





Abstract
In many animals, sperm competition and sexual conflict are thought to drive the rapid evolution of male-specific genes, especially those expressed in the testes. A potential exception occurs in the male pregnant pipefishes, where females transfer eggs to the males, eliminating testes from participating in these processes. Here, we show that testis-related genes differ dramatically in their rates of molecular evolution and expression patterns in pipefishes and seahorses (Syngnathidae) compared to other fish. Genes involved in testis or sperm function within syngnathids experience weaker selection in comparison to their orthologs in spawning and livebearing fishes. An assessment of gene turnover and expression in the testis transcriptome suggests that syngnathids have lost (or significantly reduced expression of) important classes of genes from their testis transcriptomes compared to other fish. Our results indicate that more than 50 million years of male pregnancy have removed syngnathid testes from the molecular arms race that drives the rapid evolution of male reproductive genes in other taxa.
A major goal of evolutionary genomics is to resolve the mechanisms responsible for broad patterns of genome evolution. One well-established observation regarding the evolution of genomes is that male-biased genes, especially those specific to the testis, tend to exhibit a pattern of rapid evolution and stronger signatures of selection compared to other genes in the genome (Meiklejohn et al. 2003; Zhang et al. 2004; Ellegren and Parsch 2007). The typical explanation for this pattern is that rapid evolution is driven either by sperm competition, which occurs when the ejaculates from different males compete with one another to fertilize eggs (Parker 1970, 1984), or sexual conflict, where males are selected to manipulate the reproductive interests of the females in a way that enhances short-term male fitness at the expense of female fitness (Parker 1979, 1984; Yapici et al. 2008). These mechanisms are not mutually exclusive; hence, we may expect sperm competition and sexual conflict to act in tandem in many sexually reproducing organisms.
With respect to sperm competition, we expect sexual selection to target sperm abundance, testis size, sperm characteristics, and seminal fluid proteins involved in male-male competition. In animals where females mate with multiple males per reproductive bout, sperm abundance and testis size are expected to increase as a function of the intensity of sperm competition (Parker 1990; Møller and Briskie 1995; Vahed and Parker 2012). This expectation is so pervasive that testis size has become a metric for the strength of sperm competition in a wide range of taxa, including primates, fish, and birds (Møller 1988; Stockley et al. 1997; Pitcher et al. 2005). Modification to sperm structure or size is also a common occurrence, with hallmark examples coming from Drosophila, where sperm can be 10 times the male's body length, as observed in Drosophila hydei (Pitnick and Markow 1994; Simmons 2001). Sperm competition risk is also expected to lead to an increase in abundance or recruitment of novel seminal fluid proteins, some of which may have evolved to inactivate sperm from competing males (Zbinden et al. 2003; Ramm et al. 2015; Simmons and Lovegrove 2017; Whittington et al. 2017).
The situation becomes somewhat more complex when we turn our attention to sexual conflict. In this case, males experience selection to manipulate female behavior in a way that enhances male fitness at the expense of the female's fitness (Arnqvist and Rowe 2005). Females, in turn, are selected to resist such male manipulation, potentially leading to an arms race between the reproductive interests of males and females (Arnqvist and Rowe 2005). Compelling examples of sexual conflict come from insects, where males have been shown to produce a range of effects on females, from reducing their mating receptivity, as occurs in katydids (Simmons and Gwynne 1991), to inflicting harm by increasing a female's fecundity beyond her optimal rate, a phenomenon observed in fruit flies and field crickets (Baumann 1974; Loher 1979; Fowler and Partridge 1989; Arnqvist and Nilsson 2000). These examples, along with many others, have led to an expansion of our understanding of the varied mechanisms that males use to manipulate females, where tactics include mating plugs, seminal receptivity inhibitors, anti-aphrodisiacs, seminal toxins, aggressive sperm, and infertile sperm (Arnqvist and Rowe 2005).
This wide variety and divergence of reproductive phenotypes must ultimately be reflected in changes at the level of the genome. In terms of specific protein-coding genes, both sperm competition and sexual conflict are thought to increase rates of molecular evolution and alter expression patterns as males compete with other males and evolve to overcome female defenses. A large proportion of genes involved in male reproduction appear to evolve rapidly and show signs of positive selection (Gavrilets 2000; Wyckoff et al. 2000; Swanson and Vacquier 2002; Torgerson et al. 2002). Expression patterns can also change in response to increased sperm competition risk, as evidenced by investment plasticity in ejaculate components in the presence of rival males (Fedorka et al. 2011; Simmons and Lovegrove 2017). Moreover, sperm competition and sexual conflict are also expected to lead to a burst of novel genes and gene turnover within the genome, as new genes are recruited to participate in these processes (Zhang et al. 2007; Roberts et al. 2009; Harrison et al. 2015; Whittington et al. 2017).
Although the predictions of sperm competition and sexual conflict theory have been tested extensively in species with polygamous mating systems and strong sexual selection on males, very few studies have addressed these issues in species in which sexual selection and sperm competition are expected to be weak or absent. For example, we might expect species with monogamous mating systems, self-fertilization, parthenogenesis, or sex-role reversal to show different patterns of evolution compared to species with high potential for male-male competition and sexual conflict.
Species with a low potential for sperm competition are expected to evolve several phenotypic traits, such as reduced sperm abundance and testis size, a prediction that has been upheld in monogamously selected experimental populations (Hosken and Ward 2001; Simmons and García-González 2008). Low sperm competition also might relax selection on sperm morphology, potentially resulting in abnormal structure, as observed in the eusocial naked-mole rat (Heterocephalus glaber) (van der Horst et al. 2011). Also worthy of mention are the Eurasian bullfinch (Pyrrhula pyrrhula), Azores bullfinch (Pyrrhula murina), and the Greater Bandicoot rat (Bandicota indica), which have unusual sperm shape and low testis weight, leading to the hypotheses that these traits evolved as a consequence of an evolutionary history of monogamy (Durrant et al. 2010; Thitipramote et al. 2011; Lifjeld et al. 2013). The evolution of these testis and sperm-related phenotypes might include a shift in the rate of molecular evolution in genes typically involved in testis function and the loss or reduced expression of genes with historically male-biased expression. For monogamous systems, testis-related traits (and the underlying genes) might be predicted to experience purifying selection or to be evolving neutrally, because they are either required to maintain a function or would experience relaxed selection due to a lack of sperm competition (Birkhead and Møller 1996; Bauer and Breed 2006; van der Horst and Maree 2014).
In self-fertilizing systems, a significantly smaller transcriptome and the absence of sex-biased genes is also predicted. Such a pattern has been documented in the self-fertilizing nematodes Caenorhabditis elegans and Caenorhabditis briggsae (Thomas et al. 2012), the latter of which was shown to have evolved a smaller genome and experienced the loss of some sexually important genes, such as male secreted short genes, which are involved in sperm competition and the production of sperm glycoproteins (Yin et al. 2018). Thus, some of the predictions regarding genome evolution as a consequence of sexual selection and conflict have been supported in at least one system. However, further work is necessary to strengthen our understanding of this phenomenon and to establish how broadly these predictions apply across disparate taxonomic groups and reproductive systems.
Here, we focus on pipefishes and seahorses (family Syngnathidae), in which females transfer eggs to the brood pouches of males and the male carries the developing embryos during an extended pregnancy (Paczolt and Jones 2010). All studies of parentage in pipefishes and seahorses have shown that males are always the genetic fathers of the offspring in their pouch, indicating that sperm competition is absent (Jones and Avise 1997a,b, 2001; Mccoy et al. 2001; Avise et al. 2002). Additional research has revealed the presence of traits typically associated with reduced sperm competition, including low testis weight and low sperm counts (Kvarnemo and Simmons 2004; Piras et al. 2016a). Furthermore, males possess no known mechanism to transfer sperm, seminal fluid, or any other related molecules to the female during mating, removing the testis from a role in sexual conflict. Indeed, the tables are turned, as the female transfers ovarian fluid and eggs to the male during mating (Paczolt and Jones 2010). This highly unusual mating strategy of syngnathids leads to the hypothesis that the male gonad-biased transcriptome will not be a target of selection related to sperm competition or sexual conflict, a pattern that starkly contrasts with most other vertebrates. Syngnathid fishes thus provide a unique opportunity to perform a critical test of the hypothesis that testis-related genes evolve rapidly due to sexual selection and sexual conflict. Here, we address the following two questions: (1) Is the pattern of molecular evolution in genes involved in testis function and sperm competition different in syngnathid fishes compared to fishes with more conventional reproductive strategies? And (2) how has the suite of genes expressed in the testis been altered by evolution under male pregnancy?
Methods
MOLECULAR EVOLUTION OF TESTIS GENES
We compared five members of Syngnathidae with two other teleost groups with more common reproductive modes: the livebearing fishes from Poeciliidae and the spawning fishes from Cichlidae. We used assembled genomes for five species of Syngnathidae, including two seahorses (Hippocampus comes and Hippocampus erectus) that exhibit strict monogamy and three polygamous pipefishes (Syngnathus scovelli, Syngnathus floridae, and Syngnathus acus) (Wilson et al. 2003). The annotated genome for the Gulf pipefish (S. scovelli) was obtained through the Cresko Lab website (https://creskolab.uoregon.edu/pipefish/) (Small et al. 2016). We also obtained the assembled H. erectus genome reported by Lin et al. (2017) and the assembled (but not annotated) S. floridae genome from NCBI. Annotated genomes were obtained from the NCBI Genome Database for the following species (all accession numbers for this article are listed in Table S1): H. comes, S. acus, and Pundamilia nyererei (Brawand et al. 2014); Maylandia zebra (Brawand et al. 2014; Conte and Kocher 2015); Haplochromis burtoni (also known as Astatotilapia burtoni) (Brawand et al. 2014); Oreochromis niloticus (Brawand et al. 2014); Xiphophorus maculatus (Schartl et al. 2013); and Poecilia mexicana, Poecilia latipinna, and Poecilia reticulata (Künstner et al. 2016).
