





Abstract
To determine the prevalence and in vitro electrophysiological (EP) phenotype of ultra-rare SCN5A variants of uncertain significance (VUS) identified in unexplained sudden cardiac arrest (SCA) survivors.
Retrospective review of 73 unexplained SCA survivors was used to identify all patients that underwent a form of genetic testing that included comprehensive SCN5A analysis. Ultra-rare SCN5A variants (minor allele frequency < 0.005) were adjudicated according to the 2015 American College of Medical Genetics and Genomics (ACMG) guidelines. Variants designated as VUS were expressed heterologously and characterized using the whole-cell patch clamp technique. Overall, 60/73 (82%; the average age at SCA 28 ± 12 years) unexplained SCA survivors had received SCN5A genetic testing. Of these, 5/60 (8.3%) had an ultra-rare SCN5A variant. All SCN5A variants were classified as VUS. Whereas the single SCN5A VUS (p.Asp872Asn-SCN5A) identified in an unexplained SCA survivor with PR interval prolongation and inferior early repolarization conferred a loss-of-function phenotype (46.2% reduction in peak current density; 16 ms slower recovery from inactivation), the four other SCN5A VUS (p.Glu30Gly-SCN5A, p.Gln245Lys-SCN5A, p.Pro648Leu-SCN5A, and p.Glu1240Gln-SCN5A) identified in unexplained SCA survivors without early repolarization/conduction delay were indistinguishable from wild-type Nav1.5 channels.
In the absence of a phenotype(s) potentially attributable to sodium channel dysfunction, all SCN5A VUS identified in unexplained SCA survivors conferred a wild-type EP phenotype in vitro. As the background rate of SCN5A genetic variation is not trivial, great care must be taken to avoid prioritizing genotype over phenotype when attempting to ascertain the root cause of an individual's SCA.
The majority of SCN5A rare variants identified in unexplained sudden cardiac arrest (SCA) survivors with a default diagnosis of idiopathic ventricular fibrillation (IVF) are functionally wild-type in vitro and may represent background genetic noise.
The prioritization of genotype over phenotype in the evaluation of unexplained SCA survivors with a default diagnosis of IVF can lead to potentially harmful diagnostic miscues.
Careful clinical phenotyping for evidence of underlying SCN5A-encoded Nav1.5 sodium channel dysfunction (i.e. cardiac conduction disease, inferolateral early repolarization, etc.) may prove helpful when assessing the potential pathogenicity of SCN5A rare variants identified in unexplained SCA survivors with a default diagnosis of IVF.
Introduction
Sudden cardiac arrest (SCA), defined as collapse secondary to an abrupt and unexpected cessation of heart function, is responsible for ∼15–20% of all deaths and remains a major public health issue.1 Although the majority of SCAs are attributed to the arrhythmic sequelae of coronary artery disease (∼70–75%), non-ischaemic cardiomyopathies (∼10–15%), or other structural heart disorders (i.e. congenital or valvular heart disease; ∼1–5%), as many as 10–15% of all SCA survivors have otherwise structurally normal hearts.1 In these cases, primary electrical disorders such as Brugada syndrome (BrS), catecholaminergic polymorphic ventricular tachycardia (CPVT), or long QT syndrome (LQTS) are often identified.2,3 However, recent population-based studies have shown that a thorough clinical evaluation, including advanced cardiac imaging, drug provocation studies, and genetic testing, still fails to identify an underlying structural or electrical root cause in ∼5–10% of all SCA survivors.2,4 In these unexplained SCA survivors, a default diagnosis of idiopathic ventricular fibrillation (IVF) is often rendered in accordance with current expert consensus guidelines.5
Unfortunately, in comparison to other primary cardiac arrhythmia syndromes, the genetic architecture underlying IVF remains poorly defined. Prior studies have demonstrated an association between IVF and (i) a Dutch founder risk haplotype on chromosome 7q36 that encompasses the DPP6 gene,6 (ii) loss-of-function genetic variants in the IRX3 cardiac transcription factor,7 and (iii) a mixed-function variant in CALM1-encoded calmodulin.8 In addition, American College of Medical Genetics and Genomics (ACMG) pathogenic/likely pathogenic variants in the RYR2-encoded Type 2 ryanodine receptor and SCN5A-encoded Nav1.5 cardiac sodium channel have been observed in multiple unexplained SCA/IVF cohorts.9–11 Although RYR2 loss-of-function variants have been linked to IVF,12 in many RYR2- and SCN5A-positive IVF cases, evidence of a concealed, or previously overlooked primary cardiac arrhythmia syndrome [i.e. CPVT, BrS, early repolarization syndrome (ERS), LQTS, etc.] is often present.9–11,13
At present, those functional SCN5A variants identified in unexplained SCA/IVF cases have been observed in individuals with underlying early repolarization, suggestive of underlying J-wave spectrum disorder (i.e. ERS).11,14 Furthermore, the majority of rare SCN5A missense variants identified in unexplained SCA/IVF or unexplained sudden cardiac death (SCD) cohorts lack in vitro or in vivo functional data to argue for or against their pathogenicity.9,10 As such, we sought to determine the prevalence of SCN5A rare variants in a single-centre cohort of unexplained SCA survivors with a default diagnosis of IVF as well as the clinical and in vitro cellular electrophysiological (EP) phenotypes associated with these variants.
Methods
Study design and definitions
In this Mayo Clinic Institutional Review Board-approved single-centre study, the electronic medical records of 3194 consecutive patients referred to the Mayo Clinic Genetic Heart Rhythm Clinic between January 1999 and May 2018 were reviewed retrospectively to identify all individuals referred for evaluation after experiencing a sentinel event of SCA. For the purposes of this study, SCA was defined as a witnessed or unwitnessed collapse that required external defibrillation, from either ventricular fibrillation (VF) or pulseless ventricular tachycardia, as part of the successful resuscitation efforts. An SCA was considered to be the sentinel event if no prior diagnosis or clinical suspicion for an underlying SCD-predisposing condition was documented.
After exclusion of those patients that (i) that did not experience a true SCA, (ii) suffered non-cardiac arrests, (iii) had a pre- or post-SCA diagnosis of a specific cardiac channelopathy (CPVT, Brugada syndrome, LQTS, short QT syndrome, etc.), complex congenital heart disease, ischaemic heart disease (i.e. acute coronary syndrome, anomalous coronary artery, coronary vasospasm, myocardial bridging, etc.), non-ischaemic cardiomyopathy (i.e. arrhythmogenic, dilated, hypertrophic, infiltrative, restrictive, etc.) or valvular heart disease (i.e. aortic stenosis, arrhythmogenic mitral valve prolapse, etc.), and/or (iv) those that did not undergo a form of genetic testing that included comprehensive SCN5A analysis, 60 unexplained SCA survivors with a default diagnosis of IVF were included in the final analysis (Figure 1). Additional details regarding the clinical evaluation and genetic assessment of these patients can be found in the Supplementary material online.
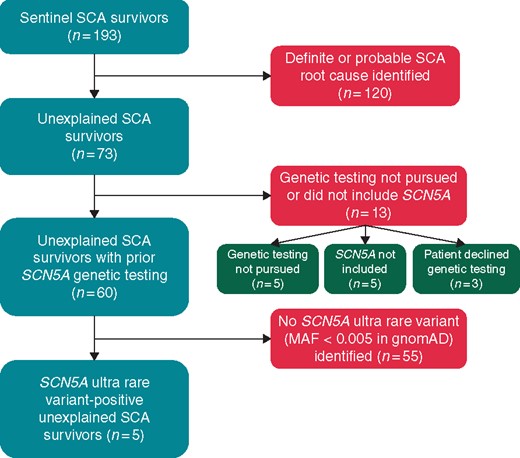
Patient selection flowchart. gnomAD, Genome Aggregation Database; MAF, minor allele frequency; SCA, sudden cardiac arrest.
