





Abstract
Polycyclic aromatic hydrocarbons (PAHs) are among the most important contaminants of groundwater. The 2- and 3-ring PAHs are of particular concern because they are water soluble in the 1–200 μg/l range and are transported with the groundwater over significant distances. Anaerobic degradation of PAH has been demonstrated in several microcosm studies with nitrate, ferric iron, or sulfate as electron acceptors and under methanogenic conditions. The biochemical degradation pathways were studied with naphthalene-degrading pure and enrichment cultures and revealed that 2-naphthoic acid is a central metabolite. Naphthalene is activated by addition of a C1-unit to generate 2-naphthoic acid, whereas methylnaphthalene is activated by addition of fumarate to the methyl group and further degraded to 2-naphthoic acid. In the central 2-naphthoic acid degradation pathway the ring system is reduced prior to ring cleavage generating e.g. 5,6,7,8-tetrahydro-2-naphthoic acid. The ring cleavage produces metabolites such as 2-carboxycyclohexylacetic acid indicating that further degradation goes via cyclohexane derivatives and not via aromatic compounds. Anaerobic degradation of PAH has also been demonstrated in situ in contaminated aquifers by identification of compound specific metabolites and using stable isotope fraction studies. Detection of specific metabolites of anaerobic PAH degradation such as naphthyl-2-methylsuccinate indicated anaerobic degradation of 2-methylnaphthalene in situ whereas 2-naphthoic acid was indicative of naphthalene and 2-methylnaphthalene degradation. Other carboxylic acids that were detected in groundwater indicated anaerobic degradation of a wide range of PAH and heterocyclic compounds. Degradation of naphthalenes in contaminated aquifers could also be confirmed by carbon stable isotope shifts in the residual substrate fraction.
1 Introduction
Aromatic hydrocarbons in the environment such as benzene, toluene, ethylbenzene, xylene (BTEX), and polycyclic aromatic hydrocarbons (PAHs) mostly originate from anthropogenic sources like mineral oil spills or former gas plants and to a minor extent from biological production in anoxic sediments, for example by cyanobacteria, or other sources such as termite nests [1–4]. Due to their importance as groundwater contaminants, there has been strong interest in the microbial degradation of BTEX and PAH. Numerous aerobic bacteria have been isolated that use aromatic hydrocarbons as sources of carbon and energy in the presence of oxygen, and the associated degradation pathways have been elucidated [5–7]. However, hydrocarbon-contaminated aquifers usually become anoxic with a redox gradient along the groundwater flow path. The major fraction of the contaminants is degraded within anoxic zones of the plume, implying that they are being degraded by anaerobic bacteria [8,9]. In the present review, we focus on the anaerobic degradation of PAHs; anaerobic degradation of BTEX is beyond the scope of this paper except in terms of the broader background. For more information on anaerobic BTEX-degradation, we refer the reader to several recently published reviews [10–13].
2 Anaerobic degradation of polycyclic aromatic hydrocarbons in microcosm studies
Mihelcic and Luthy [14] were the first to report the mineralization of naphthalene and acenaphthene in microcosm experiments under nitrate-reducing conditions. Several further studies assessed diverse aromatic compounds for biodegradation under different redox conditions, mostly with nitrate or sulfate as terminal electron acceptors. The results of some successful microcosm studies dealing with PAH degradation under anoxic conditions are summarized in Table 1. A variety of aromatic compounds were found to be biodegradable under anoxic conditions and even the degradation of higher condensed PAH such as fluoranthene has been reported [15]. So far no degradation of unsubstituted PAH has been found under methanogenic conditions, although compounds with polar substituents such as naphthol (isomer not specified) can be metabolized [14].
Microcosm studies successful in identifying anaerobic degradation of polycyclic aromatic hydrocarbons
| PAH | Electron acceptor | Inoculum | Degradation rate (μM/d) | Reference |
| Acenaphthene | Nitrate | Uncontaminated soil | 0.12 | [14] |
| Acenaphthene | Nitrate | Uncontaminated soil | 0.03 | [68] |
| Biphenyl | Nitrate | Marine sediment | 16.2; 1.5a | [17] |
| Biphenyl | Sulfate | Marine sediment | 22.7 | [17] |
| Fluorene | Sulfate | Marine sediment | 0.06 | [15] |
| Fluoranthene | Sulfate | Marine sediment | 0.014 | [15] |
| Methylnaphthalene | Sulfate | Marine sediment | 0.22 | [15] |
| Naphthalene | Nitrate | Uncontaminated soil | 1.6 | [14] |
| Naphthalene | Nitrate | Uncontaminated soil | 0.95; 1.23; 1.17 | [68] |
| Naphthalene | Nitrate | Soil | 14.1 | [69] |