Genes (n = 24) involved in spermatogenesis, sperm structure, or seminal fluid composition were selected for their expected involvement in sperm competition from previous research, gene ontology predictions, ortholog predictions, and InterPro domain predictions (McGinnis and Madden 2004; Blum et al. 2021) (Data S1). Orthologs were identified using OrthoDB (v10.1) for all members of Cichlidae and Poeciliidae under study, as well as for H. comes (Kriventseva et al. 2019). The ortholog sequence from H. comes was then reciprocally blasted (NCBI's TBLASTX and BLASTN) against genome assemblies for S. scovelli, S. floridae, S. acus, and H. erectus, which are not represented in the OrthoDB database. The best blast hits, with E-values less than 1 × 10−20, were retained as orthologs for these syngnathid species.
Amino acid sequences were aligned using the package ClustalW (Thompson et al. 1994) implemented in MEGA (version 7.0.26) (Kumar et al. 2016). A phylogenetic tree (Fig. 1a) was reconstructed using the concatenated alignment of all 24 genes using MEGA version 7.0.26 (Kumar et al. 2016). The tree was inferred by using the Maximum Likelihood method based on the JTT matrix-based model (Jones et al. 1992). Neighbor-Joining and BioNJ algorithms were applied to a matrix of pairwise distances estimated using a JTT model for the heuristic search for the initial tree, and the topology with the best log likelihood value was selected. All positions with less than 95% site coverage were eliminated, leaving a total of 10,321 positions used in the final dataset. The percentage consensus for 1000 bootstrap replicates for a clade is reported, and branch lengths are measured as the number of substitutions per site. The final topology agreed with previous studies (Sanciangco et al. 2016; Hamilton et al. 2017; Rabosky et al. 2018) and the tree was used unrooted, without branch length information, as a reference for downstream molecular evolution analyses. The tree as depicted in Figure 1a represents the rate of substitutions for the genes of interest between taxa and not a phylogenetic reconstruction based on whole-genome alignments.
The ratio of nonsynonymous to synonymous substitutions (dN/dS or ω) was estimated using models implemented in the codeml program of the PAML package (version 4.9) (Yang 2007). We ran two analyses to test the rate of molecular evolution: a site-model that allows ω to vary over different sites in a gene, and a branch model that allows ω to vary over different branches within our tree. For site-based analyses, we performed each test within each of the following three taxonomic groups: pipefishes and seahorses (S. scovelli, S. floridae, S. acus, H. comes, and H. erectus), spawning cichlids (P. nyererei, M. zebra, H. burtoni, and O. niloticus), and livebearers (X. maculatus, P. mexicana, P. latipinna, and P. reticulata). For branch models, genes were aligned across all species (including those within Syngnathidae, Poeciliidae, and Cichlidae). For both site and branch models, we estimated ω and the likelihood ratio test statistic for each possible selection model for the same set of 24 genes involved in male reproduction.
The site models (model = 0) for M0, M1a, M2a, M7, and M8 (NSsites = 0, 1, 2, 7, 8) were run for each gene, within each taxonomic group, and the model of best fit was determined through a likelihood ratio test (α = 0.05). We tested each of the three taxonomic groups separately, which provided a model of best fit and an overall ω estimate for each gene for each taxonomic group (72 total analyses). Primarily, we focused on a comparison between three models: a one-ratio model (M0) of purifying selection (ω < 1), a nearly neutral model (M1a) that includes two classes of sites (purifying selection [ω < 1] and neutral [ω = 1]), and a positive selection model (M2a) with three classes of sites (positive selection [ω > 1], purifying selection [ω < 1], and neutral [ω = 1]). Additionally, the beta model with 10 classes of sites (ω ≤ 1) (M7) and the beta and ω model with 11 classes of sites (10 with ω ≤ 1 and one with ω > 1) (M8) were also run. For genes where M8 was significant, the null hypothesis M8a (NSsites = 8, fix_omega = 1, omega = 1) was also run (Swanson et al. 2003; Wong et al. 2004). Our results concentrate on our M0-M1a-M2a comparisons, because these provide a more stringent test and are nearly identical to our M0-M7-M8-M8a comparisons (Yang et al. 2000). Results for the likelihood ratio test for all site models, as well as the dN/dS estimates for each gene, can be found in Data S1. Additionally, the dN/dS values for all genes were averaged within the three family groups and the variance among genes within each group was calculated.
The branch models (NSsites = 0) for M0 versus M2 (model = 0, 2) were run and the model of best fit was determined through a likelihood ratio test (α = 0.05). We tested the Syngnathidae branch for a different value of ω, in comparison to the other species of Cichlidae and Poeciliidae. This approach estimated two separate ω values (one for Syngnathidae and one for all other fish) and a likelihood value, which was compared against a null model (M0) that estimates a uniform ω for all fish. Results for the dN/dS estimates and the likelihood ratio tests for all branch models can be found in Data S1.
GENOME-WIDE dN/dS ESTIMATION IN SYNGNATHUS
To compare the rate of molecular evolution of the 24 genes of interest to a genome-wide expectation within the genus Syngnathus, we identified single-copy orthologs among four species of pipefish. The genome files for S. acus, S. rostellatus, and S. typhle were obtained from NCBI (Table S1), and analyzed along with the S. scovelli genome from the Cresko Lab. The genome, annotation, and protein files for S. acus were used as a reference species to run MAKER (version 3.01.03) (Cantarel et al. 2008) to annotate the genomes of the other three Syngnathus species. Syngnathus acus was used as the reference because it has the highest quality annotation of the species listed here.
After genome annotations were obtained for each of our species, OrthoFinder (version 2.5.4) (Emms and Kelly 2019) was used to align proteins and identify common orthologs between species of Syngnathus. OrthoFinder produced a list of all single-copy orthologs, which were then aligned at the protein level using MAFFT (version 7.487) (Katoh and Standley 2013). The nucleotide sequences were then retrieved for each protein alignment from the appropriate genomes using PAL2NAL (version 14) (Suyama et al. 2006). These nucleotide alignments were analyzed in codeml (PAML version 4.9) (Yang 2007) using a site model analysis, as described above (MOLECULAR EVOLUTION OF TESTIS GENES in Methods). The result was a dN/dS estimate and best fitting model of selection for each gene identified as a single copy ortholog across four members of the genus Syngnathus. Alignments with a dN/dS estimate of 999 were checked and removed (eight alignments, leaving a total of n = 13,545).
GENE TURNOVER IN GULF PIPEFISH TESTES
For this analysis, we addressed the hypothesis that patterns of gene turnover in the testes of syngnathid fishes would show a signature consistent with the loss of sperm competition in pipefishes and seahorses. We used RNA-seq data to identify testis-enriched genes in the Gulf pipefish (S. scovelli) and the Japanese pufferfish (Takifugu rubripes), which has external spawning. We selected the Japanese pufferfish for this comparison because it likely engages in sperm competition, is a member of the Percomorpha (which also includes the Syngnathidae), has a well-annotated genome, and is one of the few fish with replicated testis and ovary RNA-seq data available (Yamahira 1994). We blasted each set of highly expressed testis genes against the genome of the other species to determine the percentage of ortholog matches for highly expressed, testis-enriched genes. The proportion of matching testis orthologs was then compared to an expected distribution of ortholog matches, which was generated by randomly sampling sets of orthologs from the entire genome (without regard to testis enrichment).
We generated the S. scovelli RNA-seq data by using next-generation sequencing on testis tissue samples from seven males and ovary samples from five females. Fish were collected from the Gulf of Mexico, USA (Redfish Bay, TX) in accordance with IACUC approval (2013-0020). Only individuals that showed either a well-developed brood pouch or secondary sexual traits were used to ensure all fish were sexually mature. Fish were euthanized with an overdose of MS-222. RNA was extracted and isolated from both testis and ovary tissues using TRIzol® Reagent (Life Technologies, Carlsbad, CA) (Leung and Dowling 2005). Libraries were prepared using TruSeq mRNA Library Prep Kit version 2 by Michigan State University RTSF Genomics Core and quality was tested using Caliper GX and qPCR methods. All individuals were barcoded and sequenced individually using two lanes of Illumina HiSeq 2500 sequencing, and base calling was done with Illumina Real Time Analysis (RTA) (version 1.17.21.3). The output of RTA was demultiplexed and converted to FastQ with Illumina Bcl2fastq (version 1.8.4) resulting in 150 bp paired end reads used for downstream analyses. Raw paired-end reads from testes and ovaries for the Japanese pufferfish were obtained through the NCBI SRA library (Table S1) (Wang et al. 2017; Yan et al. 2018). Trinity (version 2.8.4) (Grabherr et al. 2011) was used for de novo assembly of testis-only reads into a separate transcriptome for both T. rubripes and S. scovelli using default parameters. For this analysis, we used a de novo assembled transcriptome instead of a reference genome for two main reasons. The first is using a de novo transcriptome ensures that we capture sequences that might not be present in the genome (Grabherr et al. 2011). This can be especially important if genomes have different assembly annotation qualities. The second is that the S. scovelli genome was not annotated using testis RNA-seq data, so although all the genes should be present within the genome, novel transcripts that are important to this specific analysis might not be (Small et al. 2016). Trimmomatic (Bolger et al. 2014) was used to trim reads inside of Trinity, using the settings SLIDINGWINDOW:4:5 LEADING:5 TRAILING:5 MINLEN:25.
