





Abstract
Methanotrophs are a fascinating group of bacteria that have the unique ability to grow on methane as their sole carbon and energy source. They appear to be widespread in nature and have been isolated from a number of different environments, including soils, sediments, freshwater, marine sediments, seawater, acid peat bogs, hot springs and cold environments such as the Antarctic. There are now eight recognised genera of methanotrophs. Methanotrophs have attracted a great deal of interest over the past 30 years since they have considerable potential for the production of bulk chemicals, fine chemicals and in bioremediation processes, such as the degradation of the groundwater pollutant trichloroethylene. More recently, they are being extensively studied in a wide variety of environments since methanotrophs play a critical role in the global methane cycle. Polymerase chain reaction-based methods have been used to study the ecology and diversity of methanotrophs. We review here molecular ecology methods that are already available or which are currently being developed for methanotrophs. These are based on 16S ribosomal RNA technology and specific amplification of ‘functional genes’, such as those encoding unique enzymes in the metabolism of these organisms including methane monooxygenase and methanol dehydrogenase.
1 Introduction
Methane oxidising bacteria (methanotrophs) are a unique group of methylotrophic bacteria that utilise methane as their sole source of carbon and energy. Some methanotrophs also utilise methanol, but none can grow on multi-carbon compounds [1]. Methanotrophs can be isolated from a wide variety of environments including soils, sediments and freshwater and are all obligately aerobic, Gram negative bacteria.
Over the past 30 years there has been considerable interest in the use of methanotrophs for the production of single cell protein and bulk chemicals, such as propylene oxide. Their potential for bioremediation processes, such as the removal of trichloroethylene from the environment, has also been explored [2]. Methanotrophs play an important role in the oxidation of methane in the natural environment, oxidising methane biologically produced in, for example, wetlands and freshwater. Methane oxidising bacteria are also believed to oxidise methane from the atmosphere (i.e. at very low concentrations, 1.7 ppm) thereby mitigating global warming due to the effects of the greenhouse gas methane [3,4].
The difficulties in using traditional culture-based techniques to study the ecology of methanotrophs have been discussed previously. In particular, the slow growth of methanotrophs, together with scavenging of non-methanotrophs on agar isolation plates, has hampered studies. However, despite the inherent problems of isolation, several new methane oxidising bacteria have recently been identified from more extreme environments. These have included thermophilic methanotrophs from hot springs [5], psychrophilic methanotrophs from the Antarctic and Siberia [6], acidophilic methanotrophs from peat bogs [7,8], and halotolerant alkaliphilic methanotrophs from soda lakes [9].
2 Classification and characterisation of methanotrophs
In a pioneering study in 1970, Whittenbury and colleagues isolated and characterised over 100 methanotroph isolates and grouped them into five proposed genera, based on their morphology, type of resting stage formed, intracytoplasmic membrane structure and other physiological characteristics [10]. The genera proposed, Methylomonas, Methylobacter, Methylococcus, Methylocystis and Methylosinus are similar to those currently accepted [11], with the exception of three new genera, Methylomicrobium[12], Methylocaldum[13] and Methylosphaera[6].
Methanotrophs can be divided into two distinct physiological groups. Type I methanotrophs (Methylomonas, Methylocaldum, Methylosphaera, Methylomicrobium and Methylobacter) assimilate formaldehyde produced from the oxidation of methane (via methanol) using the ribulose monophosphate pathway [14], contain predominantly 16-carbon fatty acids and possess bundles of intracytoplasmic membranes. Type II methanotrophs (Methylocystis and Methylosinus) utilise the serine pathway for formaldehyde assimilation, have intracytoplasmic membranes arranged around the periphery of the cell and contain predominantly 18-carbon fatty acids [1](Table 1).
Characterisation of methanotrophs
| Genus | Phylogeny | Membrane type | Major PLFA | Formaldehyde assimilation pathway | Enzyme type | Mol% G+C content of DNA |
| Methylomonas | γ Proteobacteria | Type I | 16:1 | RuMP | pMMO | 51–59 |
| Methylobacter | γ Proteobacteria | Type I | 16:1 | RuMP | pMMO | 49–54 |
| Methylomicrobium | γ Proteobacteria | Type I | 16:1 | RuMP | pMMO | 50–60 |
| Methylosphaera | γ Proteobacteria | Type I | 16:1 | RuMP | pMMO | 43–46 |
| Methylocaldum | γ Proteobacteria | Type I | 16:1 | RuMP/serine | pMMO | 56–58 |
| Methylococcus | γ Proteobacteria | Type I | 16:1 | RuMP/serine | pMMO/sMMO | 59–66 |
| Methylosinus | α Proteobacteria | Type II | 18:1 | Serine | pMMO/sMMO | 62–63 |
| Methylocystis | α Proteobacteria | Type II | 18:1 | Serine | pMMO/sMMO | 62–63 |
| Genus | Phylogeny | Membrane type | Major PLFA | Formaldehyde assimilation pathway | Enzyme type | Mol% G+C content of DNA |
| Methylomonas | γ Proteobacteria | Type I | 16:1 | RuMP | pMMO | 51–59 |
| Methylobacter | γ Proteobacteria | Type I | 16:1 | RuMP | pMMO | 49–54 |
| Methylomicrobium | γ Proteobacteria | Type I | 16:1 | RuMP | pMMO | 50–60 |
| Methylosphaera | γ Proteobacteria | Type I | 16:1 | RuMP | pMMO | 43–46 |
| Methylocaldum | γ Proteobacteria | Type I | 16:1 | RuMP/serine | pMMO | 56–58 |
| Methylococcus | γ Proteobacteria | Type I | 16:1 | RuMP/serine | pMMO/sMMO | 59–66 |
| Methylosinus | α Proteobacteria | Type II | 18:1 | Serine | pMMO/sMMO | 62–63 |
| Methylocystis | α Proteobacteria | Type II | 18:1 | Serine | pMMO/sMMO | 62–63 |
RuMP, ribulose monophosphate pathway; pMMO, particulate methane monooxygenase; sMMO, soluble methane monooxygenase.