SCN5A mammalian expression vectors and site-directed mutagenesis
The p.Glu30Gly-SCN5A, p.Gln245Lys-SCN5A, p.Pro648Leu-SCN5A, p.Asp872Asn-SCN5A, and p.Glu1240Gln-SCN5A variants of uncertain significance (VUS) were engineered by site-directed mutagenesis (QuikChange Site-Directed Mutagenesis Kit, Stratagene, La Jolla, CA, USA) using PCR technique into most common alternatively spliced SCN5A transcripts, H558/Q1077del (Genbank accession no. AY148488) of the human cardiac voltage-dependent Na+ channel α subunit SCN5A cDNA in the pcDNA3 vector (Invitrogen, Carlsbad, CA, USA). The presence of each sequence variant and integrity of the constructs was verified by direct DNA sequencing.
Heterologous expression of variant and wild-type sodium channels
1 μg SCN5A wild type or SCN5A VUS-containing (p.Glu30Gly-SCN5A, p.Gln245Lys-SCN5A, p.Pro648Leu-SCN5A, p.Asp872Asn-SCN5A, and p.Glu1240Gln-SCN5A) cDNA were co-transfected with 0.25 μg green fluorescence protein cDNA (kindly provided by Dr Gianrico Farrugia, Mayo Clinic, Rochester, MN, USA) with the use of 3 μL Lipofectamine (Invitrogen, Carlsbad, CA, USA). Transfected HEK-293 or TSA201 cells were cultured in OPTI-MEM (Gibco, Carlsbad, CA, USA) and incubated for 24 h. Cells exhibiting green fluorescence were selected for EP experiments. Each SCN5A variant was studied as a matched pair (i.e. variant compared to wild type) using multiple transfections of HEK-293/TSA201 cells from the same passage.
Electrophysiological measurements and data analysis
Standard whole-cell patch clamp technique was used to measure wild type and variant (i.e. VUS) sodium currents at room temperature (22–24°C) with the use of an Axopatch 200B amplifier, Digidata 1440A and pclamp 10 software (Axon Instruments, Sunnyvale, CA, USA). The extracellular solution contained (mmol/L): 140 NaCl, 4 KCl6, 1.8 CaCl2, 0.75 MgCl2, and five 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH adjusted to 7.4 with NaOH. The intracellular solution contained (mmol/L): 120 CsF, 20 CsCl, two ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), and five HEPES, pH adjusted to 7.4 with CsOH.1 Microelectrodes were pulled on a P-97 puller (Sutter Instruments, Novato, CA, USA) and fire polished to a final resistance of 2–3 MΩ. Series resistance was compensated by 80–85%. Currents were filtered at 5 kHz and digitized at 10 kHz with an eight-pole Bessel filter. The voltage-dependence of activation, steady-state inactivation, recovery from inactivation, and late inward sodium current (INaL) were determined using voltage-clamp protocols described in the figure legend. In all protocols, a holding potential of −100 or −120 mV and a start to start interval of 1–3 s were used. Data were analysed using Clampfit (Axon Instruments, Sunnyvale, CA, USA), Excel (Microsoft, Redmond, WA, USA), and fitted with Origin 8 or 9 (OriginLab Corporation, Northampton, MA, USA) software.
The voltage-dependence of activation curve was fitted with a Boltzmann function: GNa/GNa, max = {1 + exp [(V − V1/2)/k]}−1, where V1/2 and k are the half-maximal voltage of activation and the slope factor respectively, and GNa = INa/(V − Vrev), where Vrev is the reversal potential. The steady-state inactivation curve was fitted with a Boltzmann function: INa/INa, max = {1 + exp [(V − V1/2)/k]}−1, where V1/2 and k are the half-maximal voltage of inactivation and the slope factor, respectively. Recovery from inactivation was fitted with a two-exponential function: y = y0 + [Af exp(−t/τf)] + [As exp(−t/τs)], where Af and As indicate the fractions of the fast and the slow components of recovery from inactivation, respectively, and τf and τs indicate the time constants for recovery from fast and slow inactivation, respectively.
Statistical analysis
JMP ® Pro 10.0.0 (SAS Incorporated, Cary, NC, USA) was employed for statistical analysis. All continuous variables are presented as mean ± standard error of the mean. For clinical parameters, the Fisher’s exact test was used to compare categorical variables and the Wilcoxon rank-sum/Mann–Whitney U test was used to compare continuous variables. For cellular EP parameters, a Student’s t-test was performed to determine statistical significance between wild type and variant groups. As each SCN5A variant was studied as matched pairs (i.e. variant vs. wild type) over the course of several years, the five SCN5A variants cannot be compared directly to each other due to inherent differences in the EP properties of wild-type Nav1.5 observed at different time periods and across HEK-293/TSA201 passages. For all tests, a two-tailed P-value of ≤0.05 was considered statistically significant. All authors had full access to and take full responsibility for the integrity of the data.
Results
Baseline demographics and prevalence of SCN5A variants
Overall, 73 unrelated sentinel SCA survivors referred to the Mayo Clinic Genetic Heart Rhythm Clinic between January 1999 and May 2018 remained unexplained (i.e. no definitive or probable SCA root cause identified) following both primary/local and referral clinical evaluations (Figure 1). Amongst these 73 unexplained SCA survivors, 60 (82%) received a form of genetic testing that involved sequencing the entirety of the SCN5A gene (35 pan-cardiac gene panel/whole-exome sequencing and 25 diagnostic BrS/LQTS genetic testing) and were included in the final analysis (Figure 1). Basic demographics and details regarding the adjunct/advanced cardiovascular testing (i.e. advanced imaging, invasive testing, etc.) utilized in the evaluation of these 60 unexplained SCA survivors are provided in Table 1 and Supplementary material online, Table S1, respectively.
Baseline demographics of the Mayo Clinic Genetic Heart Rhythm Clinic unexplained sudden cardiac arrest cohort
| Unexplained SCA survivors with SCN5A genetic testing (n = 60) | |
|---|---|
| Female, n (%) | 26/60 (43%) |
| Age at presentation (years) | 28 ± 12 |
| Family history of SCA/SCD, n (%) | 11/60 (18%) |
| SCA circumstance | |
| Exertion/emotion, n (%) | 15/60 (25%) |
| Rest, n (%) | 37/60 (62%) |
| Sleep, n (%) | 7/60 (12%) |
| Not known, n (%) | 1/60 (1.7%) |
| ECG parameters | |
| PR interval (ms) | 157 ± 27 |
| QRS duration (ms) | 98 ± 17 |
| QTc (ms) | 424 ± 38 |
| Early repolarization pattern | 6/60 (10%) |
| Inferior | 2/6 (33%) |
| Lateral | 3/6 (50%) |
| Inferolateral | 1/6 (17%) |
| Echocardiography | |
| LVEF (%) | 61 ± 5 |
| LVEDD (mm) | 50 ± 5 |
| Unexplained SCA survivors with SCN5A genetic testing (n = 60) | |
|---|---|
| Female, n (%) | 26/60 (43%) |
| Age at presentation (years) | 28 ± 12 |
| Family history of SCA/SCD, n (%) | 11/60 (18%) |
| SCA circumstance | |
| Exertion/emotion, n (%) | 15/60 (25%) |
| Rest, n (%) | 37/60 (62%) |
| Sleep, n (%) | 7/60 (12%) |
| Not known, n (%) | 1/60 (1.7%) |
| ECG parameters | |
| PR interval (ms) | 157 ± 27 |
| QRS duration (ms) | 98 ± 17 |
| QTc (ms) | 424 ± 38 |
| Early repolarization pattern | 6/60 (10%) |
| Inferior | 2/6 (33%) |
| Lateral | 3/6 (50%) |
| Inferolateral | 1/6 (17%) |
| Echocardiography | |
| LVEF (%) | 61 ± 5 |
| LVEDD (mm) | 50 ± 5 |
ECG, electrocardiogram; LVEDD, left ventricular end-diastolic dimension; LVEF, left ventricular ejection fraction; QTc, heart rate-corrected QT interval; SCA, sudden cardiac arrest; SCD, sudden cardiac death.