| Naphthalene | Nitrate | Aquifer material | 0.055 | [70] |
| Naphthalene | Nitrate | River sediment | 384 | [16] |
| Naphthalene | Nitrate | Marine sediment | 35.2; 1.4a | [17] |
| Naphthalene | Ferric iron | Marine sediment | 0.05 | [71] |
| Naphthalene | Manganese (IV) | River sediment | 120 | [16] |
| Naphthalene | Sulfate | River sediment | 336b | [16] |
| Naphthalene | Sulfate | Marine sediment | 0.075 | [72] |
| Naphthalene | Sulfate | Marine sediment | 0.12 | [15] |
| Naphthalene | Sulfate | Aquifer material | 2.5 | [18] |
| Naphthalene | Sulfate | Marine sediment | 39.1 | [17] |
| Naphthalene | Nitrate or sulfatec | Aquifer material | 0.42; 0.23 | [73] |
| Naphthol | CO2 | Uncontaminated soil | 4.2 | [14] |
| Naphthol | Nitrate | Uncontaminated soil | 3.8 | [14] |
| Phenanthrene | Nitrate | Marine sediment | 3.3; 0.34a | [17] |
| Phenanthrene | sulfate | Marine sediment | 0.1 | [72] |
| Phenanthrene | Sulfate | Marine sediment | 0.11 | [15] |
| Phenanthrene | Sulfate | Marine sediment | 3.3 | [17] |
| PAH | Electron acceptor | Inoculum | Degradation rate (μM/d) | Reference |
| Acenaphthene | Nitrate | Uncontaminated soil | 0.12 | [14] |
| Acenaphthene | Nitrate | Uncontaminated soil | 0.03 | [68] |
| Biphenyl | Nitrate | Marine sediment | 16.2; 1.5a | [17] |
| Biphenyl | Sulfate | Marine sediment | 22.7 | [17] |
| Fluorene | Sulfate | Marine sediment | 0.06 | [15] |
| Fluoranthene | Sulfate | Marine sediment | 0.014 | [15] |
| Methylnaphthalene | Sulfate | Marine sediment | 0.22 | [15] |
| Naphthalene | Nitrate | Uncontaminated soil | 1.6 | [14] |
| Naphthalene | Nitrate | Uncontaminated soil | 0.95; 1.23; 1.17 | [68] |
| Naphthalene | Nitrate | Soil | 14.1 | [69] |
| Naphthalene | Nitrate | Aquifer material | 0.055 | [70] |
| Naphthalene | Nitrate | River sediment | 384 | [16] |
| Naphthalene | Nitrate | Marine sediment | 35.2; 1.4a | [17] |
| Naphthalene | Ferric iron | Marine sediment | 0.05 | [71] |
| Naphthalene | Manganese (IV) | River sediment | 120 | [16] |
| Naphthalene | Sulfate | River sediment | 336b | [16] |
| Naphthalene | Sulfate | Marine sediment | 0.075 | [72] |
| Naphthalene | Sulfate | Marine sediment | 0.12 | [15] |
| Naphthalene | Sulfate | Aquifer material | 2.5 | [18] |
| Naphthalene | Sulfate | Marine sediment | 39.1 | [17] |
| Naphthalene | Nitrate or sulfatec | Aquifer material | 0.42; 0.23 | [73] |
| Naphthol | CO2 | Uncontaminated soil | 4.2 | [14] |
| Naphthol | Nitrate | Uncontaminated soil | 3.8 | [14] |
| Phenanthrene | Nitrate | Marine sediment | 3.3; 0.34a | [17] |
| Phenanthrene | sulfate | Marine sediment | 0.1 | [72] |
| Phenanthrene | Sulfate | Marine sediment | 0.11 | [15] |
| Phenanthrene | Sulfate | Marine sediment | 3.3 | [17] |
aThe first rate is estimated for a fluidized bed reactor with continuous medium percolation, the second for a batch experiment.
bThe degradation of naphthalene was only detected after addition of 250 μM benzoate as cosubstrate to the medium.
cThe groundwater used for this experiment contained both nitrate and sulfate in significant amounts.
Microcosm studies successful in identifying anaerobic degradation of polycyclic aromatic hydrocarbons
| PAH | Electron acceptor | Inoculum | Degradation rate (μM/d) | Reference |
| Acenaphthene | Nitrate | Uncontaminated soil | 0.12 | [14] |
| Acenaphthene | Nitrate | Uncontaminated soil | 0.03 | [68] |
| Biphenyl | Nitrate | Marine sediment | 16.2; 1.5a | [17] |
| Biphenyl | Sulfate | Marine sediment | 22.7 | [17] |
| Fluorene | Sulfate | Marine sediment | 0.06 | [15] |
| Fluoranthene | Sulfate | Marine sediment | 0.014 | [15] |
| Methylnaphthalene | Sulfate | Marine sediment | 0.22 | [15] |
| Naphthalene | Nitrate | Uncontaminated soil | 1.6 | [14] |
| Naphthalene | Nitrate | Uncontaminated soil | 0.95; 1.23; 1.17 | [68] |
| Naphthalene | Nitrate | Soil | 14.1 | [69] |
| Naphthalene | Nitrate | Aquifer material | 0.055 | [70] |
| Naphthalene | Nitrate | River sediment | 384 | [16] |
| Naphthalene | Nitrate | Marine sediment | 35.2; 1.4a | [17] |
| Naphthalene | Ferric iron | Marine sediment | 0.05 | [71] |
| Naphthalene | Manganese (IV) | River sediment | 120 | [16] |
| Naphthalene | Sulfate | River sediment | 336b | [16] |
| Naphthalene | Sulfate | Marine sediment | 0.075 | [72] |
| Naphthalene | Sulfate | Marine sediment | 0.12 | [15] |
| Naphthalene | Sulfate | Aquifer material | 2.5 | [18] |
| Naphthalene | Sulfate | Marine sediment | 39.1 | [17] |
| Naphthalene | Nitrate or sulfatec | Aquifer material | 0.42; 0.23 | [73] |
| Naphthol | CO2 | Uncontaminated soil | 4.2 | [14] |
| Naphthol | Nitrate | Uncontaminated soil | 3.8 | [14] |
| Phenanthrene | Nitrate | Marine sediment | 3.3; 0.34a | [17] |
| Phenanthrene | sulfate | Marine sediment | 0.1 | [72] |
| Phenanthrene | Sulfate | Marine sediment | 0.11 | [15] |
| Phenanthrene | Sulfate | Marine sediment | 3.3 | [17] |
| PAH | Electron acceptor | Inoculum | Degradation rate (μM/d) | Reference |