Reads from both the testes and the ovaries were then mapped back to the species-specific testes transcriptome using RSEM (version 1.3.1) (Li and Dewey 2011) and the raw contig read counts were recorded. From these datasets, EBSeq (version 3.8) (Leng and Kendziorski 2019) detected differentially expressed contigs between testes and ovaries and generated a posterior probability for each contig. Using a false discovery rate (FDR) of 0.05, only contigs with a posterior probability of differential expression ≥0.95 were kept for analysis. Testis-biased fold changes were calculated per contig as the mean testis expression level divided by the mean ovary expression level. Transcripts that had an expected count of at least 500 reads and a testis-biased fold change of 2 or more were extracted and labeled as highly and differentially expressed. These cutoff values were chosen to provide a substantial sample size while ensuring we chose transcripts that were highly and differentially expressed. A total of 325 transcripts for pufferfish and 831 transcripts for pipefish were identified as highly and differentially expressed in the testis.
The transcripts from each fish were then blasted using NCBI's TBLASTX (McGinnis and Madden 2004) to their own annotated genome file to find a matching annotated gene. The annotated genome for T. rubripes was available through the NCBI Genome Database (Aparicio et al. 2002). A match was any sequence that had a blast hit with an E-value ≤ 1 × 10−20. If a transcript did not have a match in the annotated gene list, the transcript was then blasted against the whole genome. If there was a match in the whole genome file, then the transcript sequence from the de novo assembly was used in this analysis. When multiple transcripts matched the same gene, in both the annotated genome and the whole genome file, the match with the smallest E-value was used. This was done to prevent potential bias resulting from differences in paralog density or annotation quality.
The corresponding annotated sequences of the highly and differentially expressed transcripts from T. rubripes (or the transcript sequences as described above) were then blasted using NCBI's TBLASTX, to the whole genome of S. scovelli. An ortholog match was any sequence that had an alignment with an E-value ≤ 1 × 10−20. The reciprocal search for the S. scovelli highly and differentially expressed genes in the whole genome for T. rubripes was also performed. This analysis was used to calculate a percentage of orthologs present for testis-biased genes of one species in the other species’ genome.
To generate a null expectation of ortholog presence in the reciprocal comparisons between species, we sampled random sets of protein coding genes from the entire genome and performed the same analysis as described above. The number of genes in the set was identical to the number of testis-biased genes in the species of interest. We then used TBLASTX to compare these genes from the species of interest against the whole genome of the other species. The percentage of genes with significant hits was recorded, and we repeated this entire procedure 1000 times to generate a distribution for randomly chosen sets of genes. This distribution represents a null expectation for the proportion of randomly chosen genes that are present in the other species’ genome for a sample of genes equal in size to the number of testis-biased genes identified for the focal species. We then compared the proportions for testis-biased genes against this null distribution for each reciprocal comparison between pipefish and pufferfish (Fig. 2).
FUNCTIONAL EVOLUTION OF PIPEFISH TESTES
To compare the testis transcriptome of the Gulf pipefish (S. scovelli) to other related species of fish, we first obtained gonadal RNA-seq reads and genome data from six other representatives of the Percomorpha. We also obtained data from zebrafish (which is not in Percomorpha) as an outgroup. Genomes and RNA-seq data were obtained from the NCBI genome and SRA databases for the following species: T. rubripes, Oryzias latipes, P. reticulata, Paralichthys olivaceus, Nothobranchius furzeri, and Lates calcarifer (Table S1). We also obtained the zebrafish (Danio rerio) genome, with two testis and two ovary datasets (Table S1).
We trimmed the testis and ovary RNA-seq reads using Trimmomatic (with settings ILLUMINACLIP:TruSeq3-PE.fa:2:30:10:2:keepBothReads LEADING:3 TRAILING:3 MINLEN:75), and we aligned the reads against assembled genomes using HISAT2 (version 2.1.0) (Kim et al. 2015). Sorted BAM files containing the HISAT2 results were fed into StringTie (version 2.1.3b) (Pertea et al. 2015) to identify and quantify transcripts. Within each species, we ran StringTie separately for each RNA-seq dataset, merged the results into a single merged gff file, and then estimated species-specific abundances against this merged gff transcript file.
For the analysis of the top 1000 testis genes, we ranked all genes identified by StringTie by TPM (transcripts per million), from largest to smallest. For each of the top 1000 genes in terms of TPM, we extracted all transcripts assembled by StringTie. The cutoff value was chosen to provide a substantial sample size while ensuring we chose genes that were most expressed. Each transcript was converted to the protein encoded by its longest open reading frame by using the C++ program fastatoorf. To compare these transcripts against a common reference proteome, we downloaded all annotated zebrafish proteins from UniProt (Zebrafish-UP000000437_7955) and constructed a local BLAST database. We used BLASTP to compare all retained transcripts against the zebrafish proteome, using an E-value cutoff of 1 × 10−20. For each transcript with at least one hit below the E-value cutoff, we retained the zebrafish protein ID of the best hit. If multiple transcripts blasted to the same protein, duplicate entries were removed from the final list. Thus, for each species, we obtained a list of putative zebrafish orthologs that correspond to the top 1000 genes expressed in the testis transcriptome of the focal species. The number of unique genes on the final lists was 824 for P. olivaceus, 805 for T. rubripes, 746 for P. reticulata, 811 for N. furzeri, 839 for O. latipes, 711 for L. calcarifer, 919 for zebrafish, and 805 for Gulf pipefish (mean = 807.5 genes).
For the gene ontology analysis, we used the PANTHER database and associated tools (version 15.0) (Thomas et al. 2003a,b; Mi et al. 2013). We uploaded testis gene lists from each species under consideration and performed a functional classification based on the zebrafish database. We performed gene ontology analyses for molecular function and biological process by using the curated PANTHER GO-Slim databases. We also performed an analysis using the PANTHER Protein Class database for each species. Results were compiled and compared as percentages of the total hits in each database. For statistical analyses, we calculated expected proportions using the mean across the six non-pipefish Percomorphs. These proportions were used to derive expected values for Gulf pipefish and zebrafish, under the null hypothesis that these species do not differ from Percomorphs with respect to the functional classification of highly expressed testis genes. A χ2 test was used to test this hypothesis for each of the gene ontology analyses (i.e., biological process, molecular function, and PANTHER protein class). Categories with expected values less than five were lumped for the purposes of this test. In the case of a significant χ2 test, the number of genes assigned to each category was compared between Percomorphs and either pipefish or zebrafish using a one-sample, two-sided t-test, with an FDR correction at 0.05.
For the overrepresentation analysis, we used the PANTHER tools to perform a statistical overrepresentation test. This test was performed using the GO biological process complete dataset implemented in PANTHER. The zebrafish genome was used as the reference gene set, as it is an outgroup relative to all other fish under consideration here. For each species, we uploaded the list of highly expressed testis genes derived from the BLASTP analysis described above. PANTHER returns a fold-enrichment value for each gene ontology category relative to expectations based on the frequency of occurrence of genes in that category in the reference genome. We examined fold-enrichment values for all 25,888 gene ontology categories for all eight species under consideration here. We calculated mean fold-enrichment for each category across the six non-pipefish Percomorphs and compared pipefish and zebrafish against these values using a simple linear regression. For this analysis, we retained categories that had a mean of at least four genes in Percomorphs (not including pipefish), resulting in a final list of 815 gene ontology categories for comparison with Gulf pipefish and zebrafish. Clusters of genes outside the 95% prediction intervals of the linear regressions were considered to be significant outliers.
MOLECULAR EVOLUTION OF GONAD-BIASED GENES
To connect our preceding analyses together, we investigated the rate of molecular evolution of testis-biased genes from S. scovelli and T. rubripes, our representative pipefish and spawning fish. The goal was to compare testis-biased genes with ovary-biased genes, and a control set of genes that were highly and evenly expressed between both gonads. As noted above, these two species were the only species within the Percomorpha clade with replicate testis and ovary RNA-seq data.
Genes that were identified by HISAT2 (Kim et al. 2015) and quantified by StringTie (Pertea et al. 2015) (see FUNCTIONAL EVOLUTION OF PIPEFISH TESTES in Methods) were used to determine the average TPM value for each gene within testis samples and within ovary samples for S. scovelli and T. rubripes, separately. To subset genes for the gonad-biased categories, we retained genes with a four or greater fold change (i.e., a log2 fold change greater than or equal to 2) difference between testis and ovaries. To restrict attention to genes that were highly expressed, we also set an average TPM cutoff. This threshold was a minimum TPM of 100 for S. scovelli and 20 for T. rubripes. We varied the TPM threshold to keep the sample sizes for the molecular evolution analysis roughly similar across species. In particular, we lowered the TPM threshold for T. rubripes, because very few differentially expressed genes met the TPM threshold of 100. To subset genes for the evenly expressed category, we took genes with a log2 fold change between −0.3 and 0.3. For the evenly expressed genes, the TPM cutoff was a minimum of 50 for S. scovelli and a minimum of 20 for T. rubripes. Again, we varied the TPM cutoff to control the sample size. Altogether this left a list of highly expressed testis-biased genes, highly expressed ovary-biased genes, and highly and evenly expressed genes across gonads for both S. scovelli and T. rubripes.
To provide an estimate of molecular evolution for each of these identified genes, the nucleotide sequences from the three lists of genes (highly expressed testis-biased genes, highly expressed ovary-biased genes, and highly and evenly expressed genes in both gonads) as well as their respective ortholog sequences from other species within their genus were aligned. For Syngnathus, we used S. scovelli, S. rostellatus, S. typhle, and S. acus, following the same pipeline as mentioned above (see GENOME-WIDE dN/dS ESTIMATION IN SYNGNATHUS in Methods). For the Takifugu genus, we obtained the genome, annotation, and protein files for T. rubripes and T. flavidus from NCBI (Table S1). Here, single-copy orthologs were identified and aligned using the same pipeline we used for Syngnathus. Our ortholog analysis was run within Syngnathus and within Takifugu to increase the likelihood of finding orthologs between species within each genus.