Characterisation of methanotrophs
| Genus | Phylogeny | Membrane type | Major PLFA | Formaldehyde assimilation pathway | Enzyme type | Mol% G+C content of DNA |
| Methylomonas | γ Proteobacteria | Type I | 16:1 | RuMP | pMMO | 51–59 |
| Methylobacter | γ Proteobacteria | Type I | 16:1 | RuMP | pMMO | 49–54 |
| Methylomicrobium | γ Proteobacteria | Type I | 16:1 | RuMP | pMMO | 50–60 |
| Methylosphaera | γ Proteobacteria | Type I | 16:1 | RuMP | pMMO | 43–46 |
| Methylocaldum | γ Proteobacteria | Type I | 16:1 | RuMP/serine | pMMO | 56–58 |
| Methylococcus | γ Proteobacteria | Type I | 16:1 | RuMP/serine | pMMO/sMMO | 59–66 |
| Methylosinus | α Proteobacteria | Type II | 18:1 | Serine | pMMO/sMMO | 62–63 |
| Methylocystis | α Proteobacteria | Type II | 18:1 | Serine | pMMO/sMMO | 62–63 |
| Genus | Phylogeny | Membrane type | Major PLFA | Formaldehyde assimilation pathway | Enzyme type | Mol% G+C content of DNA |
| Methylomonas | γ Proteobacteria | Type I | 16:1 | RuMP | pMMO | 51–59 |
| Methylobacter | γ Proteobacteria | Type I | 16:1 | RuMP | pMMO | 49–54 |
| Methylomicrobium | γ Proteobacteria | Type I | 16:1 | RuMP | pMMO | 50–60 |
| Methylosphaera | γ Proteobacteria | Type I | 16:1 | RuMP | pMMO | 43–46 |
| Methylocaldum | γ Proteobacteria | Type I | 16:1 | RuMP/serine | pMMO | 56–58 |
| Methylococcus | γ Proteobacteria | Type I | 16:1 | RuMP/serine | pMMO/sMMO | 59–66 |
| Methylosinus | α Proteobacteria | Type II | 18:1 | Serine | pMMO/sMMO | 62–63 |
| Methylocystis | α Proteobacteria | Type II | 18:1 | Serine | pMMO/sMMO | 62–63 |
RuMP, ribulose monophosphate pathway; pMMO, particulate methane monooxygenase; sMMO, soluble methane monooxygenase.
3 Biochemistry and molecular biology of methane oxidation
The enzyme responsible for the first step in the oxidation of methane is methane monooxygenase (MMO). Two forms of this enzyme exist: a soluble, cytoplasmic enzyme complex (sMMO) and a membrane bound, particulate enzyme (pMMO). The most extensively characterised are the sMMO enzymes from Methylococcus capsulatus[15] and Methylosinus trichosporium[16]. The sMMO enzyme complex consists of three components. Protein A is a hydroxylase consisting of three subunits of approximately 60, 45 and 20 kDa, arranged in a α2β2γ2 confirmation. The α subunits contain a diiron centre believed to be the site of the monooxygenation reaction. Protein B, a coupling protein of 16 kDa, facilitates electron flow and/or the interactions between Proteins A and C. Protein C (38 kDa) is the reductase component of the sMMO and catalyses the transfer of reducing equivalents from NADH to the hydroxylase.
The first methane-specific genes cloned were the soluble methane monooxygenase genes from Methylococcus capsulatus (Bath), Methylosinus trichosporium OB3b [17] and Methylocystis sp. strain M [18]. The genes encoding sMMO are clustered on the chromosome of these organisms in the order mmoX, mmoY, mmoB, mmoZ, orfY and mmoC. The mmoX, Y and Z genes encode the α, β and γ subunits of the hydroxylase, mmoB encodes Protein B and mmoC encodes the reductase component of sMMO. The orfY gene codes for an open-reading frame of unknown function. These sequences are highly conserved at both the DNA and protein level between all three methanotrophs.
