuel, produces emulsan | ATCC |
| Aeromonas enteropelogenes DSM6394T | – | DSM |
| Escherichia coli CM2529 | – | [41] |
| Moraxella sp. SK12 | Grows on biphenyl | [42] |
| Pseudomonas corrugata LH120 | Grows on diesel fuel and naphthalene | [40] |
| Pseudomonas fluorescens VM380 | Transforms phenanthrene | K. Vanbroekhoven, unpublished |
| Pseudomonas mendocina AS302 | Resistant to heavy metals | L. Diels, unpublished |
| Pseudomonas putida BN10 | Grows on naphthalene | [43] |
| Xanthomonas campestris DSM1350 | – | DSM |
| α-Proteobacteria | ||
| Sphingomonas sp. LH128 | Grows on phenanthrene | [40] |
| Sphingomonas sp. EPA505 (DSM7526) | Grows on fluoranthene | DSM |
| β-Proteobacteria | ||
| Burkholderia sp. JS150 (DSM8530) | Grows on BTEX | DSM |
| Ralstonia metallidurans CH34 | Resistant to cadmium, cobalt and zinc ions | DSM |
| δ-Proteobacteria | ||
| Desulfovibrio desulfuricans DSM642 | Recovery of elemental palladium | DSM |
| Desulfomicrobium norvegicum DSM1741 | Utilizes sulphur | DSM |
| Actinomycetes | ||
| Mycobacterium sp. LB501T | Grows on anthracene | [16] |
| Mycobacterium sp. VM552 | Grows on pyrene and phenantrene | K. Vanbroekhoven, unpublished |
| Rhodococcus globerulus P6 | Grows on biphenyl | [44] |
| Strain | Characteristics | Origin/referencea |
| γ-Proteobacteria | ||
| Acinetobacter baumannii ATCC 19606T | – | ATCC |
| Acinetobacter baumannii DVL5014 | – | [39] |
| Acinetobacter calcoaceticus DSM30006T | Produces isoprene | DSM |
| Acinetobacter sp. LH168 | Grows on diesel fuel | [40] |
| Acinetobacter haemolyticus DSM6962T | – | DSM |
| Acinetobacter johnsonii DSM6963T | – | DSM |
| Acinetobacter junii DSM6964T | – | DSM |
| Acinetobacter lwoffii DSM2403T | – | DSM |
| Acinetobacter radioresistens DSM6976 T | Resistant to radiation, biosurfactant production | DSM |
| Acinetobacter sp. BD4 (DSM586) | Used for natural transformation, biosynthesis of polysaccharide capsule | DSM |
| Acinetobacter sp. PCI-3 (ATCC10153) | Produces sorbose | ATCC |
| Acinetobacter sp. R (ATCC27197) | Produces glutamine-asparaginase | ATCC |
| Acinetobacter sp. DSM11652 | Fixes CO2 | DSM |
| Acinetobacter venetianus RAG-1 (ATCC31012) | Grows on diesel fuel, produces emulsan | ATCC |
| Aeromonas enteropelogenes DSM6394T | – | DSM |
| Escherichia coli CM2529 | – | [41] |
| Moraxella sp. SK12 | Grows on biphenyl | [42] |
| Pseudomonas corrugata LH120 | Grows on diesel fuel and naphthalene | [40] |
| Pseudomonas fluorescens VM380 | Transforms phenanthrene | K. Vanbroekhoven, unpublished |
| Pseudomonas mendocina AS302 | Resistant to heavy metals | L. Diels, unpublished |
| Pseudomonas putida BN10 | Grows on naphthalene | [43] |
| Xanthomonas campestris DSM1350 | – | DSM |
| α-Proteobacteria | ||
| Sphingomonas sp. LH128 | Grows on phenanthrene | [40] |
| Sphingomonas sp. EPA505 (DSM7526) | Grows on fluoranthene | DSM |
| β-Proteobacteria | ||
| Burkholderia sp. JS150 (DSM8530) | Grows on BTEX | DSM |
| Ralstonia metallidurans CH34 | Resistant to cadmium, cobalt and zinc ions | DSM |
| δ-Proteobacteria | ||
| Desulfovibrio desulfuricans DSM642 | Recovery of elemental palladium | DSM |
| Desulfomicrobium norvegicum DSM1741 | Utilizes sulphur | DSM |
| Actinomycetes | ||
| Mycobacterium sp. LB501T | Grows on anthracene | [16] |
| Mycobacterium sp. VM552 | Grows on pyrene and phenantrene | K. Vanbroekhoven, unpublished |
| Rhodococcus globerulus P6 | Grows on biphenyl | [44] |
aT refers to Type strain; ATCC: American Type Culture Collection, USA; DSM: Deutsche Sammlung von Mikroorganismen, Germany.
2.3 Hydrocarbon-contaminated soil samples
Soil samples derived from four different fuel-contaminated sites, designated as sites B, P, S and F were examined with regard to Acinetobacter population diversity. Four aquifer samples (fine sand, moisture content 20.5%, pH 8.4), i.e., B4, B13, B15 and B22 originated from the kerosene-contaminated site B located near Brussels, Belgium. Sample B13 contained benzene/toluene/ethylbenzene/xylene (BTEX) at a concentration of 0.4 mg kg−1, while sample B15 contained BTEX at a concentration of 16 mg kg−1 and mineral oil at a concentration of 650 mg kg−1. Samples B4 and B22 contained no detectable levels of BTEX (< 0.5 mg kg−1) and mineral oil (< 50 mg kg−1). Aquifer samples PB1, PB2, PB3 and PB4 (silty sand, moisture content 14.5%, pH 6.3) originated from site P (Geel, Belgium) contaminated with methyl tert-butyl ether (MTBE) and benzene. Samples PB1 and PB2 originated from a location at the outbreak of the plume and samples PB3 and PB4 from a location in the centre of the plume. Sample PB1 contained 1000 mg MTBE kg−1 and 20 mg benzene kg−1 and originated from a depth of 18 m. Sample PB2 contained solely MTBE at a concentration of 600 mg kg−1 and was sampled at a depth of 15 m. Sample PB3, originating from a depth of 10 m, was contaminated with 900 mg MTBE kg−1 and 14 mg benzene kg−1, whereas sample PB4, originating from a depth of 14 m, contained no detectable benzene or MTBE concentrations. Six vadose soil samples (clay containing fine sand, moisture content 13.1%, pH 7.7), i.e., S427, S428, S429, S430, S431, and S432 were obtained from Site S (Gent, Belgium), contaminated with mineral oil. All samples contained mineral oil at concentrations of about 6000 mg kg−1. Samples S427 and S430 originated from the soil top layer (< 0.4 m), samples S428 and S431 were taken at depths between 0.4 and 1.2 m, and samples S429 and S432 originated from depths between 1.2 and 1.7 m. Soil sample F (sandy, moisture content 21%, pH 7.3) was derived from the capillary fringe of an oil refinery site near Antwerp, Belgium and was contaminated with 8000 mg mineral oil kg−1.