Baseline demographics of the Mayo Clinic Genetic Heart Rhythm Clinic unexplained sudden cardiac arrest cohort
| Unexplained SCA survivors with SCN5A genetic testing (n = 60) | |
|---|---|
| Female, n (%) | 26/60 (43%) |
| Age at presentation (years) | 28 ± 12 |
| Family history of SCA/SCD, n (%) | 11/60 (18%) |
| SCA circumstance | |
| Exertion/emotion, n (%) | 15/60 (25%) |
| Rest, n (%) | 37/60 (62%) |
| Sleep, n (%) | 7/60 (12%) |
| Not known, n (%) | 1/60 (1.7%) |
| ECG parameters | |
| PR interval (ms) | 157 ± 27 |
| QRS duration (ms) | 98 ± 17 |
| QTc (ms) | 424 ± 38 |
| Early repolarization pattern | 6/60 (10%) |
| Inferior | 2/6 (33%) |
| Lateral | 3/6 (50%) |
| Inferolateral | 1/6 (17%) |
| Echocardiography | |
| LVEF (%) | 61 ± 5 |
| LVEDD (mm) | 50 ± 5 |
| Unexplained SCA survivors with SCN5A genetic testing (n = 60) | |
|---|---|
| Female, n (%) | 26/60 (43%) |
| Age at presentation (years) | 28 ± 12 |
| Family history of SCA/SCD, n (%) | 11/60 (18%) |
| SCA circumstance | |
| Exertion/emotion, n (%) | 15/60 (25%) |
| Rest, n (%) | 37/60 (62%) |
| Sleep, n (%) | 7/60 (12%) |
| Not known, n (%) | 1/60 (1.7%) |
| ECG parameters | |
| PR interval (ms) | 157 ± 27 |
| QRS duration (ms) | 98 ± 17 |
| QTc (ms) | 424 ± 38 |
| Early repolarization pattern | 6/60 (10%) |
| Inferior | 2/6 (33%) |
| Lateral | 3/6 (50%) |
| Inferolateral | 1/6 (17%) |
| Echocardiography | |
| LVEF (%) | 61 ± 5 |
| LVEDD (mm) | 50 ± 5 |
ECG, electrocardiogram; LVEDD, left ventricular end-diastolic dimension; LVEF, left ventricular ejection fraction; QTc, heart rate-corrected QT interval; SCA, sudden cardiac arrest; SCD, sudden cardiac death.
In total, an SCN5A ultra-rare variant (minor allele frequency < 0.005 in public exomes/genomes) was identified in 8.3% (5/60) of unexplained SCA survivors. Interestingly, 2/5 (40%) were diagnosed, largely on the basis of their genetic test results, initially by local providers with an underlying cardiac sodium channelopathy (LQT3 in a p.Pro648Leu-SCN5A-positive unexplained SCA survivor and BrS in a p.Glu1240Gln-SCN5A-positive unexplained SCA survivor; details are available in the Supplementary material online). Localization of the five distinct SCN5A ultra-rare variants (p.Glu30Gly-SCN5A, p.Gln245Lys-SCN5A, p.Pro648Leu-SCN5A, p.Asp872Asn-SCN5A, and p.Glu1240Gln-SCN5A) identified in this study, as well as those functional SCN5A variants detected previously in unexplained SCA/IVF cases, on the predicted Nav1.5 protein linear topology are depicted in Figure 2. Lastly, each of the SCN5A ultra-rare variants identified in the current study were classified ambiguously as VUS in accordance with the 2015 ACMG variant classification and reporting guidelines (Table 2).
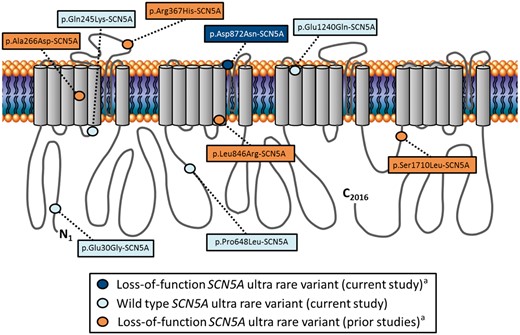
Schematic depiction of Nav1.5 linear topology illustrating the localization of SCN5A ultra rare variants observed in individuals with a default diagnosis of idiopathic ventricular fibrillation. Dark blue circle denotes a loss-of-function variant identified in the current study. Light blue circles denote wild-type variants identified in the current study. Orange circles denote loss-of-function variants identified in prior studies. aVariant(s) identified in an individual(s) with evidence of conduction delay and/or early repolarization.
SCN5A variant of uncertain significance-positive unexplained sudden cardiac arrest survivors
| ID | Sex | Age at SCA | SCA Circumstance | PR (ms) | QRS (ms) | QTc (ms) | ERP | Drug provocation | Variant(s) | gnomAD MAF | ACMG classification (criteria) | ACMG reclassification (criteria) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MC0470 | F | 52 | Rest | – | 84 | 432 | No | None | p.Glu30Gly-SCN5A | Absent | VUS (PM2 and PP3) | VUS (PM2, PP3, and BS3) |
| MC8552 | F | 49 | Rest | 180 | 84 | 456 | No | Epinephrine | p.Gln245Lys-SCN5A | 2/246 048 | VUS (PM1 and PP3) | VUS (PM1, PP3, and BS3) |
| MC5874 | M | 39 | Exertion | 170 | 88 | 436 | No | Procainamide/epinephrine | p.Pro648Leu-SCN5A | 14/276 028 | VUS (PM1) | VUS (PM1 and BS3) |
| MC2070 | M | 17 | Exertion | 230 | 140 | 448 | Yes | Procainamide/isoproterenol | p.Asp872Asn-SCN5A | 4/246 264 | VUS (PM1 and PP3) | Likely pathogenic (ii) (PS3, PM1, and PP3) |
| MC4310 | F | 19 | Rest | 150 | 98 | 454 | No | Procainamide | p.Glu1240Gln-SCN5A | 9/246 260 | VUS (PM1 and PP3) | VUS (PM1, PP3, and BS3) |
| ID | Sex | Age at SCA | SCA Circumstance | PR (ms) | QRS (ms) | QTc (ms) | ERP | Drug provocation | Variant(s) | gnomAD MAF | ACMG classification (criteria) | ACMG reclassification (criteria) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MC0470 | F | 52 | Rest | – | 84 | 432 | No | None | p.Glu30Gly-SCN5A | Absent | VUS (PM2 and PP3) | VUS (PM2, PP3, and BS3) |
| MC8552 | F | 49 | Rest | 180 | 84 | 456 | No | Epinephrine | p.Gln245Lys-SCN5A | 2/246 048 | VUS (PM1 and PP3) | VUS (PM1, PP3, and BS3) |
| MC5874 | M | 39 | Exertion | 170 | 88 | 436 | No | Procainamide/epinephrine | p.Pro648Leu-SCN5A | 14/276 028 | VUS (PM1) | VUS (PM1 and BS3) |
| MC2070 | M | 17 | Exertion | 230 | 140 | 448 | Yes | Procainamide/isoproterenol | p.Asp872Asn-SCN5A | 4/246 264 | VUS (PM1 and PP3) | Likely pathogenic (ii) (PS3, PM1, and PP3) |
| MC4310 | F | 19 | Rest | 150 | 98 | 454 | No | Procainamide | p.Glu1240Gln-SCN5A | 9/246 260 | VUS (PM1 and PP3) | VUS (PM1, PP3, and BS3) |
ACMG, American College of Medical Genetics and Genomics; BS, benign strong; F, female; gnomAD, Genome Aggregation Database; ID, identification; M, male; MAF, minor allele frequency; PM, pathogenic moderate; PP, pathogenic supporting; PS, pathogenic strong; QTc, heart-rate corrected QT interval; SCA, sudden cardiac arrest; VUS, variants of uncertain significance.