The pMMO enzyme has proved to be far more refactory to biochemical analysis [19,20]. However, the enzyme has recently been purified in active form from Methylococcus capsulatus (Bath) [21]. It consists of three putative pMMO polypeptides of 47, 27 and 25 kDa, the first two of which have previously been observed as major polypeptides present when cells were expressing a membrane-bound MMO.
The pMMO genes from Methylococcus capsulatus (Bath) have recently been cloned [22]. The genes encoding the 27 and 45 kDa pMMO polypeptides, designated pmoA and pmoB are linked on the chromosome of this organism. The predicted amino acid sequences of these genes exhibit a high degree of identity with the corresponding gene products of amoA and amoB which encode key polypeptides of the ammonia monooxygenase (AMO) from the nitrifier Nitrosomonas europaea[23]. Another gene lies 5′ of pmoA and pmoB, this is designated pmoC, encoding a putative polypeptide of around 20 kDa which may be another component of pMMO [20]. Both pmo and amo genes are present in duplicate copies in methanotrophs or nitrifiers (as detected in hybridisation experiments); however, the reason for this duplication is unknown [22,24].
The sMMO is not present in all methanotrophs and is generally observed only during growth of strains of Methylococcus or Methylosinus in copper-deficient medium. Only one Methylomonas species [25] and one Methylocystis species [18] have been reported to possess sMMO. Expression of sMMO in Methyloccocus and Methylosinus is regulated at the level of transcription by the amount of copper available for growth. At low copper-to-biomass ratios, the synthesis of sMMO is repressed while the pMMO complex is expressed [26]. The pMMO is found in all methanotrophs during copper-sufficient growth conditions and appears to require copper ions for both expression and activity. It has a relatively narrow substrate specificity but will, in addition to oxidising methane, also co-oxidise a number of short chain alkanes, alkenes and ammonia. In contrast, sMMO has a very broad substrate specificity, possessing the ability to co-oxidise a wide range of alkanes, alkenes and aromatic compounds [15]. There is no apparent advantage for a methanotroph to possess the sMMO system other than the capacity to grow in copper-deficient conditions and its ecological significance is not known.
The second enzyme of the methane oxidation pathway is methanol dehydrogenase (MDH). The mxaF gene encoding the large subunit of methanol dehydrogenase [27] is present in all Gram negative methylotrophs (e.g. methane and methanol utilisers) [28]. It appears to be highly conserved and is therefore a good indicator of the presence of these organisms in the natural environment.
4 Molecular ecology of methanotrophs
Molecular ecological studies have adopted two separate approaches for the identification of methanotrophs in the environment. The first indirect approach relies on enrichment and/or isolation with subsequent characterisation of methanotrophs from environmental samples. The second direct approach relies on polymerase chain reaction (PCR) technology and the use of phylogenetic and functional gene probes for the detection and analysis of methanotrophs directly from environmental samples without the prerequisite of cultivation. At each stage in both approaches, molecular biological analysis of the key genes (methane monooxygenase, methanol dehydrogenase and 16S rRNA) can be carried out and comparisons made to see if the same organisms that grew in culture from the samples is representative of what was actually in the environmental sample (as revealed by DNA analysis). These molecular ecological approaches are summarised in Fig. 1.

A strategy for the molecular ecological analysis of environmental samples.
Several other approaches have been used to study methanotrophs in environmental samples. Phospholipid analysis has been used successfully for the detection of methanotrophs in the wetland environment [29]. This technique relies on the fact that methanotrophs contain unusual fatty acids (16:1 and 18:1 derivatives). Phospolipid analysis is, however, limited due to a small database of fatty acid profiles from methanotrophs and the relatively high expense of equipment used in the analysis. The detection and identification of methanotrophs in environmental samples can also be attempted using antibodies either to whole cells of culturable methanotrophs or to key enzymes such as methane monooxygenase or methanol dehydrogenase. However, these methods do not guarantee that all methanotrophs may be detected and extensive control experiments with large numbers of culturable methanotrophs are necessary to validate antibody probes. Estimates of the biomass and activities of methanotrophs can be made by measuring the release of 14CO2 and incorporation of 14CH4 added to environmental samples [30]. However, this technique needs to be coupled with further methods designed to analyse bacterial community structure.
5 Phylogenetic gene probes for methanotrophs
Application of molecular biology techniques to methanotrophs has been aided considerably by the sequencing of 16S rRNA genes from a number of methanotrophs and methylotrophs (reviewed in [1]), and phylogenetic relationships within genera of methanotrophs have been examined [1](Fig. 2). Type I methanotrophs (Methylomonas, Methylomicrobium, Methylobacter, Methylocaldum, Methylosphaera and Methylococcus) cluster in the γ-subdivision of the class Proteobacteria, whereas Type II methanotrophs (Methylosinus and Methylocystis) are found in the α-2 subdivision of the Proteobacteria. The observation that methanotrophs can be separated into coherent phylogenetic clusters that share common physiological characteristics is important for the use of 16S rRNA probe technology in the study of the ecology of these bacteria. Hanson and colleagues have used this information to design oligonucleotide probes with a broad specificity for methanotrophs and methylotrophs found in the natural environment [1]. The properties of these probes are presented in Table 2.