2.4 Hydrocarbon decontamination in a laboratory microcosm experiment
Microcosms were set up in which soil F samples were treated by applying different nutrient regimes and/or oil concentrations (Table 2). The original soil samples contained 8000 mg mineral oil kg−1. In some microcosms, up to 70,000 mg kg−1 diesel fuel containing dissolved PAHs was added. Mineral nutrients consisting of NH4NO3 and K4P2O7 (Boucquillon, Belgium) dissolved in sterile H2O were added to obtain a final C:N:P ratio of 100:10:1 (w). Oleophilic nutrient, i.e., Inipol EAP-22 was applied at 10% (w) of the mineral oil content present in the samples. The Acinetobacter population was examined before treatment and after 32 (T1), 140 (T3), 237 (T4), 302 (T5), 385 (T6) and 645 (T9) days of treatment.
Treatment of soil F in soil microcosms in the laboratory biostimulation experiment
| Microcosm | Hydrocarbon content (mg kg−1) | Biostimulation treatment | |
| Mineral oil | PAHa | ||
| F | 8000 | 2.5 | – |
| FMN | 8000 | 2.5 | Mineral nutrients |
| F70MN | 70,000 | 112.5 | Mineral nutrients |
| F70HNa | 70,000 | 112.5 | Oleophilic nutrient and aeration |
| Microcosm | Hydrocarbon content (mg kg−1) | Biostimulation treatment | |
| Mineral oil | PAHa | ||
| F | 8000 | 2.5 | – |
| FMN | 8000 | 2.5 | Mineral nutrients |
| F70MN | 70,000 | 112.5 | Mineral nutrients |
| F70HNa | 70,000 | 112.5 | Oleophilic nutrient and aeration |
aPAH content is the sum content of five PAHs added, i.e., phenanthrene, fluorene, fluoranthene, anthracene, and pyrene.
Treatment of soil F in soil microcosms in the laboratory biostimulation experiment
| Microcosm | Hydrocarbon content (mg kg−1) | Biostimulation treatment | |
| Mineral oil | PAHa | ||
| F | 8000 | 2.5 | – |
| FMN | 8000 | 2.5 | Mineral nutrients |
| F70MN | 70,000 | 112.5 | Mineral nutrients |
| F70HNa | 70,000 | 112.5 | Oleophilic nutrient and aeration |
| Microcosm | Hydrocarbon content (mg kg−1) | Biostimulation treatment | |
| Mineral oil | PAHa | ||
| F | 8000 | 2.5 | – |
| FMN | 8000 | 2.5 | Mineral nutrients |
| F70MN | 70,000 | 112.5 | Mineral nutrients |
| F70HNa | 70,000 | 112.5 | Oleophilic nutrient and aeration |
aPAH content is the sum content of five PAHs added, i.e., phenanthrene, fluorene, fluoranthene, anthracene, and pyrene.
2.5 Design of an Acinetobacter 16S rRNA gene specific primer pair
A multiple alignment of Acinetobacter 16S rRNA gene sequences was constructed using the GCG Wisconsin Package (Accelrys Inc., USA). The sequences used represented all known classified Acinetobacter species. Most of the sequences used were retrieved from GenBank (National Centre for Biotechnology Information; http://www.ncbi.nlm.nih.gov/) except for the 16S rRNA gene sequences of A. venetianus RAG-1 and Acinetobacter sp. LH168, which were sequenced in our laboratory in the course of this study. Regions of 100% homology between members of the genus Acinetobacter were searched visually for primer design. The chosen specific primer set consisted of the forward primer Ac436f (5′ TTT AAG CGA GGA GGA GG 3′) and the reverse primer Ac676r (5′ ATT CTA CCA TCC TCT CCC 3′), numbered according to E. coli positioning. The selectivity of the Ac primer set was evaluated using Advanced Blast Search program (GenBank, NCBI, USA) and the Sequence Match program from Ribosomal Database Project (RDP II, USA). A 40 bp long GC rich sequence (5′ CGC CCG CCG CGC GCG GCG GGC GGG GCG GGG GCA CGG GGG G 3′) was attached to the 5′ end of the Ac436f primer in order to allow separation of the 240 bp amplified fragments by DGGE.
2.6 Total DNA extraction from axenic cultures and soil samples
Total genomic DNA was extracted from purified bacterial strains grown in 5 ml 869 medium by a protocol adapted from Krin et al. [17]. Before lysis with proteinase K and SDS, cells were subjected to a lysis step using lysozyme (2 mg ml−1) in TE buffer (10 mM Tris–HCl, 1 mM EDTA, pH 8) and RNAse (10 mg ml−1). This suspension was incubated at 37 °C for 10 min. After precipitating the genomic DNA, air dried pellets were resuspended in 500 μl bidistilled sterile H2O. For PCR purposes, the DNA concentration in the extract was measured spectrophotometrically (Ultrospec 3000UV/Visible, Pharmacia Biotech, UK) and adjusted to 100 ng μl−1.
Nucleic acids from 1 g soil were extracted according to the protocol described by Boon et al. [18]. The DNA pellet was resuspended in TE buffer and co-precipitated impurities were removed using Wizard clean-up columns (Wizard DNA Clean-up System, Promega, USA).
2.7 PCR amplification
Acinetobacter 16S rRNA gene PCR amplification (T3 thermocycler, Biometra, Germany) using primer set Ac436f and Ac676r was performed in a reaction volume of 50 μl using the following time-temperature profile. A short denaturation for 15 s at 95 °C was followed by 50 cycles of denaturation for 3 s at 95 °C, annealing for 10 s at 58 °C and elongation for 30 s at 74 °C. A final extension step of 2 min at 74 °C was included. The PCR mixture used contained 0.5 μg primer Ac436f or GC-Ac436f, 0.25 μg primer Ac676r, 200 μM of each deoxynucleoside triphosphate, 1.25 U ex Taq DNA polymerase, 5 μl 10×ex Taq buffer, and bidistilled sterile water. Approximately 100 ng of pure strain DNA or total soil DNA was added to the PCR mixture. General bacterial 16S rRNA gene primers 27f (5′ AGA GTT TGA TCC TGG CTC AG 3′) and 1492r (5′ TAC GGY TAC CTT GTT ACG ACT T 3′) were applied in the first round of the nested PCR approach as described by Johnson [19]. One microlitre DNA generated during the first PCR round was used as template for the second PCR round using primers GCAc436f and Ac676r. The quality of the soil DNA for PCR purposes was assessed by amplification of an eubacterial 16S rRNA gene fragment from the soil DNA extracts using primers GC-63f and 518r as described by El-Fantroussi et al. [20].
2.8 Denaturing gradient gel electrophoresis (DGGE)
DGGE based on the protocol described by Muyzer et al. [21] was performed on an Ingeny phor U-2 apparatus (Ingeny International BV, The Netherlands). An 8% (wt vol−1) polyacrylamide gel in Tris–Acetate–EDTA (TAE) buffer (Bio-Rad Laboratories, Germany) with a denaturing gradient of 30–60% (where 100% denaturant contains 7 M urea and 40% formamide) was loaded either with 2.5 μl PCR product derived from pure strain DNA or 10 μl PCR products derived from soil DNA. Optimal denaturing conditions were defined based on the theoretical melting temperatures of amplification fragments that were calculated with the Melt program (Melt Analysis Software, Version 1.0.1, Ingeny International BV, The Netherlands). Electrophoresis was performed for 14 h at 55 °C at a constant voltage of 130V. Gels were soaked in a fixation buffer (1× TAE containing 0.5% (v/v) acetic acid) and stained for 30 min in a TAE solution of 1× Sybr Gold nucleic acid stain (Molecular probes Europe BV, The Netherlands). The gels were photographed under UV light using a digital camera system with image software (Image Master VDS&Liscap Image Capture 1.0, Pharmacia Biotech, UK). Analysis of DGGE profiles was done with the Bionumerics software 2.5 (Applied Maths, Belgium).