| Acenaphthene | Nitrate | Uncontaminated soil | 0.12 | [14] |
| Acenaphthene | Nitrate | Uncontaminated soil | 0.03 | [68] |
| Biphenyl | Nitrate | Marine sediment | 16.2; 1.5a | [17] |
| Biphenyl | Sulfate | Marine sediment | 22.7 | [17] |
| Fluorene | Sulfate | Marine sediment | 0.06 | [15] |
| Fluoranthene | Sulfate | Marine sediment | 0.014 | [15] |
| Methylnaphthalene | Sulfate | Marine sediment | 0.22 | [15] |
| Naphthalene | Nitrate | Uncontaminated soil | 1.6 | [14] |
| Naphthalene | Nitrate | Uncontaminated soil | 0.95; 1.23; 1.17 | [68] |
| Naphthalene | Nitrate | Soil | 14.1 | [69] |
| Naphthalene | Nitrate | Aquifer material | 0.055 | [70] |
| Naphthalene | Nitrate | River sediment | 384 | [16] |
| Naphthalene | Nitrate | Marine sediment | 35.2; 1.4a | [17] |
| Naphthalene | Ferric iron | Marine sediment | 0.05 | [71] |
| Naphthalene | Manganese (IV) | River sediment | 120 | [16] |
| Naphthalene | Sulfate | River sediment | 336b | [16] |
| Naphthalene | Sulfate | Marine sediment | 0.075 | [72] |
| Naphthalene | Sulfate | Marine sediment | 0.12 | [15] |
| Naphthalene | Sulfate | Aquifer material | 2.5 | [18] |
| Naphthalene | Sulfate | Marine sediment | 39.1 | [17] |
| Naphthalene | Nitrate or sulfatec | Aquifer material | 0.42; 0.23 | [73] |
| Naphthol | CO2 | Uncontaminated soil | 4.2 | [14] |
| Naphthol | Nitrate | Uncontaminated soil | 3.8 | [14] |
| Phenanthrene | Nitrate | Marine sediment | 3.3; 0.34a | [17] |
| Phenanthrene | sulfate | Marine sediment | 0.1 | [72] |
| Phenanthrene | Sulfate | Marine sediment | 0.11 | [15] |
| Phenanthrene | Sulfate | Marine sediment | 3.3 | [17] |
aThe first rate is estimated for a fluidized bed reactor with continuous medium percolation, the second for a batch experiment.
bThe degradation of naphthalene was only detected after addition of 250 μM benzoate as cosubstrate to the medium.
cThe groundwater used for this experiment contained both nitrate and sulfate in significant amounts.
Although most of the studies were performed with inocula from marine sediments, fresh water inocula also showed potential for biodegradation of PAH. Degradation rates varied strongly, even for assays with the same compounds and electron acceptors. In general, batch experiments showed lower degradation rates than those conducted with continuous medium percolation. The highest degradation rates were observed in a sediment column after 500 days of percolation with a mineral salt medium [16]. A negative correlation between the molecular mass of the compound and its degradation rate was observed under similar experimental conditions in batch experiments [15]. There was usually no correlation between energy yield and degradation rates [16,17] even though the rates of PAH removal versus biomass production were 5–10 times higher for nitrate reducers than for sulfate reducers [17]. The results of Mihelcic and Luthy [14] are especially interesting because they showed a PAH degradation potential even in non-contaminated sites.
In summary, there seems to be a large potential for natural attenuation of PAH in diverse anoxic environments. PAH containing up to four rings can be transformed, even though it seems likely that the more condensed compounds are co-metabolized with smaller ones [15]. However, for a long time attempts to culture anaerobic PAH-degrading bacteria failed. Only recently transferable naphthalene-degrading, denitrifying, and sulfate-reducing enrichments and pure cultures have been reported [18–22].
3 Peripheral pathways of anaerobic PAH degradation leading to the central intermediate 2-naphthoic acid
So far anaerobic degradation pathways have only been investigated in detail for two polycyclic aromatic hydrocarbons. The first data on metabolites of anaerobic naphthalene degradation were published in 1997 [22]. The authors investigated a sulfate-reducing enrichment culture that degraded naphthalene and phenanthrene. The major metabolites were extracted from culture supernatants and identified by GC-MS; they were identified as 2-naphthoic acid and phenanthroic acid. When the cultures were grown in a 13C-bicarbonate-buffered medium, the label was found in the carboxyl group of 2-naphthoic acid showing that it derived from CO2. Investigation of a sulfate-reducing naphthalene-degrading enrichment culture originating from a tar oil-contaminated aquifer also revealed the incorporation of 13CO2 into the carboxyl group of the major metabolite 2-naphthoic acid [20] (Fig. 1). Both cultures were also able to grow on 2-naphthoic acid as their sole source of carbon, indicating that 2-naphthoic acid was a real intermediate of the degradation pathway and not a dead end metabolite. It remains unclear, however, whether the primary activation reaction of naphthalene is in fact a carboxylation or another single or sequence of reactions introducing a carboxyl group from CO2.