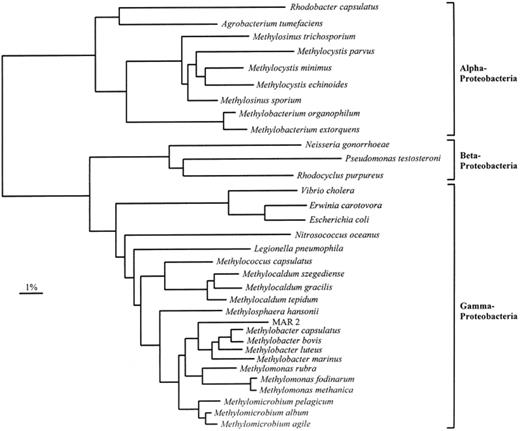
A phylogenetic tree of the 16S rRNA sequences from methanotrophs, methylotrophs and other representative bacteria belonging to the α, β, and γ subdivisions of the Proteobacteria. MAR 2 is a marine methanotroph isolate. The dendrogram shows the results from analysis using DNADIST, and the bar represents 1% sequences divergence, as determined by measuring the lengths of the horizontal lines connecting any two species.
Methanotroph functional and phylogenetic group-specific 16S rRNA probes/primers
| Primer | Sequence (5′–3′)a | Target genus | Temperature (°C) for specificity in: | Reference | |
| Colony hybridisation | PCR | ||||
| Mb 1007r | CACTCTACGATCTCTCACAG | Methylobacter | 60 | 58 | [7] |
| Mc 1005r | CCGCATCTCTGCAGGAT | Methylococcus | 55 | 54 | [7] |
| Mm 1007r | CACTCCGCTATCTCTAACAG | Methylomonas | 55 | 58 | [7] |
| Ms 1020r | CCCTTGCGGAAGGAAGTC | Methylosinus | 55 | 54 | [7] |
| fm 142f | GTTCGGAATAACTCAGGG | Serine pathway methylotrophs | 60 | NT | [7] |
| fm 64f | GAACGGTAACAGGCCTTCGG | RuMP pathway methanotrophs | 60 | NT | [7] |
| 10-γ | GGTCCGAAGATCCCCCGCTT | RuMP pathway methylotrophs | 52 | NT | [34] |
| 9-α | CCCTGAGTTATTCCGAAC | Serine pathway methylotrophs | 52 | NT | [34] |
| mxaF 1003f | GCGGCACCAACTGGGGCTGGT | All Gram negative methylotrophs | NT | 59 | [7] |
| mxaF 1561r | GGGCAGCATGAAGGGCTCCC | All Gram negative methylotrophs | NT | 59 | [7] |
| A189f | GGNGACTGGGACTTCTGG | pMMO/AMO | NT | 56 | [39] |
| A682r | GAASGCNGAGAAGAASGC | pMMO/AMO | NT | 56 | [39] |
| mmoX 882f | GGCTCCAAGTTCAAGGTCGAGC | sMMO+ methanotrophs | NT | 55 | [38] |
| mmoX 1403r | TGGCACTCGTAGCGCTCCGGCTCG | sMMO+ methanotrophs | NT | 55 | [38] |
| 16S rRNA 27f | AGAGTTTGATCMTGGCTCAG | All Eubacteria | NT | 60 | |
| 16S rRNA 1490r | TACGGYTACCTTGTTACGACTT | All Eubacteria | NT | 60 | |
| Primer | Sequence (5′–3′)a | Target genus | Temperature (°C) for specificity in: | Reference | |
| Colony hybridisation | PCR | ||||
| Mb 1007r | CACTCTACGATCTCTCACAG | Methylobacter | 60 | 58 | [7] |
| Mc 1005r | CCGCATCTCTGCAGGAT | Methylococcus | 55 | 54 | [7] |
| Mm 1007r | CACTCCGCTATCTCTAACAG | Methylomonas | 55 | 58 | [7] |
| Ms 1020r | CCCTTGCGGAAGGAAGTC | Methylosinus | 55 | 54 | [7] |
| fm 142f | GTTCGGAATAACTCAGGG | Serine pathway methylotrophs | 60 | NT | [7] |
| fm 64f | GAACGGTAACAGGCCTTCGG | RuMP pathway methanotrophs | 60 | NT | [7] |
| 10-γ | GGTCCGAAGATCCCCCGCTT | RuMP pathway methylotrophs | 52 | NT | [34] |
| 9-α | CCCTGAGTTATTCCGAAC | Serine pathway methylotrophs | 52 | NT | [34] |
| mxaF 1003f | GCGGCACCAACTGGGGCTGGT | All Gram negative methylotrophs | NT | 59 | [7] |
| mxaF 1561r | GGGCAGCATGAAGGGCTCCC | All Gram negative methylotrophs | NT | 59 | [7] |
| A189f | GGNGACTGGGACTTCTGG | pMMO/AMO | NT | 56 | [39] |
| A682r | GAASGCNGAGAAGAASGC | pMMO/AMO | NT | 56 | [39] |
| mmoX 882f | GGCTCCAAGTTCAAGGTCGAGC | sMMO+ methanotrophs | NT | 55 | [38] |
| mmoX 1403r | TGGCACTCGTAGCGCTCCGGCTCG | sMMO+ methanotrophs | NT | 55 | [38] |
| 16S rRNA 27f | AGAGTTTGATCMTGGCTCAG | All Eubacteria | NT | 60 | |
| 16S rRNA 1490r | TACGGYTACCTTGTTACGACTT | All Eubacteria | NT | 60 | |
mxaF, methanol dehydrogenase large subunit structural gene; pMMO, particulate methane monooxygenase; AMO, ammonia monooxygenase; mmoX, gene encoding the α subunit of protein A of soluble methane monooxygenase. NT, not tested.