2.9 Sensitivity of the genus-specific PCR detection
The sensitivity of the Acinetobacter genus-specific PCR detection system was evaluated by applying PCR on (i) a dilution series of genomic DNA of strain RAG-1, and (ii) DNA extracts from soils inoculated with different concentrations of strains RAG-1 or LH168. Cells were precultured on rich medium 869 until OD reached 0.5, washed twice in 0.01 M MgSO4 and used for either direct DNA extraction, or inoculation of soil samples. For the latter, strains A. venetianus RAG-1 and Acinetobacter sp. LH168 were added separately to soil F prior to DNA extraction at cell concentrations of approximately 108, 107, 106, 105, 104, 103, 102, 10 and 1 CFU g−1 soil. Extraction of genomic DNA from the culture and from soil was performed as described above. DNA from soil was extracted after incubation for 0.5 h at room temperature. The DNA concentration of the cell culture containing 108 cells was determined spectrophotometrically to be 24 μg ml−1. Genomic DNA and total soil DNA were subsequently used as template in the direct, semi-nested and nested PCR approaches as described above. PCR products were analyzed by agarose gel electrophoresis.
2.10 Cloning and sequencing of soil-amplified SSU rRNA fragments
PCR fragments obtained from soil DNA by applying the Acinetobacter specific primer pair, were cloned into pCR4-TOPO vector using the TOPO Cloning Kit as described in the manufacturer's protocol (Invitrogen SA, Belgium) without prior concentration or purification of the PCR products. Clones were randomly selected and examined for the presence of the appropriate 240 bp fragment by means of PCR using the specific Acinetobacter primers. The DGGE profile of soil clones generating appropriate fragments were compared with the initial Acinetobacter community fingerprints using DGGE. The fragments were sequenced by Westburg (Westburg, The Netherlands). The clone sequences were compared to GenBank using the advanced Blastn Search program (http://www.ncbi.nlm.nih.gov/blast).
2.11 Nucleotide sequence accession numbers
Partial Acinetobacter 16S rRNA gene sequences derived from soil obtained in this work have been deposited in the GenBank database under Accession Nos. AY207013 to AY207027. The full 16S rRNA gene sequences of strains LH168 and RAG-1 have been deposited as Accession Nos. AF542962 and AF542963, and partial 16S rRNA gene sequences of Acinetobacter strains ATCC10163, ATCC27197, DSM11652, and DSM586 as Accession Nos. AY221635 to AY221638.
3 Results
3.1 Design of a 16S rRNA gene specific primer pair for specific PCR detection of Acinetobacter sp
16S rRNA gene sequences of 33 Acinetobacter spp. were compared to 16S rRNA genes of 13 other γ-proteobacterial bacteria and to sequences of three sphingomonadaceae, belonging to the class of α-proteobacteria. A 17 bp forward oligomer Ac436f (E. coli numbering 436–452) located in the V3 variable region of the prokaryotic SSU rRNA and an 18 bp reverse primer Ac676r (E. coli numbering 676–659) positioned in the V4 variable region were selected. The Ac436f sequence showed 100% identity to 30 of the aligned Acinetobacter 16S rRNA gene sequences but had a one base pair mismatch at the 3′ end of the oligomer of the sequences of Acinetobacter baumannii strain ATCC 19606T, and Acinetobacter sp. ATCC 11171 and ATCC 17905 (Accession Nos. Z93435 (Table 3), Z93444 and Z93447, respectively). However, other A. baumannii 16S rRNA gene sequences (Accession Nos. X81660 and AJ247197) obtained from A. baumanni DSM30007, which is identical to ATCC19606T, showed 100% identity to the primer sequence. All 33 aligned Acinetobacter 16S rRNA gene sequences showed 100% identity to the reverse primer. From the 100 hits produced with Blast (n= 100), primer Ac436f was 100% identical to the corresponding region of the SSU rRNA sequences of 46 Acinetobacter sp. isolates and of 11 uncultured putative Acinetobacter spp. Of the remaining 43 16S rRNA gene sequences (also 100% identical to the Ac436f oligonucleotide sequence), 35 were sequences of uncultured putative γ-proteobacteria and the other eight sequences originated from a series of unidentified bacteria such as glacial ice bacteria, soil bacteria, one marine bacterium, and one γ-proteobacterium.
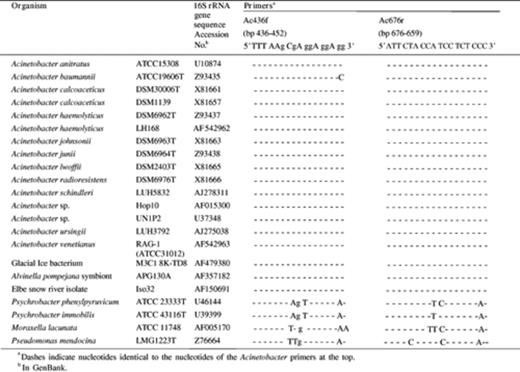
16S rRNA gene sequence homology between the designed Acinetobacter genus-specific primers and corresponding 16S rRNA gene regions of Acinetobacter species and other γ-proteobacterial strains
Similarly, results from Sequence Match showed that 17 of the 20 hits obtained were 16S rRNA gene sequences of Acinetobacter spp. and the remaining three corresponded to as yet unidentified bacteria. Blast results for primer Ac676r showed that 68 sequences of the produced alignments (100 hits) scored as Acinetobacter 16S rRNA genes, an additional nine SSU rRNA genes corresponded to uncultured Acinetobacter clones and 16 corresponded to uncultured γ-proteobacteria. Of the remaining seven sequences, four were sequences from unidentified glacial ice bacteria; one corresponded to the earlier mentioned marine bacterium, one to an Elbe river snow isolate and one to an Alvinella pompejana symbiont. All sequences showed 100% similarity to the Ac676r sequence. Based on total 16S rRNA gene sequence homology, these uncultured and unidentified bacteria might all be affiliated with the Acinetobacter genus. With Sequence Match, the 18-mer reverse primer resulted in 20 hits of which all were Acinetobacter 16S rRNA gene sequences.
The specificity of PCR amplification using the Acinetobacter primer pair was tested using DNA template from Acinetobacter species and other bacterial species (Table 1). Products of the expected size (240 bp) were only obtained with DNA from strains belonging to the Acinetobacter genus (data not shown) including A. baumannii strains DVL5014 and ATCC19606T.