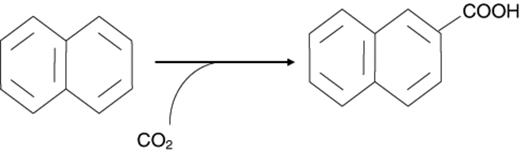
Proposed activation reaction for naphthalene by addition of CO2 to generate 2-naphthoic acid.
The sulfate-reducing freshwater enrichment culture N47 also grew on 1-naphthoic acid, 2-methylnaphthalene, cyclohexanecarboxylic acid, and acetate [20]. Since this study was performed with an enrichment culture, it is possible that organisms other than the naphthalene-degrading strain consumed these substrates.
Closer investigation showed 2-naphthoic acid to be a prominent metabolite of 2-methylnaphthalene degradation, indicating that the degradation pathways of naphthalene and 2-methylnaphthalene converge at the level of 2-naphthoic acid [23]. However, the major metabolites detected in experiments with 2-methylnaphthalene were naphthyl-2-methyl-succinic acid and naphthyl-2-methylene-succinic acid. These findings strongly suggested that the methyl group of 2-methylnaphthalene was activated by addition of fumarate. This was confirmed in cell suspension experiments in which addition of 2-methylnaphthalene and fumarate led to the formation of naphthyl-2-methyl-succinate (Fig. 2) [23]. The reaction was very sensitive to oxygen and could only be measured in the presence of the strong reducing agent titanium(III)-citrate. The analogy to anaerobic toluene degradation by denitrifying bacteria suggests that the naphthyl-2-methyl-succinate synthase reaction is also catalysed by a glycyl radical enzyme.
![Proposed scheme of the upper pathway of anaerobic 2-methylnaphthalene degradation (1*) to the central intermediate 2-naphthoic acid (8*), (2*) fumaric acid, (3*) naphthyl-2-methyl-succinic acid, (4) naphthyl-2-methyl-succinyl-CoA, (5*) naphthyl-2-methylene-succinyl-CoA, (6) naphthyl-2-hydroxymethyl-succinyl CoA, (7) naphthyl-2-oxomethyl-succinyl-CoA. Compounds marked with (*) have been identified as pure substance or free acids. Taken from [23].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/femsec/49/1/10.1016_j.femsec.2004.02.019/1/m_FEM_27_f2.jpeg?Expires=1749997236&Signature=ryyGoyQn8r9sUE7sdgpXIDrCBB4pchY17Loh4HZ7JVVoDMwRme11CHL3TUvWPA4BYXGSr5nodzqhFjyPT9tVIbdWG7yZDo8MaOSUgXX99euM79ffjI6RVP3rDo7cT63Xi-A9n54jNIsKIIkMzduFuzYJtKya5YlMMo~Oa9zFhOYM8spviooKCm-YRIM-5-6cw41t2ULFdb53IHSufS11E-C6oUQqXbQ7SWM04YSk3DXcJ5psSgArOutvlgL~i2-KvkYft0Y3P9mE-sfCnhIWi-2h2lzMj4cTwv2TVbuGmRHlhK~LxZZ-b0hxbpyc67RaygGTLp4tF0bH5oDI5xhclQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Proposed scheme of the upper pathway of anaerobic 2-methylnaphthalene degradation (1*) to the central intermediate 2-naphthoic acid (8*), (2*) fumaric acid, (3*) naphthyl-2-methyl-succinic acid, (4) naphthyl-2-methyl-succinyl-CoA, (5*) naphthyl-2-methylene-succinyl-CoA, (6) naphthyl-2-hydroxymethyl-succinyl CoA, (7) naphthyl-2-oxomethyl-succinyl-CoA. Compounds marked with (*) have been identified as pure substance or free acids. Taken from [23].
The first step in the anaerobic degradation pathway of toluene by denitrifying bacteria of the genera Azoarcus and Thauera is addition of fumarate to the methyl group of toluene [24–27]. The reaction is catalyzed by benzylsuccinate synthase, which is a novel member of the glycyl radical enzyme family as resolved from the DNA-sequence of the coding genes [27–32]. The proposed mechanism for benzylsuccinate synthase is abstraction of a hydrogen atom from the methyl group of toluene and its subsequent addition to the C-2 atom of fumarate. The hydrogen atom remains enzyme bound until the generated benzylsuccinate radical abstracts the hydrogen radical back from the enzyme yielding benzylsuccinate [24,27,33].
The next enzymatic reaction in the degradation pathway of 2-methylnaphthalene was identified to be the transfer of a CoenzymeA moiety (CoA) from succinyl-CoA to naphthyl-2-methylsuccinate [34]. The reaction was measured in both the presence and absence of oxygen with the same enzyme activities. As the thermodynamic equilibrium was on the side of the free naphthyl-2-methylsuccinate and succinyl-CoA, the backwards reaction was measured with synthesized naphthyl-2-methylsuccinyl-CoA and succinate as the substrates.