aN=A:C:G:T, S=C:G, M=C:A and Y=C:T.
Methanotroph functional and phylogenetic group-specific 16S rRNA probes/primers
| Primer | Sequence (5′–3′)a | Target genus | Temperature (°C) for specificity in: | Reference | |
| Colony hybridisation | PCR | ||||
| Mb 1007r | CACTCTACGATCTCTCACAG | Methylobacter | 60 | 58 | [7] |
| Mc 1005r | CCGCATCTCTGCAGGAT | Methylococcus | 55 | 54 | [7] |
| Mm 1007r | CACTCCGCTATCTCTAACAG | Methylomonas | 55 | 58 | [7] |
| Ms 1020r | CCCTTGCGGAAGGAAGTC | Methylosinus | 55 | 54 | [7] |
| fm 142f | GTTCGGAATAACTCAGGG | Serine pathway methylotrophs | 60 | NT | [7] |
| fm 64f | GAACGGTAACAGGCCTTCGG | RuMP pathway methanotrophs | 60 | NT | [7] |
| 10-γ | GGTCCGAAGATCCCCCGCTT | RuMP pathway methylotrophs | 52 | NT | [34] |
| 9-α | CCCTGAGTTATTCCGAAC | Serine pathway methylotrophs | 52 | NT | [34] |
| mxaF 1003f | GCGGCACCAACTGGGGCTGGT | All Gram negative methylotrophs | NT | 59 | [7] |
| mxaF 1561r | GGGCAGCATGAAGGGCTCCC | All Gram negative methylotrophs | NT | 59 | [7] |
| A189f | GGNGACTGGGACTTCTGG | pMMO/AMO | NT | 56 | [39] |
| A682r | GAASGCNGAGAAGAASGC | pMMO/AMO | NT | 56 | [39] |
| mmoX 882f | GGCTCCAAGTTCAAGGTCGAGC | sMMO+ methanotrophs | NT | 55 | [38] |
| mmoX 1403r | TGGCACTCGTAGCGCTCCGGCTCG | sMMO+ methanotrophs | NT | 55 | [38] |
| 16S rRNA 27f | AGAGTTTGATCMTGGCTCAG | All Eubacteria | NT | 60 | |
| 16S rRNA 1490r | TACGGYTACCTTGTTACGACTT | All Eubacteria | NT | 60 | |
| Primer | Sequence (5′–3′)a | Target genus | Temperature (°C) for specificity in: | Reference | |
| Colony hybridisation | PCR | ||||
| Mb 1007r | CACTCTACGATCTCTCACAG | Methylobacter | 60 | 58 | [7] |
| Mc 1005r | CCGCATCTCTGCAGGAT | Methylococcus | 55 | 54 | [7] |
| Mm 1007r | CACTCCGCTATCTCTAACAG | Methylomonas | 55 | 58 | [7] |
| Ms 1020r | CCCTTGCGGAAGGAAGTC | Methylosinus | 55 | 54 | [7] |
| fm 142f | GTTCGGAATAACTCAGGG | Serine pathway methylotrophs | 60 | NT | [7] |
| fm 64f | GAACGGTAACAGGCCTTCGG | RuMP pathway methanotrophs | 60 | NT | [7] |
| 10-γ | GGTCCGAAGATCCCCCGCTT | RuMP pathway methylotrophs | 52 | NT | [34] |
| 9-α | CCCTGAGTTATTCCGAAC | Serine pathway methylotrophs | 52 | NT | [34] |
| mxaF 1003f | GCGGCACCAACTGGGGCTGGT | All Gram negative methylotrophs | NT | 59 | [7] |
| mxaF 1561r | GGGCAGCATGAAGGGCTCCC | All Gram negative methylotrophs | NT | 59 | [7] |
| A189f | GGNGACTGGGACTTCTGG | pMMO/AMO | NT | 56 | [39] |
| A682r | GAASGCNGAGAAGAASGC | pMMO/AMO | NT | 56 | [39] |
| mmoX 882f | GGCTCCAAGTTCAAGGTCGAGC | sMMO+ methanotrophs | NT | 55 | [38] |
| mmoX 1403r | TGGCACTCGTAGCGCTCCGGCTCG | sMMO+ methanotrophs | NT | 55 | [38] |
| 16S rRNA 27f | AGAGTTTGATCMTGGCTCAG | All Eubacteria | NT | 60 | |
| 16S rRNA 1490r | TACGGYTACCTTGTTACGACTT | All Eubacteria | NT | 60 | |
mxaF, methanol dehydrogenase large subunit structural gene; pMMO, particulate methane monooxygenase; AMO, ammonia monooxygenase; mmoX, gene encoding the α subunit of protein A of soluble methane monooxygenase. NT, not tested.