3.2 Potential of the PCR–DGGE Acinetobacter specific system to differentiate between different Acinetobacter species
Primer Ac436f was combined with a 40 bp GC-clamp for DGGE purposes and used in combination with primer Ac676r to amplify 16S rRNA gene fragments from eight of the 10 known Acinetobacter species. Presently, the genus Acinetobacter contains nine species described in the Approved List of Bacterial Names, including A. calcoaceticus, A. baumannii, A. haemolyticus, A. johnsonii, A. junii, A. lwoffii, A. schindleri, A. ursingii and A. radioresistens. Di Cello et al. [6] recently introduced the species ‘Acinetobacter venetianus’ which is not yet in the Approved List of Bacterial Names. According to Vaneechoutte et al. [22]Acinetobacter strain RAG-1 also belongs to this new species. PCR products were analyzed by DGGE to examine whether or not different species or strains generated a different profile. As shown in Fig. 1, amplicons from different species could indeed be discriminated from each other by DGGE suggesting that this method might allow a rapid estimation of Acinetobacter diversity and possibly direct identification at the species level of the detected strains in environmental samples. 16S rRNA gene products of Acinetobacter strains belonging to the same species and displaying highly similar SSU RNA sequences showed a similar melting behavior and co-migrated on DGGE. Strains LH168 and A. haemolyticus DSM6962T, both displaying 98% similarity for their full-length 16S rRNA gene (Emboss, matcher), generated the same pattern. One strain, A. junii DSM6964T generated multiple DGGE bands and doublet bands were generated by A. johnsonii DSM6963T and A. lwoffii DSM2403T.
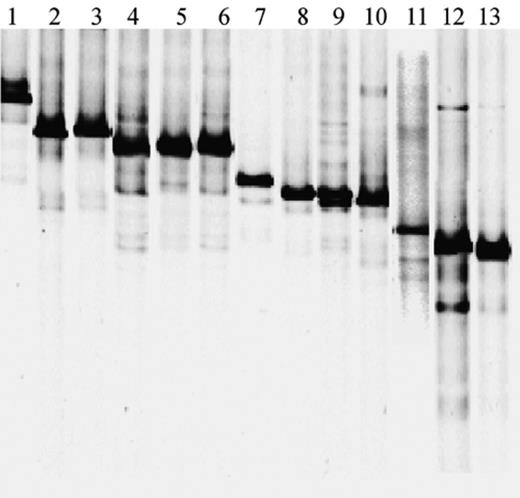
PCR–DGGE profiles of Acinetobacter 16S rRNA gene amplicons obtained from pure strains using the Acinetobacter specific primers GC-Ac436f and Ac676r. Lanes: 1, A. johnsonii DSM6963T; 2, A. calcoaceticus DSM30006T; 3, Acinetobacter sp. ATCC10153; 4, A. haemolyticus LH168; 5, Acinetobacter sp. ATCC27197; 6, A. haemolyticus DSM6962T; 7, A. venetianus RAG-1; 8, A. radioresistens DSM6976T; 9, A. lwoffii DSM2403T; 10, Acinetobacter sp. DSM586; 11, A. baumannii DVL5014, 12, A. junii DSM6964T; 13, Acinetobacter sp. DSM11652.
3.3 Sensitivity of Acinetobacter specific PCR
In order to determine the detection limit of the PCR system, two Acinetobacter strains RAG-1 and LH168 were separately inoculated into soil prior to DNA extraction. Both strains were introduced in sterilized sand at cell concentrations of 108 to 1 CFU g−1 soil. In addition, PCR was applied to decreasing concentrations of pure genomic DNA extracted from strain RAG-1. PCR amplification using the genus-specific Acinetobacter primer pair without GC clamp enabled detection down to introduced cell densities of 104 g−1 soil and to 2.4 pg of pure genomic DNA. A decrease in the detection limit was observed when the GC rich 40-mer was clamped to primer Ac436f leading to detection limit of 106 cells g−1 soil (Fig. 2(a)) and to 0.24 ng pure genomic DNA. No improvement in sensitivity was made when using a semi-nested PCR approach consisting of a first PCR amplification round using primers Ac436f and Ac676r followed by PCR amplification with primer pair GC-Ac436f/Ac676r and PCR products of the first round as template. However, improvements were made using a nested PCR approach with first round primers 27f and 1472r and a second round primers GC-Ac36f and Ac676r. This approach enabled detection of introduced cells down to approximately 10 cells g−1 soil (Fig. 2(b)) or to 0.24 fg of genomic DNA. However, when high numbers of cells were added, some non-specific DNA fragments were obtained (Fig. 2(b)).
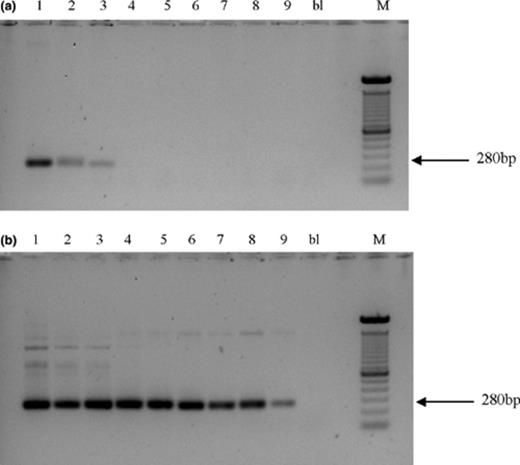
Amplification of 16S rRNA gene of Acinetobacter sp. RAG-1 introduced in soil samples using direct PCR with primers GC-Ac436f and Ac676r (a), and using the nested PCR approach (b). Legend: 108 CFU/g soil (1), 107 CFU/g soil (2), 106 CFU/g soil (3), 105 CFU/g soil (4), 104 CFU/g soil (5), 103 CFU/g soil (6), 102 CFU/g soil (7), 101 CFU/g soil (8), 1 CFU/g soil (9), blank (bl), and 100 bp marker (M).
3.4 Detection and diversity of Acinetobacter spp. in non-treated historically hydrocarbon-contaminated soils
Hydrocarbon-contaminated soil samples from different sites were examined for the presence of dominant Acinetobacter populations using the direct PCR approach applying primer pair GC-Ac436f and Ac676r followed by DGGE diversity analysis of the amplicons (Fig. 3(a)). For some soil samples, i.e., soils S429, S431, B4, and the site P samples, the presence of Acinetobacter and its diversity were also addressed using the nested-PCR approach (Fig. 3(b)).
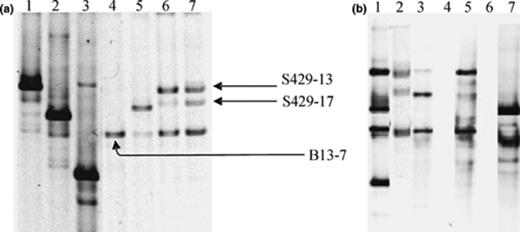
16S rRNA gene DGGE fingerprinting of Acinetobacter communities in hydrocarbon contaminated soil samples using PCR products obtained with Acinetobacter specific primers GC-Ac436f and Ac676r in the direct PCR approach (a) and the nested PCR approach (b). (a) DGGE profiles for strain A. johnsonii DSM6963T (lane 1), strain Acinetobacter sp. LH168 (lane 2), strain A. junii DSM6964T (lane 3), soil B13 (lane 4), soil B15 (lane 5), soil S428 (lane 6), soil S429 (lane 7). Cloned ‘bands’ are indicated within the soil fingerprint based on the comparison of the migration profiles of pure clones and the soil profile. (b) DGGE ladder (lane 1); DGGE profile of soil S429 obtained by the direct PCR (lane 2), and the nested PCR (lane 3); DGGE profile of soil S431 obtained by the direct PCR (lane 4), and the nested PCR (lane 5); DGGE profile of soil B4 obtained by the direct PCR (lane 6), and the nested PCR (lane 7); The DGGE ladder consists of A. johnsonii, A. haemolyticus, A. lwoffii, and A. junii.