Although the amino acid sequence of the enzyme is not yet known, the analogy with the toluene degradation pathway suggests that the CoA-transferase could be a member of the novel family III of CoA-transferases, which comprises succinyl-CoA:(R)-benzylsuccinate CoA-transferase, oxalyl-CoA:formate CoA-transferase, and (E)-cinnamoyl-CoA:(R)-phenyllactate CoA-transferase [35].
The next step in the degradation pathway of 2-methylnaphthalene is a beta-oxidation of the methylene carbon of naphthyl-2-methylsuccinyl-CoA. This reaction was measured in crude cell extracts with naphthyl-2-methylsuccinyl-CoA as the substrate and phenazinmethosulfate as electron acceptor [34]. The product of the reaction, naphthyl-2-methylenesuccinyl-CoA, hydrolyzed spontaneously with an extremely short half-life of 3 min in aqueous solutions at neutral pH, as demonstrated by studies with the synthesized reference compound. The naphthyl-2-methylenesuccinyl-CoA produced in the enzyme tests hydrolyzed to Coenzyme-A and the free acid, which was identified by HPLC-chromatography.
The further reactions in the degradation pathway are probably addition of water to the double bond at the methylene carbon to yield naphthyl-2-hydroxymethyl-succinyl-CoA and a subsequent oxidation to naphthyl-2-oxomethyl-succinyl-CoA. This compound should be cleaved in a thiolytic reaction with Coenzyme-A, generating the central intermediate 2-naphthoic acid and succinyl-CoA (Fig. 2).
Other authors have studied the anaerobic degradation of 2-methylnaphthalene with a marine sulfate-reducing enrichment culture and identified methyl-2-naphthoic acid as a metabolite, but could not identify naphthyl-2-methyl-succinate [36]. These investigators proposed that methylnaphthalene is activated by a carboxylation reaction on the aromatic ring similar to the first reaction in naphthalene degradation. This could mean either that a further pathway exists for the anaerobic degradation of 2-methylnaphthalene in addition to that of fumarate addition, or that the observed metabolite is the product of a side reaction in the naphthalene degradation pathway resulting from the addition of a C1-unit. In the freshwater culture N47 the degradation pathways for anaerobic naphthalene and 2-methylnaphthalene are indeed induced simultaneously, as shown by enzyme assays with naphthalene grown cells that contained the first three enzymes of the naphthylmethylsuccinate synthase pathway [34]. Production of methyl-2-naphthoic acid from methylnaphthalene has also been identified in supernatants of the sulfate-reducing enrichment culture N47 when grown on 2-methylnaphthalene as the substrate (data not shown).
4 The 2-naphthoic acid ring cleavage pathway in the anaerobic degradation of polycyclic aromatic hydrocarbons
One of the prominent metabolites identified in studies of anaerobic naphthalene and 2-methylnaphthalene degradation was 2-naphthoic acid. Other reduced derivatives were identified in addition to this compound such as 1,2,3,4-tetrahydro-2-naphthoic acid, 5,6,7,8-tetrahydro-2-naphthoic acid, hexahydro-2-naphthoic acid, octahydro-2-naphthoic acid, and decahydro-2-naphthoic acid (Fig. 3) [20,23,37,38]. These metabolites indicated that a reduction of the bicyclic ring system might precede ring cleavage.
![Proposed reductive 2-naphthoic acid pathway of anaerobic PAH degradation. (I) 2-naphthoic acid, (II) 5,6,7,8-tetrahydro-2-naphthoic acid, (III) hydroxydecahydro-2-naphthoic acid, (IV) β-oxo-decahydro-2-naphthoic acid, (V) C11H16O4-diacid, (VI) 2-carboxycyclohexylacetic acid. Compounds III and IV are putative intermediates that have not been identified so far. Taken from [39].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/femsec/49/1/10.1016_j.femsec.2004.02.019/1/m_FEM_27_f3.jpeg?Expires=1749997236&Signature=zaYu3XNXBfCiW6anQlkeBdVOw8D4uCn1o8evJpNoVjotV7mlPPbHrCTIOygbg1CeSsUlpIJULq8AouzzebKk60XhsAZ14KRACg6jTFkaX0be1KO6VtyyxnXxwBMsGi-vveioURUDFfFfjk5ukeS2lnSGS3sxy0wMi5NeaPLiOOLfIBLsOqlI6b1jWJQfTdbU17-rT8z3dkdJYHghDfd8FXMwzaXH7gsiSBKwN~oRJf1NOfh6gdaBbiQqNolMxfq4c7s2i5HZpDXPs3a5uFyBSKvMV29A9dn2Diuq-4YZ364k3~VpzTE8tlGRurINazKWvSAi25-iQCnJlo7yqGLcBg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Proposed reductive 2-naphthoic acid pathway of anaerobic PAH degradation. (I) 2-naphthoic acid, (II) 5,6,7,8-tetrahydro-2-naphthoic acid, (III) hydroxydecahydro-2-naphthoic acid, (IV) β-oxo-decahydro-2-naphthoic acid, (V) C11H16O4-diacid, (VI) 2-carboxycyclohexylacetic acid. Compounds III and IV are putative intermediates that have not been identified so far. Taken from [39].