SCN5A variant of uncertain significance-positive unexplained sudden cardiac arrest survivors
| ID | Sex | Age at SCA | SCA Circumstance | PR (ms) | QRS (ms) | QTc (ms) | ERP | Drug provocation | Variant(s) | gnomAD MAF | ACMG classification (criteria) | ACMG reclassification (criteria) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MC0470 | F | 52 | Rest | – | 84 | 432 | No | None | p.Glu30Gly-SCN5A | Absent | VUS (PM2 and PP3) | VUS (PM2, PP3, and BS3) |
| MC8552 | F | 49 | Rest | 180 | 84 | 456 | No | Epinephrine | p.Gln245Lys-SCN5A | 2/246 048 | VUS (PM1 and PP3) | VUS (PM1, PP3, and BS3) |
| MC5874 | M | 39 | Exertion | 170 | 88 | 436 | No | Procainamide/epinephrine | p.Pro648Leu-SCN5A | 14/276 028 | VUS (PM1) | VUS (PM1 and BS3) |
| MC2070 | M | 17 | Exertion | 230 | 140 | 448 | Yes | Procainamide/isoproterenol | p.Asp872Asn-SCN5A | 4/246 264 | VUS (PM1 and PP3) | Likely pathogenic (ii) (PS3, PM1, and PP3) |
| MC4310 | F | 19 | Rest | 150 | 98 | 454 | No | Procainamide | p.Glu1240Gln-SCN5A | 9/246 260 | VUS (PM1 and PP3) | VUS (PM1, PP3, and BS3) |
| ID | Sex | Age at SCA | SCA Circumstance | PR (ms) | QRS (ms) | QTc (ms) | ERP | Drug provocation | Variant(s) | gnomAD MAF | ACMG classification (criteria) | ACMG reclassification (criteria) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MC0470 | F | 52 | Rest | – | 84 | 432 | No | None | p.Glu30Gly-SCN5A | Absent | VUS (PM2 and PP3) | VUS (PM2, PP3, and BS3) |
| MC8552 | F | 49 | Rest | 180 | 84 | 456 | No | Epinephrine | p.Gln245Lys-SCN5A | 2/246 048 | VUS (PM1 and PP3) | VUS (PM1, PP3, and BS3) |
| MC5874 | M | 39 | Exertion | 170 | 88 | 436 | No | Procainamide/epinephrine | p.Pro648Leu-SCN5A | 14/276 028 | VUS (PM1) | VUS (PM1 and BS3) |
| MC2070 | M | 17 | Exertion | 230 | 140 | 448 | Yes | Procainamide/isoproterenol | p.Asp872Asn-SCN5A | 4/246 264 | VUS (PM1 and PP3) | Likely pathogenic (ii) (PS3, PM1, and PP3) |
| MC4310 | F | 19 | Rest | 150 | 98 | 454 | No | Procainamide | p.Glu1240Gln-SCN5A | 9/246 260 | VUS (PM1 and PP3) | VUS (PM1, PP3, and BS3) |
ACMG, American College of Medical Genetics and Genomics; BS, benign strong; F, female; gnomAD, Genome Aggregation Database; ID, identification; M, male; MAF, minor allele frequency; PM, pathogenic moderate; PP, pathogenic supporting; PS, pathogenic strong; QTc, heart-rate corrected QT interval; SCA, sudden cardiac arrest; VUS, variants of uncertain significance.
Clinical and electrocardiographic phenotype of SCN5A variant of uncertain significance-positive unexplained sudden cardiac arrest survivors
As the majority of functional SCN5A variants detected previously in cases of unexplained SCA with a default diagnosis IVF had evidence of cardiac conduction delay (i.e. PR prolongation or intraventricular conduction delay) and/or inferolateral early repolarization, we next sought to assess carefully the clinical and electrocardiographic phenotypes of the SCN5A VUS-positive unexplained SCA survivors identified in this study for evidence of an underlying cardiac sodium channelopathy [i.e. BrS, ERS, primary cardiac conduction disease (PCCD), LQTS, or multifocal ectopic Purkinje-related premature contractions (MEPPC)].
Whereas the p.Glu30Gly-SCN5A-, p.Gln245Lys-SCN5A-, p.Pro648Leu-SCN5A, and p.Glu1240Gln-SCN5A-positive unexplained SCA survivors displayed no electrocardiographic hallmarks of any SCN5A-mediated disease (Table 2), serial 12-lead electrocardiograms (ECGs) from the p.Asp872Asn-SCN5A-positive unexplained SCA survivor demonstrated consistent PR interval prolongation, non-specific intraventricular conduction delay, and a dynamic inferior early repolarization pattern (Figure 3D and Table 2). Although a Type 1 BrS pattern was not induced during procainamide challenge, the p.Asp872Asn-SCN5A-positive unexplained SCA survivor underwent a diagnostic electrophysiology study which revealed PVCs, highly fragmented QRS signals, and short-coupled premature ventricular contraction (PVC)-triggered ventricular arrhythmias originating from the tricuspid annulus, right ventricular outflow tract, and right ventricular apex that were ablated successfully. Importantly, no evidence of structural heart disease has been observed in the p.Asp872Asn-SCN5A-positive unexplained SCA survivor on computed tomography coronary angiography, cardiac magnetic resonance imaging, or serial transthoracic echocardiography over 11 years of follow-up.