Using the direct PCR approach, Acinetobacter populations were not detected in either the aerobic or anaerobic soil samples originating from site P contaminated with MTBE and benzene, whereas PCR products were obtained with the primer pair GC-63f/518r and 27f/1472r targeting the eubacterial 16S rRNA gene. No PCR product was obtained using the nested PCR approach (data not shown).
Regarding site B, Acinetobacter populations were detected in samples B13 and B15, containing relatively high BTEX and mineral oil concentrations, but not in the less contaminated samples B4 and B22 when the direct PCR approach was applied. Locations B13 and B15 seem to harbor different Acinetobacter populations (Fig. 3(a), lanes 4 and 5). In contrast with the direct PCR approach, PCR fragments were obtained with soil B4 DNA using nested PCR approach, indicating that soil B4 contains Acinetobacter populations, but present at lower cell concentrations than those in soils B13 and B15. The corresponding population in B4 seems to be different to those in soils B13 and B15 (Fig. 3(b), lane 7).
PCR products were obtained using the direct approach for samples S428 and S429 but not with samples S427, S430, S431 and S432. Similar profiles were obtained for samples S428 and S429 despite their different sampling depths (Fig. 3(a), lanes 6 and 7). The nested PCR approach generated products for both samples S429 and S431. For the S429 soil, the DGGE patterns obtained using both PCR approaches were identical (Fig. 3(b), lanes 2 and 3). The S431 soil, for which no PCR product was generated using the direct PCR approach, seems to contain an Acinetobacter community similar to those of soils S428 and S429 (Fig. 3(b), lanes 2–5) but probably present at lower cell numbers. Comparison of the Acinetobacter fingerprints generated by the direct PCR approach between samples derived from different sites revealed one common band in samples B13, S428, S429 and a faint one in sample F.
To confirm the identity of the recovered amplicons from the analyzed soil samples and to get more insight into the identity of the corresponding Acinetobacter populations, PCR fragments obtained with the direct PCR approach were cloned in pCR4-TOPO vectors. The cloned bands were reamplified with primers GC-Ac436f and Ac676r and the PCR products were positioned within the soil DGGE profile by comparing the profiles generated from the soil DNA and from individual clones (Fig. 3(a)). The clones were sequenced and the sequences were analyzed using BLAST (Table 3). All sequences showed the highest similarity with sequences of Acinetobacter 16S rRNA genes, confirming the selectivity of the primer set used. Moreover, the position of the bands on the gel matched well with the position expected from their sequence information, e.g., the sequences of clones S429–13, S429–17 and B13–7 showed highest similarity with the 16S rRNA gene of A. johnsonii, A. calcoaceticus and A. lwoffii, respectively and showed migration profiles on DGGE similar to the migration profiles of the 16S rRNA gene fragments of the corresponding type strains. Unfortunately, no sequence information was obtained for the one faint band from the population profile of soil sample F, but its co-migration with band B13–7 suggests that this band also corresponds to strains related to A. lwoffii.
3.5 Monitoring of Acinetobacter community dynamics in a laboratory biostimulation experiment
Temporal changes in Acinetobacter diversity were assessed in differently treated soil F samples using the direct or PCR approaches. Prior to treatment, soil F yielded a faint signal with the direct PCR approach (Fig. 4(a), lanes 4–5). After nested PCR on the DNA obtained from the soil F sample, a DGGE profile was obtained that was quite similar to that of soil sample F70MNT1 (Fig. 4(b), lanes 2–3) but with two predominant bands rather than one, i.e., one corresponding to the band of A. haemolyticus as in the soil F70MNT1 profile (Fig. 4(b), lane 2–3) and the other to the band of A. lwoffii (as indicated in the DGGE marker, Fig. 4(b)) (data not shown). Acinetobacter populations were not detected with either the direct or nested approach in soil sample F throughout the experiment nor when treated without nutrient amendment or mineral nutrients. In soil F, containing an additional 70,000 mg kg−1 diesel fuel and 112.5 ppm PAH and amended with mineral nutrients, an Acinetobacter population consisting of one major band in the DGGE profile could be detected after biostimulation for 32 days (Fig. 4(a), lanes 6–7). This band was also found with soil F but only using the nested PCR approach, indicating that addition of hydrocarbons and mineral nutrients particularly increased the population size of the corresponding species in the soil. At later time points, no Acinetobacter population could be detected using the direct PCR approach (data not shown). However, when the more sensitive nested PCR approach was applied on soil sample F70MN T3 (day 140), a population was obtained similar to that of F70MN T1 and to that of the initial soil (Fig. 4(b), lanes 2, 3, 6 and 7), indicating that the Acinetobacter community composition and number had returned to their initial state. Note that similar populations were obtained using either the direct or nested PCR approach on F70MN T1 DNA (Fig. 4(b), lanes 2 and 3).
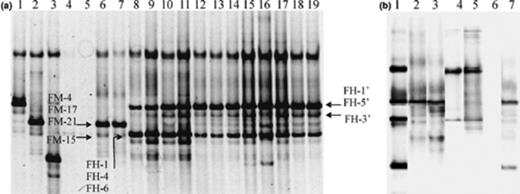
16S rRNA gene DGGE fingerprinting of Acinetobacter communities in soil F samples treated in a laboratory experiment using PCR products obtained with Acinetobacter specific primers GC-Ac436f and Ac676r in the direct PCR approach (a) and the nested PCR approach (b). (a) DGGE profiles for strain A. johnsonii DSM6963T (lane 1), strain A. haemolyticus LH168 (lane 2), strain A. junii DSM6964T (lane 3), soil F prior to treatment (lanes 4 and 5), soil F70MN T1 (lanes 6 and 7), soil F70HNa T1 (lanes 8 and 9), soil F70HNa T3 (lanes 10 and 11), soil F70HNa T4(lanes 12 and 13), soil F70HNa T5 (lanes 14 and 15), soil F70HNa T6 (lanes 16 and 17), soil F70HNa T9 (lanes 18 and 19). All soil samples were analyzed in duplicate. Subscripts T1, T3,… refer to sampling times as described in materials and methods. No Acinetobacter population was monitored in soil FMN. Cloned ‘bands’ are indicated within the soil fingerprint based on the comparison of the migration profiles of pure clones and the soil profile. (B) DGGE ladder (lane 1); DGGE profile of soil F70MN T1 obtained by the direct PCR (lane 2), and the nested PCR (lane 3); DGGE profile of soil F70HNa T3 obtained by the direct PCR (lane 4), and the nested PCR (lane 5); DGGE profile of soil F70MNT3 obtained by the direct PCR (lane 6), and the nested PCR (lane 7). The DGGE ladder consists of A. johnsonii, A. haemolyticus, A. lwoffii, and A. junii.
A clearly distinct, more diverse and stable Acinetobacter population evolved in aerated soil F70HNa, containing additional hydrocarbons and treated with Inipol EAP-22 (Fig. 4(a), lane 8–19). Similar populations were obtained using either PCR approach (Fig. 4(b), lanes 4 and 5). Interestingly, the Acinetobacter PCR–DGGE profiles obtained from soil microcosms F70HNa exhibited banding patterns similar to those obtained from soils S428 and S429. Cluster analysis on DGGE fingerprints resulted in a dendrogram composed of separate clusters according to (i) the origin of the soil or (ii) the biostimulation treatment (Fig. 5).