Although both 1,2,3,4-tetrahydro-2-naphthoic acid and 5,6,7,8-tetrahydro-2-naphthoic acid were found in supernatants of naphthalene grown cells, the reduction pathway probably proceeds via 5,6,7,8-tetrahydro-2-naphthoic acid. This is indicated by degradation experiments with the naphthalene-degrading culture N47 and tetralin as a carbon source in which 5,6,7,8-tetrahydro-2-naphthoic acid was found as the major metabolite but no 1,2,3,4-tetrahydro-2-naphthoic acid [39]. Similarly, 5,6,7,8-tetrahydro-2-naphthoic acid was identified as an important metabolite of naphthalene degradation in a marine, sulfate-reducing enrichment culture, and the investigator concluded that the degradation pathway proceeds via this intermediate [37].
For tetralin degradation, this means that the addition of the C1-unit is specific to the aromatic ring because this is the only way in which 5,6,7,8-tetrahydro-2-naphthoic acid could be produced. On the other hand, the finding that the 2-naphthoic acid degradation pathway proceeds via 5,6,7,8-tetrahydro-2-naphthoic acid indicates that ring II of 2-naphthoic acid, which is distal to the carboxyl group, is reduced first. This is important for the later ring cleavage mechanism. Due to the electron withdrawing function of carboxylic groups, the ring cleavage should be initiated in the beta-position to the carboxyl group of 5,6,7,8-tetrahydro-2-naphthoic acid. If ring cleavage were to be initiated from 1,2,3,4-tetrahydro-2-naphthoic acid, this might generate a monoaromatic compound which would than be degraded via reactions analogous to the benzoyl-CoA pathway. If the ring reduction goes via 5,6,7,8-tetrahydro-2-naphthoic acid, ring II which is distal to the carboxyl group would be reduced first and the cleavage would produce a compound with a cyclohexane ring. That this is probably the case was shown in experiments in which two ring cleavage products, with a cyclohexane ring and two carbonic acid side chains, respectively, were identified as degradation products but no monoaromatic compounds (structures V and VI in Fig. 3) [39]. One of these metabolites was 2-carboxycyclohexylacetic acid (structure VI in Fig. 3), which was probably generated by cleavage of a C2-fragment via beta-oxidation of the other metabolite, a C11H16O4-diacid (structure V in Fig. 3). Both metabolites (structures IV and V in Fig. 3) were identified in cultures grown with naphthalene, methylnaphthalene, and tetralin, indicating that the three compounds are degraded via a novel central degradation pathway with cyclohexanoic compounds as intermediates and not through the benzoyl-CoA pathway [39].
5 Other aromatic compounds that are subject to the PAH-degradation pathways
Phenanthrene is the only PAH other than naphthalene, methylnaphthalene, and tetralin that has been investigated more closely with respect to anaerobic degradation [22]. The initial activating reaction suggested for this substance was a carboxylation reaction, as phenanthroic acid was observed as a metabolite in culture supernatants.
A similar reaction may be of importance for anaerobic degradation of benzothiophene. The naphthalene-degrading sulfate-reducing freshwater culture N47 was able to degrade benzothiophene to 2-carboxybenzothiophene, 5-carboxybenzothiophene, and the reduced derivative dihydrocarboxybenzothiophene in a cometabolic reaction with naphthalene as the auxiliary substrate [40]. Conversion to the carboxylic acid was quantitative and slightly inhibited naphthalene degradation. This reaction may be of major importance in contaminated aquifers; the same metabolites were identified at a former gas plant site at relatively high concentrations of up to 500 μg l−1 (see below) [40].
Anaerobic degradation of other heterocyclic compounds such as indole and quinoline has been investigated in more detail, and we refer to a recent review on this topic [41].
6 Detection of anaerobic degradation of polycyclic aromatic hydrocarbons in situ at contaminated field sites
So far, the evidence for biodegradation of petroleum hydrocarbons in situ has been based mainly on measurements of indirect microbial activity parameters. One classical approach is the mass balance calculation between contaminants, electron acceptors, and products [42,43]. Furthermore, computer modeling that incorporates transport of contaminants and reaction stoichiometries of electron donors and acceptors can be applied [44]. Biological evidence for the degradation of pollutants can sometimes be obtained from microcosm studies with contaminated material from the field site. In such experiments, radio-labeled substrates can be amended and the successive conversion of aromatic hydrocarbons to metabolites and/or the total mineralization to CO2 shown for individual compounds (see above). There are only a very few reports of studies using the release of radio-labeled substrates into the subsurface and subsequent identification of degradation products, an approach which comes close to direct measurement of in situ degradation rates [45]. Other field biological parameters include the determination of differences and/or changes in microbial numbers, biomass, diversity, and activity within the contaminated area in relation to uncontaminated sites [46]. Very recently, the detection of potential degraders by selective enrichment of organisms (MPN counts) or 16 rDNA sequences, or the identification of degradation-related enzymes (e.g., for aerobic naphthalene degradation) by molecular tools, have been reported as further approaches to evaluate biodegradation [47–49]. However, notwithstanding the many indirect indicators identified for in situ microbial activities and degradation potentials, parameters for direct proof of in situ bioremediation remain scarce.
Direct proof of in situ biodegradation can be obtained from the detection of metabolic intermediates in field samples. The prerequisites for this approach include the knowledge that the metabolites are: (1) unequivocally and uniquely related to the parent hydrocarbon, (2) not used commercially or industrially, (3) biologically and chemically stable, and (4) released into the medium [50]. Following this approach, active anaerobic degradation in situ has been shown for toluene, and o, m, p-xylene in some field studies. Here the major targets of interest are benzylsuccinate and the corresponding methylbenzylsuccinates, which are the first intermediates resulting from fumarate addition to the terminal methyl group. These have been detected at several fuel-hydrocarbon contaminated sites [50–54]. Further possible degradation products of BTEX-like benzoate and alkylbenzoates have been detected in more than a dozen field studies [50]. However, the presence of benzoate and alkylbenzoates in groundwater does not definitively demonstrate anaerobic BTEX degradation since they could also originate from other natural precursor compounds such as amino acids.