For the genes of interest that had single-copy orthologs, we used codeml (PAML version 4.9) (Yang 2007) to implement site models as previously mentioned (MOLECULAR EVOLUTION OF TESTIS GENES in Methods). The result was a dN/dS estimate for each gene identified as gonad biased or evenly expressed within either Takifugu or Syngnathus. Genes with a dN/dS estimate higher than 10 were checked and removed if there were no synonymous differences in the alignment (thus inaccurately inflating dN/dS). Ultimately, the following number of genes produced dN/dS estimates within each category: Syngnathus testis biased (429), ovary biased (473), and evenly expressed (546); Takifugu testis biased (97), ovary biased (151), and evenly expressed (726).
Results
MOLECULAR EVOLUTION OF TESTIS GENES
The phylogeny of the taxa involved in our analysis of molecular evolution is shown in Figure 1a, and the species-specific distributions of dN/dS values from the sites models are shown in Figure 1b. These models indicate that the dN/dS of Syngnathidae (0.204 ± 0.029, mean ± SEM) is smaller but not significantly so, compared to those of Poeciliidae (0.269 ± 0.044) and Cichlidae (0.283 ± 0.041) for genes involved in testis-related processes (Mann-Whitney-Wilcoxon test, two-sided, paired, n = 24: Syngnathidae vs. Poeciliidae, P = 0.277; Syngnathidae vs. Cichlidae, P = 0.121; Poeciliidae vs. Cichlidae, P = 0.473). We also observe that the Syngnathidae orthologs show a nonsignificant reduction in variance in dN/dS (σ2 = 0.021), compared to Poeciliidae (σ2 = 0.049; Brown-Forsythe test, P = 0.242) and Cichlidae (σ2 = 0.042; P = 0.132).
![Rates of molecular evolution among Syngnathidae, Poeciliidae, and Cichlidae. (a) A maximum likelihood phylogeny for species included in our analysis of molecular evolution, with bootstrap support indicated for each node. Branch lengths are proportional to the number of substitutions per site for the set of 24 genes with an expected involvement in sperm competition or sexual conflict. For these genes, we also show for each taxon studied (b) the median dN/dS values (box plots indicate interquartile range) and (c) the percentage of genes (same set of 24 genes for each taxonomic group) that fit each model of selection as inferred by codeml. The three models are as follows: a one-ratio model (M0) of purifying selection (ω < 1), a nearly neutral model (M1a) that includes two classes of sites (purifying selection [ω < 1] and neutral [ω = 1]), and a positive selection model (M2a) with three classes of sites (positive selection [ω > 1], purifying selection [ω < 1], and neutral [ω = 1]).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/evolut/76/9/10.1111_evo.14579/3/m_evo14579-fig-0001-m.jpeg?Expires=1750579276&Signature=dnTMkW8MwR~UsL4KTWlvhwElOWu5FuvYFR5nXYqmQcmnE8~d2qxI0IYq3BXkB6xcF6vubzufWQM6IOZW8c161qUJRc9frBzoU4aO6Y3NSCJ0p3FVsbwL4qeg968aOpcsrugLJ7bku7saRFvZmVp3uj2mPt~sa2CjtP0GjSqa6nfl7UIVihlGvosEjIx982jOnAm2O2YYXVSHbDGNZFk1wd8OYqoHVnuibb5OyoGmZoU2UE4hpAoyqI4WX03qfWqZhjMvJqvxHwDdje-Gr2vPpyOsoUqX7jw-AyK7SHrbsUc~ev2p3yIyGH5WviOd3fqYwxS~xgjIf68qC5VHZpfy5Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Rates of molecular evolution among Syngnathidae, Poeciliidae, and Cichlidae. (a) A maximum likelihood phylogeny for species included in our analysis of molecular evolution, with bootstrap support indicated for each node. Branch lengths are proportional to the number of substitutions per site for the set of 24 genes with an expected involvement in sperm competition or sexual conflict. For these genes, we also show for each taxon studied (b) the median dN/dS values (box plots indicate interquartile range) and (c) the percentage of genes (same set of 24 genes for each taxonomic group) that fit each model of selection as inferred by codeml. The three models are as follows: a one-ratio model (M0) of purifying selection (ω < 1), a nearly neutral model (M1a) that includes two classes of sites (purifying selection [ω < 1] and neutral [ω = 1]), and a positive selection model (M2a) with three classes of sites (positive selection [ω > 1], purifying selection [ω < 1], and neutral [ω = 1]).
To further investigate these patterns, we estimated models of selection for each gene using likelihood ratio tests, as implemented in codeml. For each ortholog in each taxonomic group, we were able to classify its best-fitting evolutionary model as purifying selection, nearly neutral, or positive selection. Figure 1c shows the percentages of orthologs categorized into each model for Syngnathidae (20.8% purifying selection, 79.2% nearly neutral, and 0% positive selection), Poeciliidae (62.5% purifying selection, 33.3% nearly neutral, and 4.2% positive selection), and Cichlidae (58.3% purifying selection, 29.2% nearly neutral, and 12.5% positive selection). The gene-by-gene results are summarized in Data S1. Overall, these results are consistent with the idea that syngnathid fishes have experienced a simultaneous reduction in positive selection and purifying selection in their testis transcriptomes, showing a pattern of generally weaker selection acting on male reproductive function, compared to other fishes.
With respect to our branch models, we found that 41.7% (n = 10) of the genes showed a different branch rate (significance for M2) for ω between Syngnathidae and the other teleosts (Poeciliidae and Cichlidae; Data S1). In eight of these cases, ω was greater for the Syngnathidae branch (with an average ω of 0.218) than the other teleosts branches (average ω of 0.114), and in the other 2 cases ω was greater for the other teleosts (average ω of 0.277) than the Syngnathidae branch (average ω of 0.157). For these 10 genes, the Syngnathidae alignments best fit models of nearly neutral evolution (seven genes) or purifying selection (three genes).
Our genome-wide dN/dS analysis within the genus Syngnathus resulted in an average dN/dS value of 0.267 ± 0.002 (mean ± SEM). The percentage of orthologs categorized into each model of selection were as follows: 62.2% purifying selection (M0), 20.2% nearly neutral (M1a), and 17.6% positive selection (M2a). These proportions differ substantially from those observed for the 24 testis- or sperm-associated proteins examined in Syngnathidae (Fig. 1c).
GENE TURNOVER IN GULF PIPEFISH TESTES
For our second question, we examined patterns of gene turnover in syngnathid testes by comparing the presence of orthologs for testis-enriched genes between the Gulf pipefish (S. scovelli) and the Japanese pufferfish (T. rubripes). Testis-enriched transcripts of the Gulf pipefish were compared against the Japanese pufferfish genome to establish the proportion of pipefish transcripts present in the pufferfish. We also performed the reciprocal comparison, to establish the proportion of pufferfish transcripts present in the pipefish. The proportions of matches for testis transcripts were compared against a null distribution generated by conducting the same procedure a thousand times for randomly chosen sets of genes from each species’ genome (regardless of testis enrichment).
Our comparison of testis transcriptomes between the Gulf pipefish and the Japanese pufferfish showed that pipefish testis-enriched genes were significantly more likely to have orthologs in the pufferfish genome compared to subsets of randomly chosen genes (Fig. 2, bottom). In addition, the reverse pattern was seen in the reciprocal comparison, where pufferfish testis-enriched genes were less likely than randomly chosen genes to appear in the pipefish genome (Fig. 2, top). Specifically, 90.6% of pipefish testis-enriched genes were present in the pufferfish genome, whereas a mean of 85.9% of randomly chosen sets of genes had orthologs in pufferfish. However, only 75.4% of pufferfish testis-enriched genes were found in the pipefish genome, despite a mean of 92.9% for randomly chosen sets of pufferfish genes.
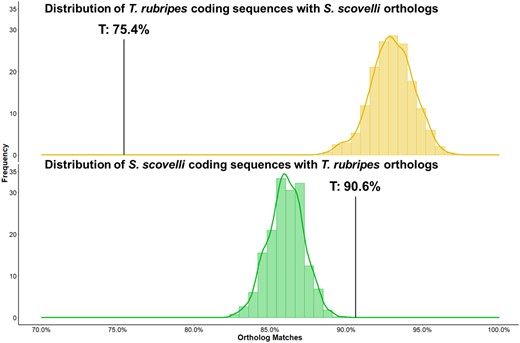
Gene turnover between Syngnathus scovelli and Takifugu rubripes testis transcriptomes. Distributions represent the expected number of sequences with orthologs in the counter-species by randomly sampling 1000 times across all coding sequences, given a sample size equal to the number of testis-biased (T) genes in the focal species. Black bars represent the percentage of highly expressed testis-biased genes with orthologs in the counter-species. Top: Pufferfish testis-biased genes are less likely than expected to be present in the pipefish genome. Bottom: Pipefish testis-biased genes are more likely than randomly chosen genes to be present in the pufferfish genome.
The difference in the distributions for reciprocal comparisons for randomly chosen genes likely arises from distinct histories of gene duplication and gene loss or differences in genome quality in terms of assembly and annotation. This would be consistent with findings on the pufferfish lineage (Tetraodon), which has been documented to have recently lost genes that arose after the teleost whole-genome duplication event, potentially leading to pufferfish speciation (Taylor et al. 2003; Kai et al. 2011). This historical pattern of genome evolution could contribute in part to the difference in null expectations and underscores the need to interpret the percentage of shared testis orthologs relative to a null distribution.