aN=A:C:G:T, S=C:G, M=C:A and Y=C:T.
When oligonucleotide probes targeting methanotrophs were labelled with digoxigenin-11-dd UTP, they could be used to detect different groups of methanotrophs present in soil samples that had been enriched for 1 month in a methane–air atmosphere [1]. Detection methods for methanotroph-specific rRNA sequences performed reliably in Northern slot blot assays with detection limits of around 10 ng of RNA from pure cultures [1]. Methanotroph-specific 16S rRNA gene probes have been designed to specifically detect sequences from the genera Methylosinus, Methylococcus, Methylobacter, Methylomonas and Methylomicrobium[7,31]. These have been applied in studies on agricultural soil, marine enrichments and peat bog DNA samples. In the acidic peat environment, probes identified novel sequences which may be from putative acidophilic methanotrophs that are closely related to Methylosinus/Methylocystis (Type II methanotrophs) [7]. The change in composition of bacterial populations of methane enrichments from the marine environment was monitored by analysing 16S rDNA libraries constructed from DNA (taken before and after enrichment) using universal eubacterial 16S rRNA primers and PCR. Methanotroph group-specific probes failed to give a signal with samples from these 16S rDNA enrichments even though the structural gene for methanol dehydrogenase mxaF was detected by PCR (see below) in the same DNA samples. The dominant 16S rRNA sequence that was present in samples only after enrichment on methane was recovered from libraries and sequenced. This 16S rRNA gene was found to show a close phylogenetic relationship to Methylomonas indicating that novel methanotrophs related to extant Methylomonas spp. were present in enrichment cultures [31].
6 Fluorescent in situ hybridisation
Fluorescent in situ hybridisation (FISH) is a powerful tool in microbial ecology, allowing the detection of specifically stained single cells in different environments, determination of their phylogenetic affiliation and the ability to quantify organisms in mixed communities without the need for isolation in pure cultures [32]. These techniques also allow the possibility of detection of previously uncultured organisms. Labelled oligonucleotide probes can be designed and used to detect microorganisms at the kingdom-, genus- and species-specific levels in a variety of environments including pure cultures, environmental enrichments, biofilms, and soils [33].
Few studies have specifically looked at the detection of methanotrophs with fluorochrome labelled probes. Fluorochrome labelling of probes designated 9-α and 10-γ (see Table 2) was successfully used for differentiating bacteria in the methylotroph subdivisions and classifying a previously unidentified methanotroph [34] and also to enumerate methylotrophs in a natural freshwater environment [35].
Based on the 16S rDNA sequence of a putative marine methanotroph, two oligonucleotide probes (Mm650 and Mm850, Table 2) were synthesised with a rhodamine fluorescent label attached to their 5′ termini. FISH was then used to confirm the abundance of this organism in enrichment cultures. Furthermore, these fluorescently labelled probes have been used to determine the response of this marine methanotroph to changes in culture conditions, thereby allowing the optimisation of enrichment culture conditions and isolation strategies for this organism [36].
Recently, we have designed fluor-labelled oligonucleotides which specifically detect and discriminate between Type I and Type II methanotrophs. These indocarbocyanine (Cy3)-labelled probes have been used to confirm the identity of methanotrophs in pure cultures (Fig. 3A,B) and environmental enrichments (Fig. 3C,D). Detection of methanotrophs in enrichment cultures correlate well with their genetic fingerprints determined by PCR and RFLP analysis. Other fluorochrome-labelled probes are under investigation for the detection of methanotrophs at the individual genus level. A Methylococcus-specific Cy3-labelled probe has been designed which shows excellent specificity in extensive testing on control organisms and in enrichment cultures (Fig. 3E,F).