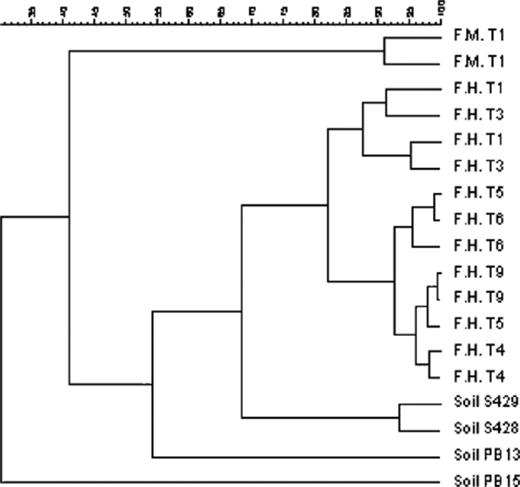
UPGMA clustering of soil DGGE fingerprints using the pearson moment-based similarity coefficient. Similarity percentages between Acinetobacter microbial populations using the PCR–DGGE specific system are indicated above. F.M. and F.H. correspond to microcosms F70MN and F70HNa while T1, T3, T4, T5, T6, T9 indicate the corresponding sampling time. Samples analysed in duplicate are indicated with the same name.
The 16S rRNA gene products obtained from the treated soils were sequenced after direct cloning of the PCR products in pCR4-TOPO vectors. PCR products reamplified from the clones were positioned on the DGGE profile by comparing soil DGGE profiles with those generated from soil clones (Fig. 4(a)). Sequences of 10 clones obtained from soil-derived DNA were analyzed by BLAST (Table 4). All sequences exhibited high levels of identity to sequences of strains from the genus Acinetobacter. The highly contaminated F soil treated with mineral nutrient showed an Acinetobacter population dominated at day 32 by A. haemolyticus (band FM-4/FM-17/FM-21). The fainter band (FM-15) was identified as A. lwoffii. Highly contaminated soil F treated with oleophilic nutrients, however, was dominated by Acinetobacter bacteria whose 16S rRNA gene sequences were most closely related to the 16S rRNA gene sequence of strains related to A. lwoffii (band FH-1/FH-4/FH-6), A. johnsonii (band FH-1′/FH-5′) and A. calcoaceticus (band FH-3′). In all cases, as for the other soils, congruency was found between the identity of the 16S rRNA gene band and its migration behavior on the DGGE gel.
BLAST analysis of 16S rRNA gene sequences obtained by PCR from the hydrocarbon-contaminated soil samples using primer pair GC-Ac436f/Ac676r
| DGGE band | Origin | Soil contamination and/or treatment | Most closely related strain (% sequence identity) | Accession Nos. in GenBank |
| B13–7 | Aquifer B13 | Kerosene (BTEX) | Acinetobacter sp. LUH4547 (100%) | AJ301674 |
| S429–13 | Soil S429 | Mineral oil | Acinetobacter johnsonii p152 (98%) | AF188300 |
| S429–17 | Soil S429 | Mineral oil | Unidentified Bacterial sp. Boom-3m–01 (96%) | Z73451 |
| FM-15 | Soil F70MN | Diesel fuel/PAHs; after 32 days of treatment by mineral nutrient | Acinetobacter lwoffii ATCC17925 (99%) | Z93441 |
| FM-17 | Soil F70MN | Diesel fuel/PAHs; after 32 days of treatment by mineral nutrient | Acinetobacter haemolyticus DSM6962T (100%) | X81662 |
| FM-21 | Soil F70MN | Diesel fuel/PAHs; after 32 days of treatment by mineral nutrient | Acinetobacter haemolyticus DSM6962T (100%) | X81662 |
| FM-4 | Soil F70MN | Diesel fuel/PAHs; after 32 days of treatment by mineral nutrient | Acinetobacter haemolyticus LH168 (99%) | AF542926 |
| FH-1 | Soil F70HNa | Diesel fuel/PAHs; after 32 days of treatment by oleophilic nutrient/aeration | Acinetobacter lwoffii ATCC17925 (99%) | U10875 |
| FH-4 | Soil F70HNa | Diesel fuel/PAHs; after 32 days of treatment by oleophilic nutrient/aeration | Acinetobacter lwoffii A382 (100%) | AF188302 |
| FH-6 | Soil F70HNa | Diesel fuel/PAHs; after 32 days of treatment by oleophilic nutrient/aeration | Acinetobacter lwoffii A382 (100%) | AF188302 |
| FH-1′ | Soil F70HNa | Diesel fuel/PAHs; after 645 days of treatment by oleophilic nutrient/aeration | Acinetobacter sp. LY1 (99%) | AJ007008 |
| FH-3′ | Soil F70HNa | Diesel fuel/PAHs; after 645 days of treatment by oleophilic nutrient/aeration | Unidentified Bacterial sp. Boom-3m–01 (97%) | Z73451 |
| FH-5′ | Soil F70HNa | Diesel fuel/PAHs; after 645 days of treatment by oleophilic nutrient/aeration | Acinetobacter sp. LY1 (98%) | AJ007008 |
| DGGE band | Origin | Soil contamination and/or treatment | Most closely related strain (% sequence identity) | Accession Nos. in GenBank |
| B13–7 | Aquifer B13 | Kerosene (BTEX) | Acinetobacter sp. LUH4547 (100%) | AJ301674 |
| S429–13 | Soil S429 | Mineral oil | Acinetobacter johnsonii p152 (98%) | AF188300 |
| S429–17 | Soil S429 | Mineral oil | Unidentified Bacterial sp. Boom-3m–01 (96%) | Z73451 |
| FM-15 | Soil F70MN | Diesel fuel/PAHs; after 32 days of treatment by mineral nutrient | Acinetobacter lwoffii ATCC17925 (99%) | Z93441 |
| FM-17 | Soil F70MN | Diesel fuel/PAHs; after 32 days of treatment by mineral nutrient | Acinetobacter haemolyticus DSM6962T (100%) | X81662 |
| FM-21 | Soil F70MN | Diesel fuel/PAHs; after 32 days of treatment by mineral nutrient | Acinetobacter haemolyticus DSM6962T (100%) | X81662 |
| FM-4 | Soil F70MN | Diesel fuel/PAHs; after 32 days of treatment by mineral nutrient | Acinetobacter haemolyticus LH168 (99%) | AF542926 |
| FH-1 | Soil F70HNa | Diesel fuel/PAHs; after 32 days of treatment by oleophilic nutrient/aeration | Acinetobacter lwoffii ATCC17925 (99%) | U10875 |
| FH-4 | Soil F70HNa | Diesel fuel/PAHs; after 32 days of treatment by oleophilic nutrient/aeration | Acinetobacter lwoffii A382 (100%) | AF188302 |
| FH-6 | Soil F70HNa | Diesel fuel/PAHs; after 32 days of treatment by oleophilic nutrient/aeration | Acinetobacter lwoffii A382 (100%) | AF188302 |
| FH-1′ | Soil F70HNa | Diesel fuel/PAHs; after 645 days of treatment by oleophilic nutrient/aeration | Acinetobacter sp. LY1 (99%) | AJ007008 |