Recently, field data for metabolites from anaerobic degradation of PAH have been reported from three sites [54,55]. In addition to succinate derivatives of monoaromatic hydrocarbons, n-alkanes, and cyclic alkanes, Gieg and Suflita [54] identified 1- and 2-naphthoic acid and methyl- and dimethylnaphthoic acid, all of which are metabolites of the anaerobic degradation of naphthalene and methylnaphthalenes, as well as 1,2,3,4-tetrahydro- and 5,6,7,8-tetrahydro-2-naphthoic acid, in two out of six aquifers investigated (Table 2). Investigations at a gasoline-affected site revealed the presence of 2-naphthoic acid (2-NA), a tetrahydro-2-naphthoic acid which was not further identified, a hexahydro-2-naphthoic acid, and a methylnaphthoic acid (Table 2) [55].
Metabolites resulting from anaerobic degradation of polycyclic aromatic hydrocarbons detected in groundwater samples from different field sites
| PAH metabolites | Concentration (μg l−1) | Putative precursor compound(s) | ||
| (A) | (B) | (C) | ||
| Naphthyl-2-methylsuccinic acid | 2–13 | 2-Methylnaphthalene | ||
| 1-Naphthoic acid | n.d. | <0.1–18 | 1-Methylnaphthalene | |
| 2-Naphthoic acid | Max 7.3 | Max 4 | 0.2–45 | Naphthalene, 2-methylnaphthalene |
| Methylnaphthoic acid | 2–7 | n.d. | Different methylnaphthalenes | |
| Dimethylnaphthoic acid | 1.4–2.7 | |||
| 1,2,3,4-Tetrahydro-2-naphthoic acid | 1.2–3.2 | n.d. | 0.9–10 | |
| 5,6,7,8-Tetrahydro-2-naphthoic acid | 0.1–5* | Naphthalene, 2-methylnaphthalene | ||
| Hexahydro-2-naphthoic acid | n.d. | Naphthalene, 2-methylnaphthalene | ||
| PAH metabolites | Concentration (μg l−1) | Putative precursor compound(s) | ||
| (A) | (B) | (C) | ||
| Naphthyl-2-methylsuccinic acid | 2–13 | 2-Methylnaphthalene | ||
| 1-Naphthoic acid | n.d. | <0.1–18 | 1-Methylnaphthalene | |
| 2-Naphthoic acid | Max 7.3 | Max 4 | 0.2–45 | Naphthalene, 2-methylnaphthalene |
| Methylnaphthoic acid | 2–7 | n.d. | Different methylnaphthalenes | |
| Dimethylnaphthoic acid | 1.4–2.7 | |||
| 1,2,3,4-Tetrahydro-2-naphthoic acid | 1.2–3.2 | n.d. | 0.9–10 | |
| 5,6,7,8-Tetrahydro-2-naphthoic acid | 0.1–5* | Naphthalene, 2-methylnaphthalene | ||
| Hexahydro-2-naphthoic acid | n.d. | Naphthalene, 2-methylnaphthalene | ||
Metabolites resulting from anaerobic degradation of polycyclic aromatic hydrocarbons detected in groundwater samples from different field sites
| PAH metabolites | Concentration (μg l−1) | Putative precursor compound(s) | ||
| (A) | (B) | (C) | ||
| Naphthyl-2-methylsuccinic acid | 2–13 | 2-Methylnaphthalene | ||
| 1-Naphthoic acid | n.d. | <0.1–18 | 1-Methylnaphthalene | |
| 2-Naphthoic acid | Max 7.3 | Max 4 | 0.2–45 | Naphthalene, 2-methylnaphthalene |
| Methylnaphthoic acid | 2–7 | n.d. | Different methylnaphthalenes | |
| Dimethylnaphthoic acid | 1.4–2.7 | |||
| 1,2,3,4-Tetrahydro-2-naphthoic acid | 1.2–3.2 | n.d. | 0.9–10 | |
| 5,6,7,8-Tetrahydro-2-naphthoic acid | 0.1–5* | Naphthalene, 2-methylnaphthalene | ||
| Hexahydro-2-naphthoic acid | n.d. | Naphthalene, 2-methylnaphthalene | ||
| PAH metabolites | Concentration (μg l−1) | Putative precursor compound(s) | ||
| (A) | (B) | (C) | ||
| Naphthyl-2-methylsuccinic acid | 2–13 | 2-Methylnaphthalene | ||
| 1-Naphthoic acid | n.d. | <0.1–18 | 1-Methylnaphthalene | |
| 2-Naphthoic acid | Max 7.3 | Max 4 | 0.2–45 | Naphthalene, 2-methylnaphthalene |
| Methylnaphthoic acid | 2–7 | n.d. | Different methylnaphthalenes | |
| Dimethylnaphthoic acid | 1.4–2.7 | |||
| 1,2,3,4-Tetrahydro-2-naphthoic acid | 1.2–3.2 | n.d. | 0.9–10 | |
| 5,6,7,8-Tetrahydro-2-naphthoic acid | 0.1–5* | Naphthalene, 2-methylnaphthalene | ||
| Hexahydro-2-naphthoic acid | n.d. | Naphthalene, 2-methylnaphthalene | ||
Investigations at a former gasworks site using GC-MS analysis revealed a number of metabolites originating from anaerobic degradation of naphthalene and alkyl-naphthalenes (Table 2) [40]. Furthermore, two products of a putative carboxylation of the heterocyclic compound benzothiophene – 5-carboxybenzothiophene and dihydrocarboxybenzothiophene – were found in almost every groundwater well at the site [40,56]. In another study, liquid chromatography (LC) followed by electrospray ionization (ESI) and tandem mass spectrometry (MS-MS) was applied directly to groundwater samples and revealed the presence of naphthyl-2-methylsuccinic acid, which is the first intermediate in the anaerobic degradation of 2-methylnaphthalene under sulfate-reducing conditions [57]. In all three studies, 2-naphthoic acid (2-NA) was the major metabolite in terms of both distribution and concentration. This might be the result of its role as a central metabolite in the degradation pathways of several PAHs.