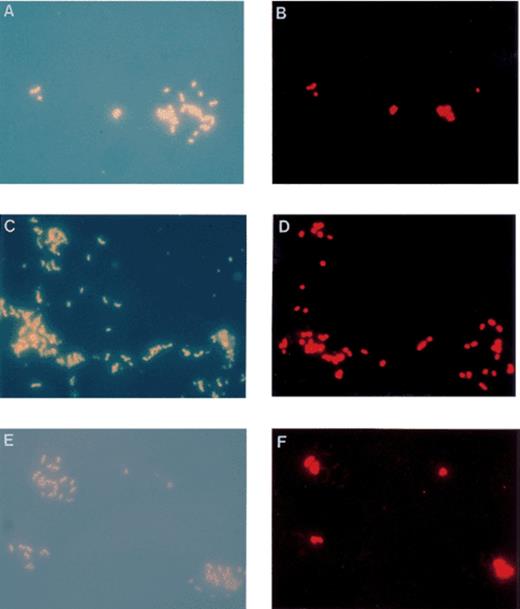
Epifluorescence photomicrographs of DAPI stained cells (left) and the corresponding in situ hybridisation with Cy3 labelled 16S rDNA probes (right). Photomicrographs represent the same field of view. (A and B) Methylococcus capsulatus (Bath) and Methylocystis parvus (OBBP) mixed culture. In situ hybridisation with a type I methanotroph specific probe (MG-64), highlighting detection of the Methylococcus cells and not Methylocystis cells. (C and D) Environmental soil enrichment of methanotrophs. In situ hybridisation with the MG-64 type I methanotroph specific probe. (E and F) Methylococcus capsulatus (Bath) and Methylocystis parvus (OBBP) mixed culture. In situ hybridisation with a Methylococcus genus specific probe (MC-1020), specifically detecting Methylococcus cells and not Methylocystis cells.
Although FISH provides a means to detect microorganisms in their natural environment without the need for cultivation, limitations do exist. Probe conferred fluorescence is directly correlated with the number of copies of rRNA in the cell, which in turn is dependent on cell growth rates [37]. Within the environment, lower cell growth rates lead to low detection sensitivity of individual cells. Moreover, environments that exhibit high background fluorescence can interfere with specific detection of bacteria [33].
7 Functional gene probes for methanotrophs
The high degree of identity between sMMO genes has enabled the design of PCR primers which specifically amplify each of the five sMMO structural genes [38] such that amplification of each of these genes is now possible from cultured methanotrophs containing sMMO and from total DNA from a variety of different freshwater, estuarine, soil and wetland samples. Subsequent cloning and DNA sequencing of a number of these PCR products has proved that methanotroph-specific sMMO-encoding DNA had been amplified from environmental DNA samples and that these genes are very similar to, but not identical to known sMMO gene sequences [38].
The PCR technology developed with sMMO genes is useful for studying wetlands or contaminated aquifers where methanotrophs containing sMMO might dominate in a copper-depleted environment. However, a better functional gene probe for these organisms would be one based on the pMMO, present in all extant methanotrophs.
Sequence data on pmo and amo genes have allowed the design of degenerate PCR primers which will specifically amplify a 525 bp internal DNA fragment of these genes from a variety of methanotrophs and nitrifiers. Representatives of each of the phylogenetic groups of methanotrophs (α and γ Proteobacteria) and ammonia oxidising nitrifiers (β and γ Proteobacteria) were included. Phylogenetic analysis of these data gives phylogenies similar to that of the 16S rRNA from these bacteria (Fig. 4) and suggest that the pMMO and AMO may be evolutionarily related enzymes, despite their different physiological roles in these bacteria [39].

Phylogenetic analysis of the derived amino acid sequences of pmoA genes from methanotrophs and amoA genes from nitrifiers. The dendrogram shows the results from analysis using PROTDIST, and the bar represents 3% sequences divergence, as determined by measuring the lengths of the horizontal lines connecting any two species.
The degenerate oligo PCR primers described above have now been used successfully to specifically amplify pmo/amo sequences from DNA isolated from a wide variety of environments including seawater, freshwater, agricultural and forest soils and wetlands. Sequencing of these PCR products and examination of derived amino acid sequences of the PmoA/AmoA now allows identification of these sequences as those originating from an α, β or γ representative of the methane oxidising Proteobacteria. Sequences from putative acidophilic methanotrophs were detected in peat samples using the pmo/amo primer set [40]. At present, an extensive data base of sequences from extant methanotrophs and nitrifiers is being established which will aid further molecular ecological studies. More recently PCR primers which specifically amplify amoA sequences have been designed [41] and pmoA specific primers are currently being designed (Murrell et al., unpublished). These new primers may help assess the relative roles of methanotrophs and nitrifiers in the cycling of methane in a number of interesting environments including seawater, agricultural and forest soils.
Another potentially useful marker is the mxaF gene. PCR primers which specifically amplify a 550 bp fragment of mxaF have been used to extend the database of mxaF sequences from methanotrophs and methylotrophs and to identify mxaF sequences in marine, soil and wetland samples [28,31,38]. The mxaF gene has also been used as a hybridisation probe in conjunction with restriction fragment length polymorphism (RFLP) to distinguish between a number of different methanotrophs and methylotrophs. The mxaF gene is not specific for methanotrophs, however.