FUNCTIONAL EVOLUTION OF PIPEFISH TESTES
To examine the evolutionary changes in the testes of Syngnathidae since the origin of male pregnancy, we compared the testis transcriptome of the Gulf pipefish to six other percomorph species, as well as to zebrafish (D. rerio), an outgroup species. The results of the GO analysis are shown in Figures S1–S3. There were slight but nonsignificant differences between the Gulf pipefish and other fish taxa with respect to these GO categories (Fig. S2). For the protein class analysis, we used the PANTHER Protein Class database (version 15.0). This analysis revealed an increase in expression of genes encoding calcium-binding proteins, membrane traffic proteins, and chaperones for the Gulf pipefish in comparison to the other percomorph species (Fig. 3a). Gulf pipefish also show a dramatically lower number of genes categorized as protein modifying enzymes (Fig. 3a). Notably, the zebrafish transcriptome does not differ significantly from the Percomorphs (excluding pipefish) with respect to any of these categories (Fig. 3a), suggesting that Gulf pipefish testes differ from typical fish testes in terms of the types of proteins encoded by highly expressed genes.
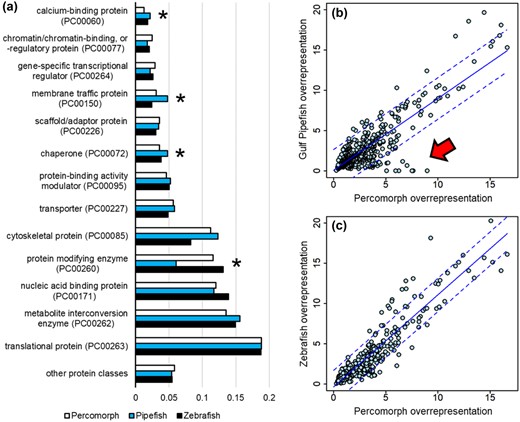
Functional evolution of pipefish testes. (a) Gene ontology (GO) analysis using the PANTHER protein-class database on highly expressed Gulf pipefish testis genes. Pipefish differ significantly from other Percomorphs (P = 0.019), whereas zebrafish do not differ from Percomorphs (P = 0.628). Asterisks indicate tests that are significant at an FDR of 0.05 (one-sample, two-sided t-test). (b) PANTHER overrepresentation analysis comparing pipefish against mean Percomorph values for GO categories (with a mean ≥4 genes in Percomorphs). (c) PANTHER overrepresentation analysis for zebrafish compared to Percomorphs. In panels (B) and (C), lines indicate linear regression (solid) and 95% prediction intervals (dashed). A noticeable cluster of GO categories is below the 95% prediction interval in pipefish (red arrow, Table 1).
The results of our comparative overrepresentation analysis are shown in Figure 3b,c. The scatterplot indicates a cluster of GO categories that are strongly overrepresented in most Percomorphs (more than threefold enrichment) but are not overrepresented or are missing from Gulf pipefish (Fig. 3b, red arrow). The comparison of zebrafish to Percomorphs contains no such cluster, indicating that the reduction of genes in these GO categories is unique to the Gulf pipefish testis. We examined the functions of these outlying genes (Fig. 3b), as well as the protein-modifying enzymes that were apparently lost from the highly expressed category in the pipefish testis transcriptome (Fig. 3a). Beginning with the protein-modifying enzymes, we compared gene counts for each of the subcategories of protein-modifying enzymes between Gulf pipefish and the other taxa (Table 1, Panel A). Gulf pipefish show a reduction in genes classified as various types of proteases, nonreceptor serine/threonine protein kinases, and ubiquitin-protein ligases relative to the other species. The overrepresentation analysis reveals a suite of GO categories that have evolved reduced expression in Gulf pipefish relative to other Percomorphs and zebrafish (Table 1, Panel B). These GO categories are all related to the assembly and function of the sperm cell's flagellum, or cell division and meiosis.
Underrepresented PANTHER protein classes and GO biological process categories in Gulf pipefish testes compared to other fishes
| A. PANTHER Protein Class | Gulf Pipefish Gene Count | Mean Percomorph Gene Count | Zebrafish Gene Count |
|---|---|---|---|
| Cysteine protease (PC00081) | 3 | 7.3 | 7 |
| Metalloprotease (PC00153) | 2 | 4.5 | 9 |
| Nonreceptor serine/threonine protein kinase (PC00167) | 3 | 10.5 | 10 |
| Protease (PC00190) | 9 | 4.7 | 3 |
| Protein phosphatase (PC00195) | 2 | 1.8 | 0 |
| Serine protease (PC00203) | 2 | 6.5 | 16 |
| Ubiquitin-protein ligase (PC00234) | 7 | 15.5 | 20 |
| Protein-Modifying Enzyme (PC00260) | 0 | 0.3 | 0 |
| B. Gene Ontology Biological Process | Gulf Pipefish Enrichment | Mean Percomorph Enrichment | Zebrafish Enrichment |
| Axonemal dynein complex assembly (GO:0070286) | 0 | 7.64 | 13.08 |
| Cilium or flagellum-dependent cell motility (GO:0001539) | 0 | 8.98 | 7.35 |
| Cilium-dependent cell motility (GO:0060285) | 0 | 8.98 | 7.35 |
| Microtubule-based protein transport (GO:0099118) | 0 | 6.27 | 3.63 |
| Protein transport along microtubule (GO:0098840) | 0 | 6.27 | 3.63 |
| Intraciliary transport (GO:0042073) | 0 | 7.52 | 4.51 |
| Chromosome organization involved in meiotic cell cycle (GO:0070192) | 0 | 5.09 | 6.16 |
| Homologous chromosome segregation (GO:0045143) | 0 | 5.97 | 8.96 |
| Axoneme assembly (GO:0035082) | 0.68 | 7.04 | 9.59 |
| Transport along microtubule (GO:0010970) | 0.42 | 3.75 | 1.48 |
| Meiotic cell cycle process (GO:1903046) | 0.41 | 3.08 | 3.92 |
| Meiotic nuclear division (GO:0140013) | 0.48 | 3.47 | 4.62 |
| Mitotic sister chromatid segregation (GO:0000070) | 0.64 | 4.23 | 2.82 |
| Meiosis I (GO:0007127) | 0.6 | 3.63 | 5.22 |
| Meiosis I cell cycle process (GO:0061982) | 0.55 | 3.32 | 4.77 |
| Microtubule-based movement (GO:0007018) | 0.65 | 3.79 | 3.27 |
| Microtubule bundle formation (GO:0001578) | 1.17 | 6.63 | 8.19 |
| Cilium movement (GO:0003341) | 0.95 | 5.12 | 6.63 |
| Nuclear division (GO:0000280) | 0.7 | 3.74 | 3.88 |
| Mitotic spindle organization (GO:0007052) | 0.85 | 4.49 | 4.45 |
| Epithelial cilium movement involved in extracellular fluid movement (GO:0003351) | 1 | 5.22 | 9.68 |
| Extracellular transport (GO:0006858) | 1 | 5.22 | 9.68 |
| Meiotic chromosome segregation (GO:0045132) | 0.67 | 3.41 | 4.69 |
| A. PANTHER Protein Class | Gulf Pipefish Gene Count | Mean Percomorph Gene Count | Zebrafish Gene Count |
|---|---|---|---|
| Cysteine protease (PC00081) | 3 | 7.3 | 7 |
| Metalloprotease (PC00153) | 2 | 4.5 | 9 |
| Nonreceptor serine/threonine protein kinase (PC00167) | 3 | 10.5 | 10 |
| Protease (PC00190) | 9 | 4.7 | 3 |
| Protein phosphatase (PC00195) | 2 | 1.8 | 0 |
| Serine protease (PC00203) | 2 | 6.5 | 16 |
| Ubiquitin-protein ligase (PC00234) | 7 | 15.5 | 20 |
| Protein-Modifying Enzyme (PC00260) | 0 | 0.3 | 0 |
| B. Gene Ontology Biological Process | Gulf Pipefish Enrichment | Mean Percomorph Enrichment | Zebrafish Enrichment |
| Axonemal dynein complex assembly (GO:0070286) | 0 | 7.64 | 13.08 |
| Cilium or flagellum-dependent cell motility (GO:0001539) | 0 | 8.98 | 7.35 |
| Cilium-dependent cell motility (GO:0060285) | 0 | 8.98 | 7.35 |
| Microtubule-based protein transport (GO:0099118) | 0 | 6.27 | 3.63 |
| Protein transport along microtubule (GO:0098840) | 0 | 6.27 | 3.63 |
| Intraciliary transport (GO:0042073) | 0 | 7.52 | 4.51 |
| Chromosome organization involved in meiotic cell cycle (GO:0070192) | 0 | 5.09 | 6.16 |
| Homologous chromosome segregation (GO:0045143) | 0 | 5.97 | 8.96 |
| Axoneme assembly (GO:0035082) | 0.68 | 7.04 | 9.59 |
| Transport along microtubule (GO:0010970) | 0.42 | 3.75 | 1.48 |
| Meiotic cell cycle process (GO:1903046) | 0.41 | 3.08 | 3.92 |
| Meiotic nuclear division (GO:0140013) | 0.48 | 3.47 | 4.62 |
| Mitotic sister chromatid segregation (GO:0000070) | 0.64 | 4.23 | 2.82 |
| Meiosis I (GO:0007127) | 0.6 | 3.63 | 5.22 |
| Meiosis I cell cycle process (GO:0061982) | 0.55 | 3.32 | 4.77 |
| Microtubule-based movement (GO:0007018) | 0.65 | 3.79 | 3.27 |
| Microtubule bundle formation (GO:0001578) | 1.17 | 6.63 | 8.19 |
| Cilium movement (GO:0003341) | 0.95 | 5.12 | 6.63 |
| Nuclear division (GO:0000280) | 0.7 | 3.74 | 3.88 |
| Mitotic spindle organization (GO:0007052) | 0.85 | 4.49 | 4.45 |
| Epithelial cilium movement involved in extracellular fluid movement (GO:0003351) | 1 | 5.22 | 9.68 |
| Extracellular transport (GO:0006858) | 1 | 5.22 | 9.68 |
| Meiotic chromosome segregation (GO:0045132) | 0.67 | 3.41 | 4.69 |
Note: (A) Gulf pipefish testes have significantly fewer protein-modifying enzymes (broken-down by PANTHER protein class) than those of other fishes. (B) GO categories, with fold-enrichment values, that were significantly low outliers for Gulf pipefish testes in the PANTHER overrepresentation analysis (Fig. 3b, arrow). Shown are categories with a mean of at least threefold overrepresentation in Percomorphs, and at least fivefold more overrepresentation in Percomorph transcriptomes, on average, relative to the pipefish transcriptome.