| FH-3′ | Soil F70HNa | Diesel fuel/PAHs; after 645 days of treatment by oleophilic nutrient/aeration | Unidentified Bacterial sp. Boom-3m–01 (97%) | Z73451 |
| FH-5′ | Soil F70HNa | Diesel fuel/PAHs; after 645 days of treatment by oleophilic nutrient/aeration | Acinetobacter sp. LY1 (98%) | AJ007008 |
BLAST analysis of 16S rRNA gene sequences obtained by PCR from the hydrocarbon-contaminated soil samples using primer pair GC-Ac436f/Ac676r
| DGGE band | Origin | Soil contamination and/or treatment | Most closely related strain (% sequence identity) | Accession Nos. in GenBank |
| B13–7 | Aquifer B13 | Kerosene (BTEX) | Acinetobacter sp. LUH4547 (100%) | AJ301674 |
| S429–13 | Soil S429 | Mineral oil | Acinetobacter johnsonii p152 (98%) | AF188300 |
| S429–17 | Soil S429 | Mineral oil | Unidentified Bacterial sp. Boom-3m–01 (96%) | Z73451 |
| FM-15 | Soil F70MN | Diesel fuel/PAHs; after 32 days of treatment by mineral nutrient | Acinetobacter lwoffii ATCC17925 (99%) | Z93441 |
| FM-17 | Soil F70MN | Diesel fuel/PAHs; after 32 days of treatment by mineral nutrient | Acinetobacter haemolyticus DSM6962T (100%) | X81662 |
| FM-21 | Soil F70MN | Diesel fuel/PAHs; after 32 days of treatment by mineral nutrient | Acinetobacter haemolyticus DSM6962T (100%) | X81662 |
| FM-4 | Soil F70MN | Diesel fuel/PAHs; after 32 days of treatment by mineral nutrient | Acinetobacter haemolyticus LH168 (99%) | AF542926 |
| FH-1 | Soil F70HNa | Diesel fuel/PAHs; after 32 days of treatment by oleophilic nutrient/aeration | Acinetobacter lwoffii ATCC17925 (99%) | U10875 |
| FH-4 | Soil F70HNa | Diesel fuel/PAHs; after 32 days of treatment by oleophilic nutrient/aeration | Acinetobacter lwoffii A382 (100%) | AF188302 |
| FH-6 | Soil F70HNa | Diesel fuel/PAHs; after 32 days of treatment by oleophilic nutrient/aeration | Acinetobacter lwoffii A382 (100%) | AF188302 |
| FH-1′ | Soil F70HNa | Diesel fuel/PAHs; after 645 days of treatment by oleophilic nutrient/aeration | Acinetobacter sp. LY1 (99%) | AJ007008 |
| FH-3′ | Soil F70HNa | Diesel fuel/PAHs; after 645 days of treatment by oleophilic nutrient/aeration | Unidentified Bacterial sp. Boom-3m–01 (97%) | Z73451 |
| FH-5′ | Soil F70HNa | Diesel fuel/PAHs; after 645 days of treatment by oleophilic nutrient/aeration | Acinetobacter sp. LY1 (98%) | AJ007008 |
| DGGE band | Origin | Soil contamination and/or treatment | Most closely related strain (% sequence identity) | Accession Nos. in GenBank |
| B13–7 | Aquifer B13 | Kerosene (BTEX) | Acinetobacter sp. LUH4547 (100%) | AJ301674 |
| S429–13 | Soil S429 | Mineral oil | Acinetobacter johnsonii p152 (98%) | AF188300 |
| S429–17 | Soil S429 | Mineral oil | Unidentified Bacterial sp. Boom-3m–01 (96%) | Z73451 |
| FM-15 | Soil F70MN | Diesel fuel/PAHs; after 32 days of treatment by mineral nutrient | Acinetobacter lwoffii ATCC17925 (99%) | Z93441 |
| FM-17 | Soil F70MN | Diesel fuel/PAHs; after 32 days of treatment by mineral nutrient | Acinetobacter haemolyticus DSM6962T (100%) | X81662 |
| FM-21 | Soil F70MN | Diesel fuel/PAHs; after 32 days of treatment by mineral nutrient | Acinetobacter haemolyticus DSM6962T (100%) | X81662 |
| FM-4 | Soil F70MN | Diesel fuel/PAHs; after 32 days of treatment by mineral nutrient | Acinetobacter haemolyticus LH168 (99%) | AF542926 |
| FH-1 | Soil F70HNa | Diesel fuel/PAHs; after 32 days of treatment by oleophilic nutrient/aeration | Acinetobacter lwoffii ATCC17925 (99%) | U10875 |
| FH-4 | Soil F70HNa | Diesel fuel/PAHs; after 32 days of treatment by oleophilic nutrient/aeration | Acinetobacter lwoffii A382 (100%) | AF188302 |
| FH-6 | Soil F70HNa | Diesel fuel/PAHs; after 32 days of treatment by oleophilic nutrient/aeration | Acinetobacter lwoffii A382 (100%) | AF188302 |
| FH-1′ | Soil F70HNa | Diesel fuel/PAHs; after 645 days of treatment by oleophilic nutrient/aeration | Acinetobacter sp. LY1 (99%) | AJ007008 |
| FH-3′ | Soil F70HNa | Diesel fuel/PAHs; after 645 days of treatment by oleophilic nutrient/aeration | Unidentified Bacterial sp. Boom-3m–01 (97%) | Z73451 |
| FH-5′ | Soil F70HNa | Diesel fuel/PAHs; after 645 days of treatment by oleophilic nutrient/aeration | Acinetobacter sp. LY1 (98%) | AJ007008 |
4 Discussion
In this study, we describe the development of an Acinetobacter genus-specific PCR–DGGE system for direct and culture-independent analysis of the diversity of Acinetobacter populations in environmental samples. The specificity of the Acinetobacter primer set was demonstrated by performing PCR on DNA from pure bacterial cultures, which resulted in the production of amplicons solely from Acinetobacter strains. Despite the 3′ end mismatch observed with A. baumannii 16S rRNA gene sequence of strain ATCC19606T, which might decrease amplification efficiency, a 240 bp PCR product was obtained with the Ac436f/Ac676r primer set. However, other A. baumannii 16S rRNA gene sequences described for the A. baumannii type strain DSM30007T, which is identical to ATCC19606T, do not show mismatches with the forward primer Ac436f. Therefore, we assume that either the published sequence showing a 3′ mismatch represents a variant of one of the seven copies of the rRNA gene in A. baumannii or that the mismatch is due to sequence errors.