The conclusions that can be drawn from the detection of degradation products in field samples strongly depend on our understanding of the related degradation pathways. The presence and accumulation of metabolites may serve as evidence that the parent compound has been altered in situ, but at uncertain times and locations in the past [51,58]. However, the successful detection of metabolites from degradation of PAH at several sites demonstrates that the specific anaerobic pathways studied in the laboratory are present and active in contaminated aquifers.
A further approach to identifying biodegradation of individual aromatic hydrocarbon compounds in situ is to look for a shift in the stable carbon or hydrogen isotope ratio, as the degradation of organic compounds is in some cases accompanied by stable isotope effects. The isotopically lighter molecules are preferentially degraded by microorganisms, so that the residual contaminant fraction becomes isotopically enriched in 13C [59–64]. Shifts in the stable carbon isotope ratio in contaminated aquifers have been shown to result solely from biodegradation; abiotic processes such as dilution and adsorption did not cause isotope effects [59,63]. However, the degree of isotope fractionation during degradation depends on the first enzymatic step in the degradation pathway [65]. A pronounced isotope effect has been shown for several alkylbenzenes, where the first enzymatic reaction takes place at the methyl group [59,60,65]. On the other hand, a much smaller isotope effect was observed for the degradation of the unsubstituted aromatic hydrocarbon benzene [66].
Stable isotope fractionation data for PAHs are rare. No stable carbon isotope fractionation was found during the degradation of fluoranthene, but the extent of degradation in these experiments was too low to allow any significant fractionation to be detected, if indeed any occurred [67]. A moderate carbon isotope fractionation (shift of 1.2‰) has been observed for naphthalene in an anoxic contaminant plume fuelled by a 30-year-old coal tar source [63]. A similar qualitative proof of in situ anaerobic naphthalene degradation was also obtained in a recent study at a former gasworks site. A decline in naphthalene concentration accompanied a shift in the stable isotope ratio δ13C of 3‰. Only moderate carbon isotope fractionation of 1.4‰ and 2.3‰, respectively, was detected for 1- and 2-methylnaphthalene, which were both present in much lower concentrations than naphthalene. The maximum isotope shifts that can be expected for a single compound along a groundwater flow path are shown by the example of toluene, the best studied aromatic hydrocarbon with respect to anaerobic degradation and isotope fractionation, for which isotope fractionation in the range of 10‰ has been recorded [59,63].
7 Outlook
The information acquired so far on the anaerobic degradation of polycyclic aromatic hydrocarbons shows definitely that at least naphthalene and other two-ring PAHs can be used as the sole source of carbon and energy under sulfate-, iron-, and nitrate-reducing conditions. It remains unclear whether three or more-ring PAHs are used as growth substrates or they are only co-metabolised. Nevertheless, the elucidation of the first degradation pathways of naphthalene and methylnaphthalene, and associated identification of important metabolites, have provided the basis for identifying the presence of biodegradation of PAHs in situ in natural environments such as contaminated aquifers.
The methods for cultivating anaerobic microorganisms with PAHs as sole carbon and energy source have been established in the last years, however, more bacterial strains need to be isolated and their degradation pathways studied. Not all degradation pathways are necessarily the same. Moreover, so far only few sulfate-reducing organisms have been studied in detail and the degradation pathways of e.g. denitrifying organisms might be different as well. In addition, it is uncertain if the organisms isolated so far really have the major function in the field or they are merely organisms that grew fastest under the applied cultivation conditions. Molecular studies might elucidate in the future which strains or microbial communities are the major degrading organisms in contaminated aquifers. The main challenge for future research is probably to identify the limitations of biodegradation in contaminated aquifers. The question why some contaminants are only poorly degraded although the organisms that could degrade the compounds can be isolated from the aquifer and degradation activities can be detected by compound-specific metabolites or stable isotope fractionation is still unsolved. Answers to such questions could greatly improve bioremediation or risk assessment of contaminated aquifers in the future.
Acknowledgements
The authors thank two anonymous reviewers for helpful comments. Parts of this work were supported by the Deutsche Forschungsgemeinschaft (Grant Schi 180/7) and the German Bundesministerium für Bildung und Forschung (Grant 02WT0022).
References

{kind=link}
{kind=link}
{kind=link}