Underrepresented PANTHER protein classes and GO biological process categories in Gulf pipefish testes compared to other fishes
| A. PANTHER Protein Class | Gulf Pipefish Gene Count | Mean Percomorph Gene Count | Zebrafish Gene Count |
|---|---|---|---|
| Cysteine protease (PC00081) | 3 | 7.3 | 7 |
| Metalloprotease (PC00153) | 2 | 4.5 | 9 |
| Nonreceptor serine/threonine protein kinase (PC00167) | 3 | 10.5 | 10 |
| Protease (PC00190) | 9 | 4.7 | 3 |
| Protein phosphatase (PC00195) | 2 | 1.8 | 0 |
| Serine protease (PC00203) | 2 | 6.5 | 16 |
| Ubiquitin-protein ligase (PC00234) | 7 | 15.5 | 20 |
| Protein-Modifying Enzyme (PC00260) | 0 | 0.3 | 0 |
| B. Gene Ontology Biological Process | Gulf Pipefish Enrichment | Mean Percomorph Enrichment | Zebrafish Enrichment |
| Axonemal dynein complex assembly (GO:0070286) | 0 | 7.64 | 13.08 |
| Cilium or flagellum-dependent cell motility (GO:0001539) | 0 | 8.98 | 7.35 |
| Cilium-dependent cell motility (GO:0060285) | 0 | 8.98 | 7.35 |
| Microtubule-based protein transport (GO:0099118) | 0 | 6.27 | 3.63 |
| Protein transport along microtubule (GO:0098840) | 0 | 6.27 | 3.63 |
| Intraciliary transport (GO:0042073) | 0 | 7.52 | 4.51 |
| Chromosome organization involved in meiotic cell cycle (GO:0070192) | 0 | 5.09 | 6.16 |
| Homologous chromosome segregation (GO:0045143) | 0 | 5.97 | 8.96 |
| Axoneme assembly (GO:0035082) | 0.68 | 7.04 | 9.59 |
| Transport along microtubule (GO:0010970) | 0.42 | 3.75 | 1.48 |
| Meiotic cell cycle process (GO:1903046) | 0.41 | 3.08 | 3.92 |
| Meiotic nuclear division (GO:0140013) | 0.48 | 3.47 | 4.62 |
| Mitotic sister chromatid segregation (GO:0000070) | 0.64 | 4.23 | 2.82 |
| Meiosis I (GO:0007127) | 0.6 | 3.63 | 5.22 |
| Meiosis I cell cycle process (GO:0061982) | 0.55 | 3.32 | 4.77 |
| Microtubule-based movement (GO:0007018) | 0.65 | 3.79 | 3.27 |
| Microtubule bundle formation (GO:0001578) | 1.17 | 6.63 | 8.19 |
| Cilium movement (GO:0003341) | 0.95 | 5.12 | 6.63 |
| Nuclear division (GO:0000280) | 0.7 | 3.74 | 3.88 |
| Mitotic spindle organization (GO:0007052) | 0.85 | 4.49 | 4.45 |
| Epithelial cilium movement involved in extracellular fluid movement (GO:0003351) | 1 | 5.22 | 9.68 |
| Extracellular transport (GO:0006858) | 1 | 5.22 | 9.68 |
| Meiotic chromosome segregation (GO:0045132) | 0.67 | 3.41 | 4.69 |
| A. PANTHER Protein Class | Gulf Pipefish Gene Count | Mean Percomorph Gene Count | Zebrafish Gene Count |
|---|---|---|---|
| Cysteine protease (PC00081) | 3 | 7.3 | 7 |
| Metalloprotease (PC00153) | 2 | 4.5 | 9 |
| Nonreceptor serine/threonine protein kinase (PC00167) | 3 | 10.5 | 10 |
| Protease (PC00190) | 9 | 4.7 | 3 |
| Protein phosphatase (PC00195) | 2 | 1.8 | 0 |
| Serine protease (PC00203) | 2 | 6.5 | 16 |
| Ubiquitin-protein ligase (PC00234) | 7 | 15.5 | 20 |
| Protein-Modifying Enzyme (PC00260) | 0 | 0.3 | 0 |
| B. Gene Ontology Biological Process | Gulf Pipefish Enrichment | Mean Percomorph Enrichment | Zebrafish Enrichment |
| Axonemal dynein complex assembly (GO:0070286) | 0 | 7.64 | 13.08 |
| Cilium or flagellum-dependent cell motility (GO:0001539) | 0 | 8.98 | 7.35 |
| Cilium-dependent cell motility (GO:0060285) | 0 | 8.98 | 7.35 |
| Microtubule-based protein transport (GO:0099118) | 0 | 6.27 | 3.63 |
| Protein transport along microtubule (GO:0098840) | 0 | 6.27 | 3.63 |
| Intraciliary transport (GO:0042073) | 0 | 7.52 | 4.51 |
| Chromosome organization involved in meiotic cell cycle (GO:0070192) | 0 | 5.09 | 6.16 |
| Homologous chromosome segregation (GO:0045143) | 0 | 5.97 | 8.96 |
| Axoneme assembly (GO:0035082) | 0.68 | 7.04 | 9.59 |
| Transport along microtubule (GO:0010970) | 0.42 | 3.75 | 1.48 |
| Meiotic cell cycle process (GO:1903046) | 0.41 | 3.08 | 3.92 |
| Meiotic nuclear division (GO:0140013) | 0.48 | 3.47 | 4.62 |
| Mitotic sister chromatid segregation (GO:0000070) | 0.64 | 4.23 | 2.82 |
| Meiosis I (GO:0007127) | 0.6 | 3.63 | 5.22 |
| Meiosis I cell cycle process (GO:0061982) | 0.55 | 3.32 | 4.77 |
| Microtubule-based movement (GO:0007018) | 0.65 | 3.79 | 3.27 |
| Microtubule bundle formation (GO:0001578) | 1.17 | 6.63 | 8.19 |
| Cilium movement (GO:0003341) | 0.95 | 5.12 | 6.63 |
| Nuclear division (GO:0000280) | 0.7 | 3.74 | 3.88 |
| Mitotic spindle organization (GO:0007052) | 0.85 | 4.49 | 4.45 |
| Epithelial cilium movement involved in extracellular fluid movement (GO:0003351) | 1 | 5.22 | 9.68 |
| Extracellular transport (GO:0006858) | 1 | 5.22 | 9.68 |
| Meiotic chromosome segregation (GO:0045132) | 0.67 | 3.41 | 4.69 |
Note: (A) Gulf pipefish testes have significantly fewer protein-modifying enzymes (broken-down by PANTHER protein class) than those of other fishes. (B) GO categories, with fold-enrichment values, that were significantly low outliers for Gulf pipefish testes in the PANTHER overrepresentation analysis (Fig. 3b, arrow). Shown are categories with a mean of at least threefold overrepresentation in Percomorphs, and at least fivefold more overrepresentation in Percomorph transcriptomes, on average, relative to the pipefish transcriptome.
MOLECULAR EVOLUTION OF GONAD-BIASED GENES
To tie together both our initial questions, we investigated the pattern of molecular evolution on the suite of genes expressed in the testis of pipefish. We calculated dN/dS estimates for genes that were highly expressed and testis biased, ovary biased, or evenly expressed between testis and ovaries for S. scovelli and compared these results to those for a percomorph with more conventional reproduction (T. rubripes). The following median dN/dS values were obtained for each of these categories: Syngnathus testis biased (n = 429, median = 0.218), ovary biased (n = 473, 0.224), and evenly expressed (n = 546, 0.160); Takifugu testis biased (n = 97, 0.399), ovary biased (n = 151, 0.253), and evenly expressed (n = 726, 0.224) (Fig. 4; Table S2). In Syngnathus, the rate of molecular evolution of testis-biased genes did not differ from that of ovary-biased genes (Mann-Whitney-Wilcoxon test, two-sided, unpaired: P = 0.840). This result contrasts with our analysis involving Takifugu, where testis-biased genes exhibited a significantly higher dN/dS value compared to ovary-biased genes (P ≤ 0.001). In fact, the testis-biased genes of Takifugu differed significantly from both ovary-biased genes and evenly expressed genes (P ≤ 0.001), which did not differ significantly from one another (Takifugu ovary biased vs. evenly expressed: P = 0.2029). For Syngnathus, however, both testis-biased genes and ovary-biased genes showed significantly elevated dN/dS values compared to evenly expressed genes (Syngnathus testis biased vs. evenly expressed, and ovary biased vs. evenly expressed: P ≤ 0.001).