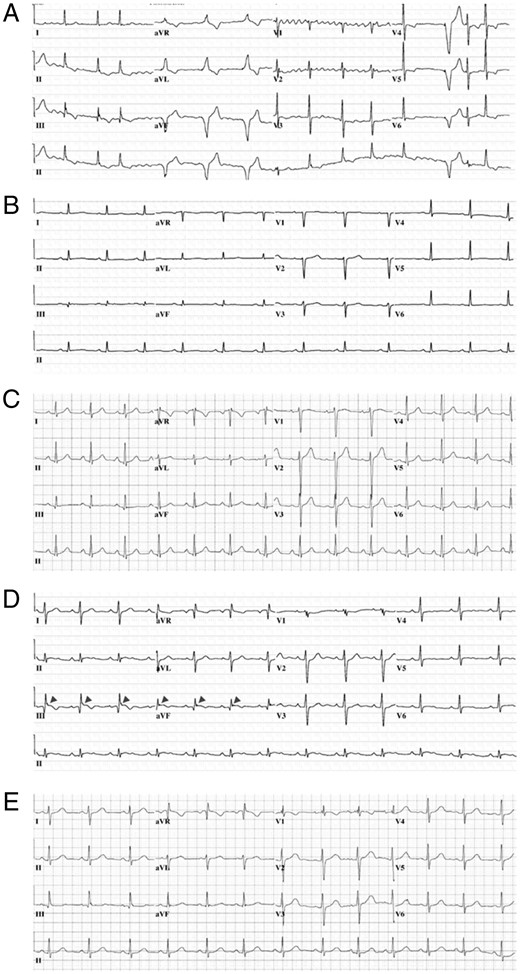
Baseline 12-lead ECGs obtained during the referral evaluation of SCN5A ultra rare variant-positive unexplained SCA survivors. (A) Representative 12-lead ECG from p.Glu30Gly-SCN5A-positive unexplained SCA survivor. (B) Representative 12-lead ECG from p.Gln245Lys-SCN5A-positive unexplained SCA survivor. (C) Representative 12-lead ECG from p.Pro648Leu-SCN5A-positive unexplained SCA survivor. (D) Representative 12-lead ECG from p.Asp872Asn-SCN5A-positive unexplained SCA survivor. (E) Representative 12-lead ECG from p.Glu1240Gln-SCN5A-positive unexplained SCA survivor. ECG, electrocardiogram; SCA, sudden cardiac arrest.
Representative 12-lead ECGs for each of the five SCN5A VUS-positive unexplained SCA survivors are shown in Figure 3 and complete case descriptions are contained within the Supplementary material online.
Characterization of SCN5A variants of uncertain significance identified in unexplained sudden cardiac arrest survivors
Given the PR interval prolongation, intraventricular conduction delay, inferior early repolarization pattern, and short-coupled ventricular arrhythmias observed in the p.Asp872Asn-SCN5A-positive unexplained SCA survivor, we hypothesized that p.Asp872Asn-SCN5A may confer a BrS- or ERS-like loss-of-function EP phenotype. Typical tracings of voltage-dependent activation from wild type and p.Asp872Asn-SCN5A are depicted in Figure 4A with holding potential at −100 mV to various depolarization potentials (see figure legend for details). Analysis of the current–voltage relationship revealed that sodium current densities were significantly reduced by p.Asp872Asn-SCN5A across the voltage from −30 to +70 mV compared to wild type (P < 0.05; Figure 4B). At −20 mV, the sodium current density was significantly reduced by 44.2% from −651.2 ± 63.1 (wild type, n = 10) to −363.3 ± 51.5 pA/pF (p.Asp872Asn-SCN5A, n = 10, P < 0.05; Figure 4B and Table 3). The voltage-dependence of activation and steady-state inactivation remained unchanged (Figure 4C and D and Table 3). However, p.Asp872Asn-SCN5A revealed slower recovery from fast component of inactivation from 6.6 ± 0.4 ms (wild type, n = 7) to 9.8 ± 0.8 ms (p.Asp872Asn-SCN5A, n = 7, P < 0.05) and slow component of inactivation from 54.1 ± 3.0 ms (wild type, n = 7) to 70.1 ± 4.8 ms (p.Asp872Asn-SCH5A, n = 7, P < 0.05), respectively (Figure 4E and Table 3). The total reduction in Nav1.5 current density (∼23–25% in a heterozygous state) and slower recovery from inactivation conferred by p.Asp872Asn-SCN5A are consistent with mild Nav1.5 loss-of-function indicating that p.Asp872Asn-SCN5A may, at a minimum, be contributing to the p.Asp872Asn-SCN5A-positive unexplained SCA survivor’s clinical picture.
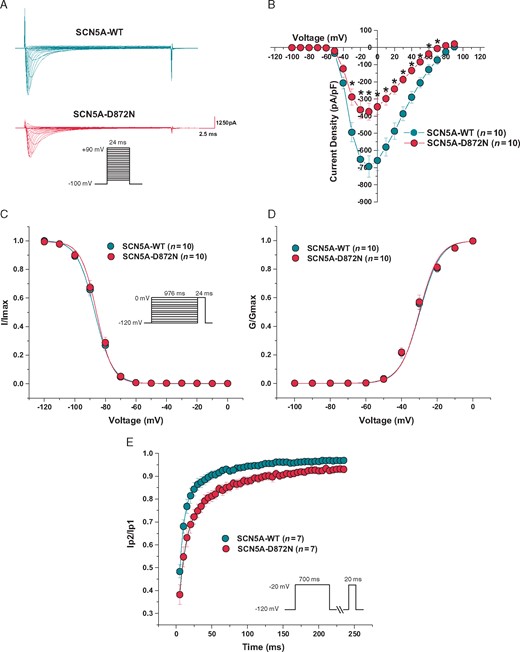
Functional characterization of the p.Asp872Asn-SCN5A ultra rare variant identified in an unexplained SCA survivor with subtle inferior early repolarization. (A) Representative whole-cell tracings determined from a holding potential of −100 mV to testing potential of +90 mV in 10 mV increments with 24 ms duration. (B) Current–voltage relationship. (C) Steady-state inactivation curves. (D) Voltage-dependence of activation curves. (E) Recovery from inactivation from a holding potential of −120 mV to pre-pulse of −20 mV with 700 ms duration and with increased recovery interval followed by a test pulse of −20 mV with 20 ms duration. All values represent mean ± standard error of the mean. *P < 0.05 vs. SCN5A-WT. D872N, p.Asp872Asn-SCN5A ultra rare variant; G/Gmax, normalized conductance; I/Imax, normalized sodium current; SCA, sudden cardiac arrest; WT, wild type.