8 Denaturing gradient gel electrophoresis
Another technique to analyse microbial community diversity without using culture based methods is denaturing gradient gel electrophoresis (DGGE). DGGE utilises sequence variations in PCR-amplified DNA fragments of identical length and separates the fragments based on their different mobility in polyacrylamide gels of increasing denaturant gradient [42]. DGGE banding patterns provide direct visualisation of dominant bacterial populations allowing a rapid and comprehensive way to survey DNA and RNA sequence diversity in a wide range of environments. Separated bands within DGGE gels can be excised, re-amplified by PCR and sequenced which provides phylogenetic affiliation of community members and eliminates the requirement of cloning and associated problems.
DGGE has been used to investigate microbial diversity in a number of different terrestrial and aquatic environments [43–44,45]. These methods are used for the differential analysis of 16S rDNA fragments and more recently, PCR amplified fragments of functional genes such as the [NiFe] hydrogenase gene of Desulfovibrio spp. [44] and the mxaF methanol dehydrogenase gene of methylotrophs. Two mxaF primers (see Table 2) have been used in DGGE to investigate and identify new Hyphomicrobium spp. from different habitats including biofilms, groundwater and lake water [45]. Although considerable sequence variation existed, mxaF gene fragments exhibited similar electrophoretic mobility, making identification of environmental isolates impossible without sequence analysis and verification. Recently, specifically amplified 16S rRNA fragments have been used to successfully analyse the β-subdivision of ammonia-oxidising bacterial populations in coastal sand dune samples [46].
The reports of analysis of methanotroph diversity in different environments using DGGE analysis have to date been few. However, universal 16S rDNA primers have been used to amplify a 200 bp product (position 341–534 in 16S rDNA of Escherichia coli) from species of rhizobia and methanotrophs in an effort to assess the ability of this technique to resolve fragments amplified from phylogenetically related bacterial groups [47]. However, resolution of these PCR products was found to be difficult. This may be a common problem with the number of fragments visualised on DGGE gels in many cases giving an underestimate of the actual microbial community diversity. Recently Jensen and co-workers [48] used the universal 16S rDNA primer set combined with DGGE to assess the diversity of agricultural soil enrichments grown with methane as the sole carbon and energy source. Sequencing of individual DGGE bands identified a high diversity of methylotrophic populations, with most sequences clustered distantly on a branch within the α-Proteobacteria, suggesting the presence of previously undescribed methylotrophs.
Recent work in our laboratory using a universal primer set (position 341–918 in 16S rDNA of E. coli) [43] has demonstrated good DGGE resolution of amplified DNA fragments from a number of representative genera of methanotrophs. Current investigation of various environmental sites is underway using both the 16S rDNA primer set and the mxaF functional gene primer set to investigate both total bacterial and methylotrophic bacterial diversity. The use of DGGE techniques to investigate the diversity in the environment of pmoA in methanotrophs is also being evaluated. However, due to sequence degeneracy in the primers (Table 2), multiple banding patterns on DGGE gels were observed for amplified methanotroph and nitrifier pmoA sequences (Bourne et al. unpublished). Development of new pmoA-specific primers is currently underway.
9 Future prospects
While molecular microbial ecology is being used with considerable success, there are limitations to these techniques [49], including DNA extraction, PCR bias [50] and cloning. However, the ever increasing database of methanotroph-specific DNA or RNA sequences, when coupled with PCR technology, now allows a variety of molecular ecological techniques to be applied directly to these bacteria. A number of important areas for research regarding the biological cycling of methane can now be examined or re-examined. Key questions still to be answered include: the relative importance of Type I and Type II methanotrophs, particularly those containing the additional methane oxidation system sMMO, in certain environments such as peat bogs and aquifers; the roles of ammonia oxidising nitrifiers and methanotrophs in the oxidation of methane in agricultural and forest soils and the effects of both long and short-term ammonium fertilisation on the capacity of these soils to oxidise methane [4]; the significance of gene duplication of pmo and amo genes in methanotrophs and nitrifiers and do they encode enzymes with different kinetic properties which may confer an advantage or be more important under certain environmental growth conditions; and which organisms are responsible for anaerobic methane oxidation in certain environments [30]. Methanotroph symbioses with wetland plants and marine invertebrates [1] can be further examined by fluorescence in situ hybridisation techniques using suites of methanotroph/methylotroph-specific probes. Which organisms in soils are responsible for methane oxidation at low (ambient, atmospheric) concentrations of methane – the so called ‘high affinity methanotrophs’? Does their enzyme system differ from that in extant methanotrophs and is it a pMMO or a sMMO-like enzyme? Molecular ecology techniques, coupled with methane oxidation measurements, will almost certainly allow the detection and isolation of novel and exciting methanotrophs with unique physiology and biochemistry from the natural environment.
Acknowledgements
The authors acknowledge funding from the NERC, BBSRC, AFRC and the EU for the work from this laboratory summarised in this review.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}