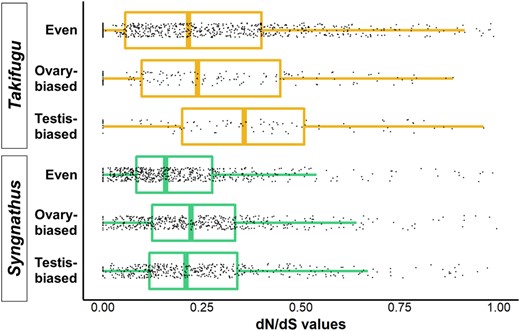
Molecular evolution of gonad-biased genes in Syngnathus and Takifugu. Using testis and ovary RNA-seq data from S. scovelli and T. rubripes, we identified genes that were highly and evenly expressed across gonads (even), and genes that were highly expressed in one gonad (ovary-biased and testis-biased). The dN/dS value was estimated using alignments from the genomes of species within Syngnathus (S. scovelli, S. acus, S. rostellatus, S. typhle) and Takifugu (T. rubripes and T. flavidus). The box plots report the median dN/dS value and the interquartile range for genes within each of the six groups.
Discussion
The study of reproductive genes and the proteins they encode has been strongly motivated by arguments related to sperm competition and sexual conflict (Zhang et al. 2007; Harrison et al. 2015; Yin et al. 2018). The underlying notion, which is consistent with much of the published literature, is that male-male competition or an arms race between the sexes drives rapid evolution of the proteins mediating these processes (Simmons 2001; Swanson and Vacquier 2002; Lüpold et al. 2016; Dean et al. 2017; Civetta and Ranz 2019; Liao et al. 2019). However, most of the tests of these ideas have been in species with considerable potential for strong selection on male reproductive function and conflict between the sexes, with a few notable exceptions, which we will discuss further below. Here, we provide a critical test of these predictions by studying molecular evolution and gene turnover in a sexually reproducing, male-pregnant vertebrate species where the testes have no opportunity to play a role in either sperm competition or sexual conflict. Our results support the interpretation that rapid evolution of male reproductive proteins and rapid gene turnover in the testis is indeed driven by sexual selection and conflict, and that the syngnathid fishes differ from the normal pattern of testis evolution because male pregnancy has diminished the role of the male gonad in sexual selection and sexual conflict.
MOLECULAR EVOLUTION OF TESTIS-RELATED GENES
Our analysis of dN/dS revealed modest values for all species under consideration here. The mean dN/dS values for the 24 testis-associated genes were most similar for cichlids and livebearers, and slightly lower but not significantly so for pipefish. The variance in dN/dS among genes was lower for pipefish than for cichlids and livebearers, but again this result was not statistically significant. None of the genes in this study displayed exceptionally large values of dN/dS, as have been observed in other studies of genes involved in male reproduction. For instance, Wyckoff et al. (2000) studied 18 genes directly involved in male reproduction in primates and found that five of these genes (28%) had dN/dS values >1. Other studies of specific gene families involved in male reproduction, such as the CRISP (cysteine-rich secretory proteins) genes and Adam (a disintegrin and metalloprotease) genes in rodents, also found evidence of positive selection, in some cases with very large dN/dS values (Grayson and Civetta 2013; Vicens and Treviño 2018). However, the study of CRISP genes, like ours, found multiple genes with evidence of positive selection despite having dN/dS values <1.
Regardless of the absolute values of dN/dS, the more compelling comparison involves the scrutiny of codeml-estimated models of selection in syngnathids relative to other fishes. Our results for cichlids and poeciliids are remarkably similar. Both groups show a majority of genes (around 60%) with a pattern of purifying selection (i.e., the one-ratio model in codeml was the best fit). About a third of the genes in each group were best fit by a nearly neutral model, and a small number of genes in each group were best described as positively selected (4% and 13% of genes, respectively, in livebearers and cichlids). Thus, in these taxa, codeml classified about two thirds of genes as experiencing some form of selection, either positive or negative. The results for syngnathids were dramatically different. None of these genes were categorized as positively selected in syngnathids and only 20% showed evidence of purifying selection. Rather, the vast majority of genes (79%) best fit a nearly neutral model (i.e., a two-ratio model with a mixture of sites either experiencing purifying selection or evolving according to neutrality). All 24 genes were present in the de novo Trinity assembly based on the RNA reads from S. scovelli testes (section GENE TURNOVER IN GULF PIPEFISH TESTES), so they are expressed at some level in pipefish testes. We interpret these results as an indication that selection on testis-expressed genes, be it positive or negative selection, is generally weaker in syngnathid fishes compared to other percomorphs.
The branch models from codeml also were consistent with this interpretation of reduced selection on testis-related genes in syngnathids compared to other taxa. Out of the 24 genes analyzed, 10 showed a significantly different rate of molecular evolution in syngnathid fishes compared to poeciliids and cichlids. For eight of these genes, the syngnathid branch had an elevated dN/dS, consistent with a relaxation of purifying selection in the syngnathid group. The other two genes had reduced dN/dS values in syngnathids, a pattern that could be consistent with several possibilities, such as an increase in purifying selection in syngnathids or a reduction of positive selection on some parts of the proteins in syngnathids. Regardless, many genes in the branch analysis are consistent with the emerging pattern that selection on testis-associated genes is weaker in the syngnathid lineage relative to other percomorphs.
These findings are particularly interesting when compared to the genome-wide molecular evolution estimates, which show that members of Syngnathidae do not have an unusually low baseline of molecular evolution across their genome. For instance, recent studies have examined the strength of selection on genes related to other complex adaptive phenotypes important to the evolutionary success of syngnathids. One example of such a trait involves the independent evolution of spiny body plates, a predator deterrent, across the seahorse phylogeny (Li et al. 2021). A comparison between spiny and nonspiny seahorse lineages revealed 37 genes that were under strong positive selection and had likely roles in teleost skin and scale development (Li et al. 2021). The independent appearance of complex morphological phenotypes such as this, the high diversification rate within the Hippocampus clade (Li et al. 2021), and the accelerated rate of nucleotide evolution within H. comes in comparison to other teleost genomes (Lin et al. 2016) demonstrate the potential for accelerated evolution of advantageous traits within syngnathids and are in blunt contrast with our findings within the testis-related genes.
GENE TURNOVER IN SYNGNATHID TESTES
We predicted that weaker selection acting upon testes in the pipefish should lead to a loss of orthologs over time (Whittington et al. 2017; Yin et al. 2018). Under this scenario, an ancestral pipefish, shortly after the evolution of male pregnancy, would have found itself in a situation where its testes produced more sperm than necessary to fertilize the eggs in its possession. Its testes also would have been producing unnecessary sperm-related substances that would have aided in sperm competition before the evolution of male pregnancy. Presumably, selection in a pregnant male would act to reduce the number of sperm and to cease the production of potentially costly reproductive substances that had once been involved in sperm competition but are no longer needed. Consequently, the genes involved in these traits would be subjected to genetic drift or active selection to reduce male energetic expenditures. The result would be a streamlined testis transcriptome that only harbors genes essential to produce just enough sperm to fertilize the eggs received during mating. This scenario agrees well with empirical observations from various syngnathid testes, where phenotypes such as unusually small testis size and low sperm density, along with semicystic spermatogenesis, are predicted to reduce the cost of sperm production (Wilson et al. 2003; Kvarnemo and Simmons 2004; Biagi et al. 2016; Piras et al. 2016b).
We chose to test this prediction by comparing the genes upregulated in the Gulf pipefish testis to those upregulated in the testis of the pufferfish (T. rubripes). We found that 90.6% of the testis-upregulated syngnathid genes were present in the pufferfish genome. This percentage is higher than the expectation based on randomly chosen genes (Fig. 2, bottom). Our interpretation of this result is that the syngnathids have retained genes that are essential for testis function, because syngnathid males, like males of other fish, must still produce sperm to reproduce. These genes, as they are essential for reproduction, have also been retained by pufferfish, resulting in nearly all testis-enriched genes in syngnathids having orthologs in the pufferfish genome.
In contrast, the reciprocal comparison, which involved looking for testis-upregulated pufferfish genes in the pipefish genome, resulted in a substantially smaller proportion of hits (75.4%). This value is much less than the proportion of hits observed for randomly chosen genes (Fig. 2, top). This result indicates that many testis-upregulated genes in pufferfish either have sequences that are not similar enough to be identified by our blast-based comparison or are missing altogether in pipefish. This pattern can be interpreted in several different ways, all of which are consistent with a reduction of selection acting on the testes of syngnathids. Syngnathids may have simply lost some genes that are involved in sperm competition. Alternatively, stronger selection acting upon the testes of the pufferfish could have led to an increase in recruitment of novel genes (Harrison et al. 2015; Whittington et al. 2017; Schmitz et al. 2020). Another possibility is that rapid evolution of testis-expressed pufferfish genes, driven in part by sperm competition or sexual conflict, could have resulted in so much sequence evolution that the ortholog was no longer identifiable in our cross-species comparison. All these interpretations are consistent with a relaxation of selection on genes associated with syngnathid testes, much like the results of our molecular evolution investigation.
FUNCTIONAL GENOMICS OF PIPEFISH TESTES
Our analyses of gene ontology and overrepresentation revealed intriguing changes during the evolution of the syngnathid testis. The most noteworthy change detected by our gene ontology analysis was a loss of protein-modifying enzymes, cysteine and serine proteases, and metalloproteases, in the highly expressed testis genes of the Gulf pipefish relative to other percomorphs and our zebrafish outgroup. Interestingly, these types of proteases are heavily involved in testis development, spermatogenesis, sperm capacitation, and sperm-egg binding (Gurupriya and Roy 2017). Cysteine proteases are involved in sperm capacitation (Lee et al. 2018) and serine proteases are a major component of the acrosome (Klemm et al. 1991). Metalloproteases have been shown to be involved in mouse testis development (Guyot et al. 2003) and are associated with sperm quality and ejaculate volume in dogs and humans (Shimokawa et al. 2002; Tentes et al. 2007). Within syngnathids, a zinc-dependent metalloprotease named patristacin has been found to be highly expressed in the brood pouch of pregnant males, suggesting a novel role for metalloproteases in male pregnancy (Harlin-Cognato et al. 2006; Whittington et al. 2015; Small et al. 2016; Lin et al. 2017). The patristacin gene family has further expanded through duplication within syngnathids, with specific patristacin genes showing distinct regulation patterns within the brood pouch depending on the stage of pregnancy (Lin et al. 2016, 2017; Small et al. 2016). In this regard, metalloproteases have an integral role to brood pouch function, and might have a less pivotal role in the function of the testis.