Cellular electrophysiological parameters for SCN5A ultra rare variants identified in unexplained sudden cardiac arrest survivors
| SCN5A construct | Current density at −20 mV (pA/pF) | Inactivation V1/2 (mV) | Activation V1/2 (mV) | Recovery from inactivation (ms) fast, slow | Functional interpretation |
|---|---|---|---|---|---|
| Wild type | −505.2 ± 71.7 | −79.4 ± 0.9 | −35.9 ± 0.9 | 6.8 ± 0.5, 68.5 ± 5.4 | Wild type |
| n = 10 | n = 10 | n = 10 | n = 7 | ||
| p.Glu30Gly | −553.6 ± 85.9 | −76.8 ± 0.5a | −35.7 ± 0.9 | 5.9 ± 0.4, 73.4 ± 4.5 | |
| n = 11 | n = 12 | n = 12 | n = 9 | ||
| Wild type | −834.6 ± 84.4 | −81.2 ± 0.7 | −37.0 ± 1.4 | 7.0 ± 0.4, 64.0 ± 3.0 | Wild type |
| n = 13 | n = 13 | n = 13 | n = 10 | ||
| p.Gln245Lys | −702.7 ± 103.2 | −80.5 ± 1.0 | −40.7 ± 1.5 | 7.7 ± 0.6, 59.2 ± 4.4 | |
| n = 12 | n = 12 | n = 12 | n = 9 | ||
| Wild type | −367.1 ± 61.8 | −88.3 ± 0.3 | −31.4 ± 1.3 | 11.6 ± 0.5, 58.5 ± 4.3 | Wild type |
| n = 13 | n = 13 | n = 13 | n = 7 | ||
| p.Pro648Leu | −440.7 ± 70.7 | −86.9 ± 0.5 | −35.2 ± 1.5a | 10.1 ± 0.6, 73.3 ± 4.4a | |
| n = 13 | n = 11 | n = 13 | n = 8 | ||
| Wild type | −651.2 ± 63.1 | −87.0 ± 0.75 | −29.7 ± 1.2 | 6.6 ± 0.4, 54.1 ± 3.0 | Loss-of-function |
| n = 10 | n = 10 | n = 10 | n = 7 | ||
| p.Asp872Asn | −363.3 ± 51.5a | −85.8 ± 0.62 | −30.0 ± 1.3 | 9.8 ± 0.8a, 70.1 ± 4.8a | |
| n = 10 | n = 10 | n = 10 | n = 7 | ||
| Wild type | −356.2 ± 60.0 | −88.0 ± 1.8 | −36.5 ± 1.5 | 11.7 ± 0.6, 65.3 ± 5.4 | Wild type |
| n = 15 | n = 15 | n = 15 | n = 7 | ||
| p.Glu1240Gln | −420.1 ± 52.0 | −87.1 ± 0.8 | −33.2 ± 1.2 | 10.7 ± 0.9, 87.1 ± 6.4a | |
| n = 16 | n = 16 | n = 16 | n = 7 |
| SCN5A construct | Current density at −20 mV (pA/pF) | Inactivation V1/2 (mV) | Activation V1/2 (mV) | Recovery from inactivation (ms) fast, slow | Functional interpretation |
|---|---|---|---|---|---|
| Wild type | −505.2 ± 71.7 | −79.4 ± 0.9 | −35.9 ± 0.9 | 6.8 ± 0.5, 68.5 ± 5.4 | Wild type |
| n = 10 | n = 10 | n = 10 | n = 7 | ||
| p.Glu30Gly | −553.6 ± 85.9 | −76.8 ± 0.5a | −35.7 ± 0.9 | 5.9 ± 0.4, 73.4 ± 4.5 | |
| n = 11 | n = 12 | n = 12 | n = 9 | ||
| Wild type | −834.6 ± 84.4 | −81.2 ± 0.7 | −37.0 ± 1.4 | 7.0 ± 0.4, 64.0 ± 3.0 | Wild type |
| n = 13 | n = 13 | n = 13 | n = 10 | ||
| p.Gln245Lys | −702.7 ± 103.2 | −80.5 ± 1.0 | −40.7 ± 1.5 | 7.7 ± 0.6, 59.2 ± 4.4 | |
| n = 12 | n = 12 | n = 12 | n = 9 | ||
| Wild type | −367.1 ± 61.8 | −88.3 ± 0.3 | −31.4 ± 1.3 | 11.6 ± 0.5, 58.5 ± 4.3 | Wild type |
| n = 13 | n = 13 | n = 13 | n = 7 | ||
| p.Pro648Leu | −440.7 ± 70.7 | −86.9 ± 0.5 | −35.2 ± 1.5a | 10.1 ± 0.6, 73.3 ± 4.4a | |
| n = 13 | n = 11 | n = 13 | n = 8 | ||
| Wild type | −651.2 ± 63.1 | −87.0 ± 0.75 | −29.7 ± 1.2 | 6.6 ± 0.4, 54.1 ± 3.0 | Loss-of-function |
| n = 10 | n = 10 | n = 10 | n = 7 | ||
| p.Asp872Asn | −363.3 ± 51.5a | −85.8 ± 0.62 | −30.0 ± 1.3 | 9.8 ± 0.8a, 70.1 ± 4.8a | |
| n = 10 | n = 10 | n = 10 | n = 7 | ||
| Wild type | −356.2 ± 60.0 | −88.0 ± 1.8 | −36.5 ± 1.5 | 11.7 ± 0.6, 65.3 ± 5.4 | Wild type |
| n = 15 | n = 15 | n = 15 | n = 7 | ||
| p.Glu1240Gln | −420.1 ± 52.0 | −87.1 ± 0.8 | −33.2 ± 1.2 | 10.7 ± 0.9, 87.1 ± 6.4a | |
| n = 16 | n = 16 | n = 16 | n = 7 |
P < 0.05 vs. wild type.
Cellular electrophysiological parameters for SCN5A ultra rare variants identified in unexplained sudden cardiac arrest survivors
| SCN5A construct | Current density at −20 mV (pA/pF) | Inactivation V1/2 (mV) | Activation V1/2 (mV) | Recovery from inactivation (ms) fast, slow | Functional interpretation |
|---|---|---|---|---|---|
| Wild type | −505.2 ± 71.7 | −79.4 ± 0.9 | −35.9 ± 0.9 | 6.8 ± 0.5, 68.5 ± 5.4 | Wild type |
| n = 10 | n = 10 | n = 10 | n = 7 | ||
| p.Glu30Gly | −553.6 ± 85.9 | −76.8 ± 0.5a | −35.7 ± 0.9 | 5.9 ± 0.4, 73.4 ± 4.5 | |
| n = 11 | n = 12 | n = 12 | n = 9 | ||
| Wild type | −834.6 ± 84.4 | −81.2 ± 0.7 | −37.0 ± 1.4 | 7.0 ± 0.4, 64.0 ± 3.0 | Wild type |
| n = 13 | n = 13 | n = 13 | n = 10 | ||
| p.Gln245Lys | −702.7 ± 103.2 | −80.5 ± 1.0 | −40.7 ± 1.5 | 7.7 ± 0.6, 59.2 ± 4.4 | |
| n = 12 | n = 12 | n = 12 | n = 9 | ||
| Wild type | −367.1 ± 61.8 | −88.3 ± 0.3 | −31.4 ± 1.3 | 11.6 ± 0.5, 58.5 ± 4.3 | Wild type |
| n = 13 | n = 13 | n = 13 | n = 7 | ||
| p.Pro648Leu | −440.7 ± 70.7 | −86.9 ± 0.5 | −35.2 ± 1.5a | 10.1 ± 0.6, 73.3 ± 4.4a | |
| n = 13 | n = 11 | n = 13 | n = 8 | ||
| Wild type | −651.2 ± 63.1 | −87.0 ± 0.75 | −29.7 ± 1.2 | 6.6 ± 0.4, 54.1 ± 3.0 | Loss-of-function |
| n = 10 | n = 10 | n = 10 | n = 7 | ||
| p.Asp872Asn | −363.3 ± 51.5a | −85.8 ± 0.62 | −30.0 ± 1.3 | 9.8 ± 0.8a, 70.1 ± 4.8a | |
| n = 10 | n = 10 | n = 10 | n = 7 | ||
| Wild type | −356.2 ± 60.0 | −88.0 ± 1.8 | −36.5 ± 1.5 | 11.7 ± 0.6, 65.3 ± 5.4 | Wild type |
| n = 15 | n = 15 | n = 15 | n = 7 | ||
| p.Glu1240Gln | −420.1 ± 52.0 | −87.1 ± 0.8 | −33.2 ± 1.2 | 10.7 ± 0.9, 87.1 ± 6.4a | |
| n = 16 | n = 16 | n = 16 | n = 7 |
| SCN5A construct | Current density at −20 mV (pA/pF) | Inactivation V1/2 (mV) | Activation V1/2 (mV) | Recovery from inactivation (ms) fast, slow | Functional interpretation |
|---|---|---|---|---|---|
| Wild type | −505.2 ± 71.7 | −79.4 ± 0.9 | −35.9 ± 0.9 | 6.8 ± 0.5, 68.5 ± 5.4 | Wild type |
| n = 10 | n = 10 | n = 10 | n = 7 | ||
| p.Glu30Gly | −553.6 ± 85.9 | −76.8 ± 0.5a | −35.7 ± 0.9 | 5.9 ± 0.4, 73.4 ± 4.5 | |
| n = 11 | n = 12 | n = 12 | n = 9 | ||
| Wild type | −834.6 ± 84.4 | −81.2 ± 0.7 | −37.0 ± 1.4 | 7.0 ± 0.4, 64.0 ± 3.0 | Wild type |
| n = 13 | n = 13 | n = 13 | n = 10 | ||
| p.Gln245Lys | −702.7 ± 103.2 | −80.5 ± 1.0 | −40.7 ± 1.5 | 7.7 ± 0.6, 59.2 ± 4.4 | |
| n = 12 | n = 12 | n = 12 | n = 9 | ||