The detection of Acinetobacter strains in soil by PCR using this primer pair was 104 cells g−1 of soil. The attachment of a GC clamp to the forward primer in order to achieve separation of the fragments on DGGE resulted in a 100-fold increase in this detection limit, suggesting that problems might be encountered when exploring the diversity of Acinetobacter in soils containing low numbers of Acinetobacter cells, when a direct PCR approach is used. Leys et al. (unpublished results) previously showed that the attached GC-clamp might play a crucial role in the processes controlling sensitive PCR detection. Few authors report the limit of detection of their PCR system when using GC-clamped primers for DGGE purposes. Salles et al. [23] reported a detection limit 5 × 105 CFU g−1 soil for Burkholderia species using a similar direct PCR–DGGE detection method. This number is only slightly lower than found in this study, where the detection limit could not be decreased by a semi-nested PCR procedure as reported by Salles et al. [23], Heilig et al. [24], and Rossi et al. [25]. However, the detection limit was reduced to at least 10 cells g−1 soil by a nested PCR approach in which the bacterial 16S rRNA gene was amplified in a first round to reduce the background of non-16S rDNA followed by a PCR with the GC-clamped Acinetobacter primer set in a second PCR round. However, when high numbers of Acinetobacter cells were added, some larger non-specific fragments were co-amplified. These fragments might distort analysis of the population profile in subsequent DGGE analysis. However, these bands were faint and did not produce additional bands in the DGGE profile. In addition, we never observed these additional bands after applying the nested PCR on DNA from environmental samples. Moreover, DGGE analysis of Acinetobacter 16S rRNA gene amplicons generated by either the direct or nested PCR approach from the environmental samples generated highly similar DGGE profiles. In contrast, in some recent experiments, some additional bands were occasionally observed using the nested PCR approach (Vanbroekhoven, unpublished results), as might be expected because of its increased sensitivity. The usefulness of the nested PCR approach was demonstrated by the fact that we detected Acinetobacter populations in some of the environmental samples, which did not show Acinetobacter populations using the direct PCR approach.
While in theory DGGE-analysis might allow differentiation to the species or even the strain level, due to sequence variations of as little as one base pair within the amplified 16S rRNA fragment, effective separation of amplified fragments of some species such as Mycobacterium spp. (N. Leys et al., submitted for publication) and Burkholderia spp. [23] with very similar 16S rRNA gene sequences is not always evident on DGGE. Clearly, practical resolution of the DGGE technique is at the species or genus level or intermediate, depending on the gene conservation level of the genus under investigation. In our case, differentiation up to species level seems feasible as DGGE analysis of the resulting amplicons allowed clear discrimination between amplicons of different Acinetobacter species. As such this might indicate that direct identification at the species level of the detected Acinetobacter strains in environmental samples is possible by comparing the migration profiles with profiles of known species. However, classification of environmental isolates belonging to the genus Acinetobacter clearly needs revision, as the Acinetobacter genus presently consists of nine taxonomic clusters and is almost exclusively based on clinical isolates. Therefore, it can be expected that phylogenetic analysis of environmental isolates will lead to the definition of new species with more closely related 16S rRNA gene sequences impairing the discriminatory power of our Acinetobacter specific PCR–DGGE approach.
Acinetobacter junii provided a multiple-band DGGE profile, as also observed by others for Burkholderia cepacia[23], Bifidobacterium adolescentis[26], Legionella[27], and Paenibacillus polymyxa[28]. In the case of P. polymyxa and B. adolescentis, this was due to the presence of multiple 16S rRNA gene copies with minor base pair substitutions. This might be the case for some Acinetobacter strains, as bacteria belonging to this genus are known to contain multiple 16S rRNA gene copies, but no report exists on the sequence divergence of these rRNA genes [29]. In Legionella, the multiple-band pattern was explained by the occurrence of several melting domains in the PCR products obtained by a specific primer pair, but this explanation seems unlikely since, in our study, multiple banding patterns were only obtained with A. junii. Another explanation is formation of heteroduplexes composed of amplification products of different templates (e.g., two intra-strain divergent 16S rRNA gene fragments), as found by Satokari et al. [26] for B. adolescentis. Since these heteroduplexes are less stable, they melt under weaker denaturing conditions. Doublet profiles with two bands close to each other were observed for A. johnsonii and A. lwoffii. According to Satokari et al. [26] and Nübel et al. [28], such profiles might be explained by termination of the elongation reaction during PCR caused by the GC clamp, due to hairpin formation leading to DNA molecules with slightly different migration behavior. We are currently cloning relevant bands from multiple banding patterns for sequencing which will allow us to gain more insight in the observed patterns.
Soil samples contaminated with the organic pollutants BTEX and mineral oil were analyzed for the presence of Acinetobacter populations initially using the direct PCR approach. As the limit of detection of this approach was 106 cells g−1 soil, the obtained signal would represent major Acinetobacter populations present in the examined soils. For some soil samples, the nested PCR approach was also applied. As evidenced by DGGE analysis and clone sequencing of the PCR amplicons, three 16S rRNA genes were generally recovered, which were related to 16S rRNA gene sequences of A. haemolyticus, A. lwoffii and A. johnsonii. However, the population composition seemed to be dependent on environmental conditions. For example, two different soil samples from the BTEX-contaminated site B harbored slightly different Acinetobacter populations. Using the nested PCR approach, a different community was recovered from the third sample. Comparison of soil Acinetobacter fingerprints from the samples originating from site F resulted in different population profiles depending on the treatment. Acinetobacter amplicons were only detected during the experiment in soils that had received additional hydrocarbons (70,000 mg kg−1). Following amendment with mineral nutrients, a transient increase in A. haemolyticus seemed to occur. Nested PCR indicates that this population seems to reside in soil F but at a lower concentration. In contrast, 32 days after treatment with an oleophilic nutrient, an Acinetobacter community apparently increased which consisted mainly of strains related to A. lwoffii and A. johnsonii, that remained stable for a further 613 days. Interestingly, hydrocarbon removal in the aerated Inipol-amended microcosms F70HNa increased from 6% after 140 days of treatment to 65% after 645 days.
The population pattern derived from soil F, originally sampled from the capillary fringe near an oil floating layer, was very similar to that of soils derived from site S, contaminated by mineral oil. Apparently, the pollution and/or treatment to which soil samples were subjected might have a greater impact on the community profile than the geological location of the soil. As such, in all cases and depending on the environmental conditions, our soils were dominated by Acinetobacter bacteria related to A. haemolyticus, A. lwoffii and A. johnsonii. Similarly, Gallego et al. [13], based on culture-dependent techniques, observed the dominant presence of A. haemolyticus and A. lwoffii strains in diesel-contaminated soils treated by inorganic nutrients. Well-described isolates of A. lwoffi[30,31], A. haemolyticus and A. johnsonii are in most cases isolates of clinical or human sources. Other authors reported the occurrence of A. calcoaceticus[32–34], A. baumannii[35] and some unclassified Acinetobacter strains [36–38] in hydrocarbon-contaminated environments, using culture-dependent techniques. Combining the developed PCR–DGGE approach with conventional culture-dependent approaches could offer the possibility to study unculturable and culturable Acinetobacter sp. and to study their relevance in hydrocarbon contaminated environments.
In conclusion, the described PCR–DGGE approach was valuable for rapidly analyzing the Acinetobacter populations in environmental samples. The application of the newly designed specific primer pair combined with the discriminatory power of DGGE will undoubtedly lead to new insights in Acinetobacter populations present in hydrocarbon-contaminated soils and a wide range of other habitats.
Acknowledgements
This research was funded by a scholarship granted to K.V. by the Flemish Institute for Technological Research (VITO).
References
Author notes
Dept. of Bacteriology, Veterinary & Agrochemical Research Institute, Groeselenberg 99, B-1180, Brussels, Belgium.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}