Our gene ontology analysis also showed a reduced number of nonreceptor serine/threonine protein kinases in pipefish testes compared to those of other fishes. This category of genes has been shown to play a role in sperm capacitation (O'Flaherty et al. 2004), and specific kinases, such as protein kinase A (PKA), can be found in the acrosomal cap and sperm flagellum in mammals (Pariset and Weinman 1994; Visconti et al. 1997). PKA activity is also involved in a series of events that leads to tyrosine phosphorylation, which is necessary for other processes within a mature spermatozoon (O'Flaherty et al. 2004; Signorelli et al. 2012; Gangwar and Atreja 2015).
The final category of gene with substantially reduced representation in pipefish testes relative to other fish testes is ubiquitin-protein ligases. These enzymes attach ubiquitin molecules to target proteins and mark them for degradation by the proteasome, and male mice knockouts of specific ubiquitin-protein ligases have nonmature spermatozoa and reduced fertility (Rodriguez and Stewart 2007). This ubiquitin-proteasome system has also been implicated in capacitation, sperm-zona pellucida penetration, elimination of defective sperm, and destruction of sperm mitochondria (Amaral et al. 2014; Zigo et al. 2019). Overall, our gene ontology analysis indicates that pipefish testes have experienced a reduction in expression of genes involved in the development and function of spermatozoa and possibly development of testes.
The overrepresentation analysis suggests a similar evolutionary pattern and is best interpreted in light of the comparison of pipefish with other percomorphs. For each individual species, the analysis indicates gene ontology classes that are represented at a higher rate in the transcriptome compared to the expectation based on the frequency of the gene ontology class in the whole genome (in this case the zebrafish genome). The scatterplot of overrepresented gene ontology categories in pipefish versus other percomorphs allowed us to identify a cluster of gene categories that were overrepresented in percomorphs but not in pipefish. By also comparing results for zebrafish, where we saw no such cluster, we were able to establish that this lack of overrepresentation in pipefish was restricted to the syngnathid lineage. The gene ontology categories that were overrepresented in the testis transcriptomes of percomorphs but not in the testes of pipefish are nearly all involved in two main functions: meiosis and the assembly of the sperm flagellum (Table 1, Panel B). The gene ontology categories associated with microtubules could be involved in either meiosis or the sperm flagellum, as microtubules are structural components of flagella and are also involved in movement of the chromosomes during cell division (Simerly et al. 1995). Extracellular transport is not obviously involved in either meiosis or the flagellum, but this is a higher level category that contains multiple cilium-related lower level categories, suggesting that this result may also be due to a lack of genes in the pipefish dataset associated with the flagellum.
Overall, the gene ontology and overrepresentation analyses paint a clear picture of aspects of the pipefish testis transcriptome that differentiate it from the transcriptomes of other percomorphs. Pipefish express fewer genes involved in processes related to sperm function, such as capacitation and sperm-egg interactions. In addition, pipefish testes show a pattern of reduced expression of genes related to meiosis and to the function of the sperm flagellum. It is important to note that our analysis targeted the top 1000 expressed genes in the testis of each species under consideration, so a lack of a gene in our analysis indicates that it is no longer expressed at a high enough level to be in the top 1000 testis-expressed genes, not necessarily that it is absent from the genome. Thus, our interpretation is that the pipefish testes have evolved to produce such a low number of sperm that many of the genes related to sperm development and function are now either absent or still necessary, but expressed at such low levels that they are no longer a major component of the pipefish testis transcriptome (such as those responsible for the formation of the flagellum), a pattern that differs markedly from the majority of other percomorphs. This interpretation is supported by empirical findings that show most syngnathids have sperm with a cylindrically shaped head and a long flagellum, properties that correlate with internal fertilization (Watanabe et al. 2000; Van Look et al. 2007; Piras et al. 2016b). These adaptive traits likely point toward sperm moving to unite with the eggs through the viscous ovarian fluids the female has deposited into the male (Watanabe et al. 2000; Van Look et al. 2007; Piras et al. 2016b). However, various syngnathids have been documented with both extremely low density of sperm and low testis weight (Watanabe et al. 2000; Kvarnemo and Simmons 2004; Van Look et al. 2007; Dzyuba et al. 2008).
GENOME-WIDE MOLECULAR EVOLUTION OF GONAD-BIASED GENES
To tie together our initial questions and analyses, we investigated the pattern of molecular evolution in the suite of genes expressed in the testis of Syngnathus and compared these results to those for genes that were ovary biased and evenly expressed between both gonads. We repeated this comparison using Takifugu to allow a comparison between pipefish and a taxonomic group with a more conventional reproductive mode. The dN/dS values in Syngnathus testis-biased genes did not differ significantly from dN/dS values for ovary-biased genes, and both were elevated in comparison to values for genes evenly expressed across gonads. This pattern differed substantially from that observed in Takifugu, where testis-biased genes differed significantly from both ovary-biased and evenly expressed genes. This result further supports the idea that testis-biased pufferfish genes have a faster rate of molecular evolution than pipefish in part due to sperm competition or sexual conflict. Syngnathid fish remarkably differ from the normal pattern of testis evolution, where testis-biased genes show a slightly elevated rate of molecular evolution most likely due to their tissue-specific nature, but not because testis are under stronger selection than ovaries (Duret and Mouchiroud 2000).
IMPLICATIONS AND CONCLUSIONS
Our study is noteworthy because it is the first to address the evolution of genes related to male fertility and the testis transcriptome in sex-role-reversed syngnathid fishes, which, as noted above, have no potential to engage in sperm competition. We find a pattern consistent with gene loss, a relaxation of selection, and a reduction in expression of genes involved in the key functions of the testis, such as sperm production, sperm capacitation, and acrosome assembly. At face value, this result affirms the general pattern in the literature that sexual selection and sexual conflict are driving forces behind the rapid evolution of male-biased genes in most taxa.
Syngnathid fishes, although unusual in having male pregnancy, are not the only species in which we might expect a reduction in the efficacy of selection on male reproductive function. Not much work has been done in this arena, but the few studies available have contributed to a better understanding of how deviations from a typical gonochoristic system with high potential for sexual selection on males can affect the evolution of gonads and male-biased genes at the level of the genome. For instance, Pauletto et al. (2018) studied the molecular evolution of reproductive genes in a sequentially hermaphroditic fish, the gilthead sea bream (Sparus aurata), which first matures as male and then transitions to female. They found that all sex-biased genes showed an elevated rate of dN/dS relative to unbiased genes and that female-biased genes were evolving more rapidly than male-biased genes, a reversal of the pattern observed in most other sexually reproducing species. The authors attribute this pattern to a change in the nature of selection on female function as a result of hermaphroditism. Another example of a non-gonochoristic system contributing to our understanding of genome evolution comes from Caenorhabditis nematodes. Several studies have compared genome evolution between selfing and outcrossing species of Caenorhabditis. These studies show that selfing species tend to lose genes over time, particularly those that were historically strongly sex biased (Thomas et al. 2012; Yin et al. 2018). Much like the present study, these studies also suggest that sexual selection and conflict are extremely important mechanisms of genome evolution in a wide array of taxa, while also highlighting that much work remains to be done and that an effort should be made to address these issues in taxa with diverse modes of reproduction.
As genetic information becomes more accessible, more comparative genomics and transcriptomics studies like these might help shape our predictions and illuminate specific gene families that might expand or contract in the face of different selection intensities. Clades that have both sexual and asexual lineages that have recently branched might prove particularly useful, such as Poecilia that includes the Amazon molly (P. formosa) that reproduces by gynogenesis (Schartl et al. 1995) or the several species of parthenogenic lizards where multiple asexual branches can be compared (Cole 1975; Murphy et al. 2000). In addition, sex-role-reversed taxa, such as other syngnathid species and sex-role-reversed birds, still have much to offer to this research enterprise (Fritzsche et al. 2021). By drawing on a wide variety of taxa and studying them with the plethora of modern research tools now available, future work should be able to definitively resolve the impacts of sexual selection and sexual conflict on genome evolution.
AUTHOR CONTRIBUTIONS
BDJ and AGJ assembled the transcriptomes and performed all the genomic analyses included in this project. APA, CMS, ER, and SPF collected the pipefish samples and performed the RNA extractions and sequencing for this project. CHR aided with the alignments used for the molecular evolution analysis. BDJ and AGJ wrote this article and included input from all the other authors.
ACKNOWLEDGMENTS
Funding was provided by a grant from the National Science Foundation (DEB-1953170) awarded to AGJ.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
DATA ARCHIVING
Data generated or analyzed during this study, except for RNA-seq reads, are included in this published article (and its Supporting Information). RNA-seq reads generated in this study are available in NCBI's SRA database under BioProject PRJNA850415. Additional data are available from the corresponding author upon request. Access to program fastatoorf can be found on GitHub: https://github.com/JonesLabIdaho/FastaToORF.
LITERATURE CITED
———. (
———. (
———. (
———. (
Associate Editor: W. Swanson
Handling Editor: A. McAdam

{kind=link}
{kind=link}
{kind=link}
{kind=link}