| Wild type | −367.1 ± 61.8 | −88.3 ± 0.3 | −31.4 ± 1.3 | 11.6 ± 0.5, 58.5 ± 4.3 | Wild type |
| n = 13 | n = 13 | n = 13 | n = 7 | ||
| p.Pro648Leu | −440.7 ± 70.7 | −86.9 ± 0.5 | −35.2 ± 1.5a | 10.1 ± 0.6, 73.3 ± 4.4a | |
| n = 13 | n = 11 | n = 13 | n = 8 | ||
| Wild type | −651.2 ± 63.1 | −87.0 ± 0.75 | −29.7 ± 1.2 | 6.6 ± 0.4, 54.1 ± 3.0 | Loss-of-function |
| n = 10 | n = 10 | n = 10 | n = 7 | ||
| p.Asp872Asn | −363.3 ± 51.5a | −85.8 ± 0.62 | −30.0 ± 1.3 | 9.8 ± 0.8a, 70.1 ± 4.8a | |
| n = 10 | n = 10 | n = 10 | n = 7 | ||
| Wild type | −356.2 ± 60.0 | −88.0 ± 1.8 | −36.5 ± 1.5 | 11.7 ± 0.6, 65.3 ± 5.4 | Wild type |
| n = 15 | n = 15 | n = 15 | n = 7 | ||
| p.Glu1240Gln | −420.1 ± 52.0 | −87.1 ± 0.8 | −33.2 ± 1.2 | 10.7 ± 0.9, 87.1 ± 6.4a | |
| n = 16 | n = 16 | n = 16 | n = 7 |
P < 0.05 vs. wild type.
Conversely, given the lack of a clinical/electrocardiographic phenotype consistent with SCN5A-mediated disease, we hypothesized that the p.Glu30Gly-SCN5A, p.Gln245Lys-SCN5A, p.Pro648Leu-SCN5A, and p.Glu1240Gln-SCN5A may represent innocuous background genetic noise. Not surprisingly, p.Glu30Gly-SCN5A, p.Gln245Lys-SCN5A, p.Pro648Leu-SCN5A, and p.Glu1240Gln-SCN5A resulted in minor cellular EP changes (Table 3), consistent with wild-type Nav1.5 channel function. Detailed cellular EP parameters for all SCN5A VUS detected in the present study are provided in Table 3.
Functional re-adjudication of SCN5A variants of uncertain significance detected in unexplained sudden cardiac arrest survivors
Lastly, in light of the respective cellular EP phenotypes elucidated for p.Glu30Gly-SCN5A, p.Gln245Lys-SCN5A, p.Pro648Leu-SCN5A, p.Asp872Asn-SCN5A, and p.Glu1240Gln-SCN5A, we sought to re-adjudicate the SCN5A VUS identified in unexplained SCA survivors. Application of the 2015 ACMG ‘functional data’ criteria [i.e. pathogenic strong 3 (PS3) for the presence of functional studies supportive of a damaging effect on a gene/gene product or benign strong 3 (BS3) for the presence of functional studies that show no damaging effect on a gene/gene product], resulted in the promotion of p.Asp872Asn-SCN5A to likely pathogenic (ii) status. Despite their wild-type-like functional status, p.Glu30Gly-SCN5A, p.Gln245Lys-SCN5A, p.Pro648Leu-SCN5A, and p.Glu1240Gln-SCN5A remain classified ambiguously as VUS according to a strict interpretation of the 2015 ACMG variant classification and reporting guidelines.
Discussion
SCN5A: truly an idiopathic ventricular fibrillation -susceptibility gene?
The association between SCN5A and IVF traces back to the discovery of four distinct loss-of-function SCN5A genetic variants in patients with either a history of SCA or inducible VF with programmed electrical stimulation.11,14 After examination of available phenotypic data for the IVF-associated p.Ala266Asp-SCN5A, p.Arg367His-SCN5A, p.Leu846Arg-SCN5A, and p.Ser1710Leu-SCN5A loss-of-function genetic variants reported previously (Figure 2; orange circles), it is clear that these variants were identified in patients that would not meet the current Heart Rhythm Society/European Heart Rhythm Association expert consensus definition of IVF,5 largely due to the presence of inferolateral early repolarization consistent with a diagnosis of ERS.15
Consistent with these prior studies, the only functional/likely pathogenic SCN5A rare variant (p.Asp872Asn-SCN5A) identified in the current study was observed in an unexplained SCA survivor with notched J-point elevation ≥0.1 mV with a horizontal ST segment in ≥2 inferior leads (Figure 3D) and a history of short-coupled PVC-triggered ventricular arrhythmias consistent with a diagnosis of ERS (Shanghai ERS score of 6.5 points). At first glance, the discovery of a fifth SCN5A loss-of-function variant in a patient with a clinical picture consistent with ERS may seem to strengthen the gene-disease association between SCN5A and ERS. However, it is important to note that (i) the prevalence of early repolarization in the general population is relatively high, (ii) no studies, including the present one, have demonstrated the co-segregation of an SCN5A variant with an ERS/ERP phenotype, and (iii) the SCN5A-positive ERS cases described to date appear to also have concomitant cardiac conduction defects (PR prolongation, intraventricular conduction delay, etc.). Although the SCN5A loss-of-function variants detected in these ERS cases may well be contributory, in light of evidence regarding the polygenic basis of the closely related BrS,16 the lack of co-segregation, and variable strength of cellular EP phenotypes observed suggests additional genetic and/or environmental factors are likely involved. As such, the association between SCN5A and ERS remains weak at best.
In contrast, no evidence of underlying BrS1, LQT3, PCCD, MEPPC, or a mixed/complex sodium cardiac channelopathy was observed in the four unexplained SCA survivors with functionally wild-type SCN5A rare variants. However, only 2/4 of these patients (both females without underlying evidence of a spontaneous Type I and non-Type I Brugada ECG pattern; Table 2) underwent sodium channel blocker (SCB) provocation and the remaining patients underwent procainamide provocation due to the unavailability of ajmaline in the USA. Although two presumably pathogenic SCN5A truncating variants (p.Phe851fs*19-SCN5A and p.Arg1958*-SCN5A) were identified in unexplained SCA survivors with a default diagnosis of IVF in the Canadian Cardiac Arrest Survivors with Preserved Ejection Fraction Registry (CASPER),10 no additional truncating or functional missense SCN5A variants have been reported, to date, in unexplained SCA survivors.
Therefore, there is currently insufficient clinical and molecular evidence to suggest the presence of a distinct SCN5A-mediated IVF subtype. Although further investigation is needed, careful clinical phenotyping of all SCN5A-positive unexplained SCA survivors, including the judicious use of SCB provocation and diagnostic electrophysiology study, may prove helpful in identifying those unexplained SCA survivors with subtle evidence of Nav1.5 dysfunction (i.e. conduction abnormalities, early repolarization, etc.) indicative of the presence of an underlying cardiac sodium channelopathy.
Background genetic noise: the Achilles heel of genetic testing in unexplained sudden cardiac arrest/sudden cardiac death
Loss- or gain-of-function SCN5A variants underlie 20–30% of BrS and 5–10% of LQTS, respectively. However, interpretation of any given SCN5A rare variant, even when identified in an individual with a clinically definite BrS or LQTS phenotype, is complicated substantially by the 2–4% background rate of rare, and presumably innocuous, SCN5A genetic variation in the general population. While as many as 1 in 10 (10%) SCN5A variants identified in probable/definitive BrS cases and one in five (20%) SCN5A variants identified in probable/definitive LQTS cases may represent ‘false positives’, the majority of SCN5A variants encountered in individuals with more ambiguous clinical phenotypes, including unexplained SCA/IVF, are likely to represent background genetic noise.
Not surprisingly, once the functional p.Asp872Asn-SCN5A observed in an unexplained SCA survivor with evidence of underlying cardiac conduction disease and inferior early repolarization was removed, the rate of functionally wild type, ultra-rare SCN5A variants (4/59; 6.7%) observed in unexplained SCA survivors in this study approaches the inherent background rate of rare SCN5A genetic variation observed in the general population (2–5%). In this context, it comes as little surprise that the remaining four SCN5A VUS (p.Glu30Gly-SCN5A, p.Gln245Lys-SCN5A, p.Pro648Leu-SCN5A, and p.Glu1240Gln-SCN5A) were found to be functionally wild-type-like, at least in vitro, and identified in unexplained SCA survivors without evidence of an underlying cardiac sodium channelopathy.
Nevertheless, 2/4 (50%; p.Pro648Leu-SCN5A and p.Glu1240Gln-SCN5A) of these functionally wild-type VUS were reported erroneously to the ordering provider as likely ‘disease-causative’ on the basis of their prior identification in BrS and/or LQTS case-derived variant compendia subject to the aforementioned rate of background genetic noise.17 Unfortunately, these potential ‘false positive’ commercial genetic test results led to the misdiagnosis of the p.Pro648Leu-SCN5A- and p.Glu1240Gln-SCN5A-positive unexplained SCA survivors as LQT3 and BrS1, respectively.
As such, this study highlights the primary danger associated with the pursuit of clinical genetic testing in individuals with unexplained SCA/IVF. Multiple prior studies have shown that the yield of ACMG-classified likely pathogenic/pathogenic variants (range 2–12%) is far exceeded by the yield of ACMG-classified VUS (range 15–28%) in the SCD-predisposing cardiac channelopathy and cardiomyopathy-susceptibility genes.3,9,10 Unfortunately, for health care professionals struggling to determine the root cause of an individual’s SCA, it is often difficult to look past a VUS in a Clinical Genome Resource-proclaimed ‘definitive’ evidence gene such as SCN5A, let alone a variant labelled as ‘disease-causative’, ‘likely pathogenic/pathogenic’, or ‘clinically actionable’ on a commercial genetic testing report. This has led to the pragmatic recommendation that genetic testing in unexplained SCA/IVF survivors is best pursued in the context of dedicated cardiovascular genetics clinics with the resources and expertise needed to interpret, reappraise, and act judiciously on the genetic testing results of unexplained SCA with a default diagnosis of IVF.3,10
Lastly, regardless of when or where genetic testing in unexplained SCA/IVF is pursued; this study serves as an important reminder of the vital role that clinical phenotype should play in variant adjudication. Unlike this study, the vast majority of health care professionals will not have the luxury of characterizing functionally the next VUS in SCN5A or other SCD-predisposing cardiac channelopathy-susceptibility gene. Furthermore, phenotype-enhanced variant classification approaches such as that recently described for CPVT18 will likely have little to no utility in unexplained SCA survivors.
As such, this study highlights the importance of scrutinizing a patient’s clinical phenotype, with an eye towards identifying evidence of a subclinical cardiac channelopathy, when a genetic test returns with an ambiguous, but potentially impactful, finding such as a SCN5A VUS. If no evidence of an underlying cardiac channelopathy emerges, it is critical to remember, particularly in light of the substantial background genetic noise observed in genes such as SCN5A, that phenotype trumps genotype in the process of rendering a clinical diagnosis. As such, in the absence of a phenotype suspicious for an established sodium cardiac channelopathy, even in individuals who have experienced a SCA, the identification of an ultra-rare SCN5A variant should never be the sole basis for a clinical diagnosis of BrS1, LQT3, or any other cardiac sodium channelopathy.
Limitations
Although this study demonstrates clearly the potential pitfalls of prioritizing genotype over phenotype in the assessment of unexplained SCA survivors, it is not without limitation. First and foremost, only 60% of SCN5A variant-positive unexplained SCA survivors in this study underwent SCB provocation and those that did were challenged with procainamide. Although ajmaline is not currently available in the United States, given recent data that suggests ajmaline is far superior to procainamide in provoking a Type I Brugada ECG pattern19, we cannot rule out the possibility, albeit remote, that any of these patients have concealed BrS. Secondly, it is widely known that heterologous expression systems fail to recapitulate the native cardiomyocyte environment. For SCN5A in particular, there are clear examples of variants (i.e. p.Asp1275Asn-SCN5A) where the EP phenotype observed in vitro differs markedly from that observed in vivo.20 Therefore, without the use of patient-specific induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) and/or a murine model capable of more completely recapitulating the native cardiomyocyte environment, we cannot rule out definitively a more pronounced effect of the identified SCN5A variants in vivo. Nevertheless, the absence of a co-segregating phenotype amongst variant-positive relatives and higher than anticipated minor allele frequencies in the Genome Aggregation Database argue strongly against p.Pro648Leu-SCN5A and p.Glu1240Gln-SCN5A being disease-causative. However, further study of p.Glu30Gly-SCN5A, p.Gln245Lys-SCN5A, and p.Asp872Asn-SCN5A in patient-derived iPSC-CMs may help to assess further their contribution, or lack thereof, to disease.
Conclusion
In the absence of a clinical phenotype (i.e. cardiac conduction defects, J-wave abnormalities, QTc prolongation, etc.) potentially attributable to dysfunction of the SCN5A-encoded Nav1.5 cardiac sodium channel, all ultra-rare SCN5A genetic variants identified in unexplained SCA survivors with a default diagnosis of IVF had a wild-type-like in vitro cellular EP phenotype. As the background rate of rare and likely innocuous genetic variants in SCN5A is not trivial, great care must be taken to avoid potentially harmful diagnostic miscues associated with the erroneous prioritization of genotype over phenotype when attempting to ascertain the root cause of an individual’s SCA.
Conflict of interest: M.J.A. is a consultant for Audentes Therapeutics, Boston Scientific, Gilead Sciences, Invitae, Medtronic, MyoKardia, St. Jude Medical, Biotronik and Daiichi Sankyo. M.J.A. and Mayo Clinic are involved in an equity/royalty relationship with AliveCor, Blue Ox Health Corporation, and Stemonix. However, none of these entities were involved in this study. The other authors have no conflict of interest to declare.
Funding
This work was supported by the Mayo Clinic Windland Smith Rice Comprehensive Sudden Cardiac Death Program.

{kind=link}
{kind=link}
{kind=link}
{kind=link}