





Abstract
Characterization of the cardiac phenotype associated with the novel LMNA nonsense mutation c.544C>T, p.Q182*, which we have identified in a large five-generation family.
A family tree was constructed. Clinical data [arrhythmia, syncope, sudden cardiac death (SCD), New York Heart Association (NYHA) class] were collected from living and deceased family members. DNA of 23 living family members was analysed for mutations in LMNA. Additionally, dilated cardiomyopathy multi-gene-panel testing and whole exome sequencing were performed in some family members to identify potential phenotype-modifiers. In this five-generation family (n = 65), 17 SCDs occurred at 49.3 ± 10.0 years. Furthermore, we identified eight additional mutation-carriers, seven symptomatic (44 ± 13 years), and one asymptomatic (44 years). First signs of disease [sinus bradycardia with atrioventricular (AV)-block I°] occurred at 36.5 ± 8.1 years. Paroxysmal atrial fibrillation (AF) (onset at 41.8 ± 5.7 years) rapidly progressed to permanent AF (46.2 ± 9.8 years). Subsequently, AV-conduction worsened, syncope, pacemaker-dependence, and non-sustained ventricular tachycardia (43.3 ± 8.2 years) followed. Ventricular arrhythmia caused SCD in patients without implantable cardioverter-defibrillator (ICD). Patients protected by ICD developed rapidly progressive heart failure (45.2 ± 10.6 years). A different phenotype was seen in a sub-family in three patients with early onset of rapidly decompensating heart failure and only minor prior arrhythmia-related symptoms. One patient received high-urgency heart transplantation (HTX) at 32 years, while two died prior to HTX. One of them developed lethal peripartum-associated heart failure. Possible disease-modifiers were identified in this ‘heart failure sub-family’.
The novel LMNA nonsense mutation c.544C>T causes a severe arrhythmogenic phenotype manifesting with high incidence of SCD in most patients; and in one sub-family, a distinct phenotype with fast progressing heart failure, indicating the need for early consideration of ICD-implantation and listing for heart-transplantation.
The novel LMNA nonsense mutation c.544C>T, p.Q182* in exon 3 is identified and characterized.
Two distinct, highly malignant disease phenotypes (‘arrhythmogenic’ vs. ‘heart-failure’) are observed in the affected family.
PSMB10 and FDX1L variants are identified as potential disease-modifiers.
For the first time, a LMNA mutation is linked to a pregnancy-induced severe heart-failure.
A comprehensive comparison of the phenotype of all known LMNA nonsense mutations and all LMNA missense mutations in exon 3 is provided.
Introduction
Inherited, familial forms of dilated cardiomyopathy (DCM) account for 50% of DCM cases.1 More than 40 genes have been linked to DCM, causing defects in various cellular compartments and pathways such as the nuclear envelope, the contractile apparatus, the Z-disk, and calcium handling.2
Mutations in the LMNA gene, encoding the Lamin A/C protein, are among the most common causes of familial DCM.3 These laminopathies can lead to a variety of clinical symptoms such as heart failure, conduction disorders, atrial fibrillation (AF), ventricular tachycardia and fibrillation (VT/VF), sudden cardiac death (SCD), and (skeletal) muscle dystrophies.4 Lamin A/C is an integral part of the nuclear lamina, which has a key function in the stability of the nuclear envelope, chromatin organization, and DNA-repair.5 Additionally, lamins play a major role in transmitting mechanical signals to the nuclear core, transport through the nuclear envelope, and regulation of transcription.6
In 1999, Fatkin et al.7 analysed 85 members of five different families with familial DCM associated with conduction defect (DCM-CD) and described a ‘classical’ time course of the disease with early rhythm abnormalities later followed by heart failure. Thereafter, a pronounced variability in age at onset, disease severity, and clinical (arrhythmia vs. heart failure) phenotype was identified in DCM-CD caused by different LMNA mutations, partly linked to different types of mutations such as missense or nonsense mutations.8,9
We, here, describe a new LMNA nonsense mutation in a large family associated with a highly arrhythmogenic phenotype and compare its clinical phenotype to other known laminopathies.
Methods
The study was approved by the Institutional Ethical Committee on Human Research at the authors’ institution. All patients gave written informed consent for participation in this study, blood sampling, isolation of DNA, molecular genetic testing, and provision of their clinical data.
Clinical data
The family tree was created based on information provided by family members and parish records. As family members are distributed throughout Europe (Slovenia, Germany, Austria, Switzerland, Spain, and Great Britain), blood samples were collected by the authors or by patient’s general practitioners. Clinical data from the index patient and other living mutation-carriers (n = 8) were collected using a standardized questionnaire (inquiring symptoms, hospitalization, contact with physicians, change in medication, interventions) that was sent to patients every 6 months for 4 years. In addition, medical reports and examination results [electrocardiogram (ECG), Holter-ECG, pacemaker/implantable cardioverter-defibrillator (ICD) interrogations, echocardiography] of both living and deceased family members were collected.
Genetic analyses
Blood samples of 23 living family members were obtained and analysed. The genomic DNA of exon 3 of LMNA of the 23 patients was amplified by polymerase chain reaction (PCR) using forward primer forward primer (5′…TGTTCTGTGACCCCTTTTCC…3′) and reverse primer (3′…TAACCTGGGAGCTGAGTGCT…5′) designed with the software Primer3Plus.10 Since the LMNA mutation c.544C>T creates a restriction site for MaeIII, the LMNA exon 3 PCR products were digested with this restriction enzyme. All positive samples were sequenced to verify the existence of the mutation.
To further identify modifying (genetic) variants that might contribute to phenotype differences, DCM multi-gene-panel analysis was carried out for patients IV:7 and V:16 (see Supplementary material online, Table S1), and whole exome sequencing (WES) was performed for two patients and one healthy relative from the ‘arrhythmia’ sub-family (IV:7, IV:8, V:10) and for one patient and two healthy relatives from the ‘heart failure’ sub-family (V:16, V:17, V:18) (see Supplementary material online, Method for details; Table 1).
Results of whole exome sequencing
| Gene | Chrom. | Mutation (DNA) // (protein) | Exon | Genotype | Impact of mutation | ExAC-MAF/ GnomAD | Gene Function | Associated phenotypes | References | |
|---|---|---|---|---|---|---|---|---|---|---|
| MITD1 | Microtubule interacting and trafficking domain containing 1 | chr2 | c.511C>T // Q/* | Exon 5 | Heterozygous | Stop codon | NA/NA | Regulation of abscission, final step of cytokinesis | – | 11 |
| CASD1 | CAS1 domain containing 1 | chr7 | c.693_696dup // A/AY* | Exon 8 | Heterozygous | Frameshift variant | NA/NA | 9-O-acetylation of sialic acids | – | 12 |
| IRF5 | Interferon regulatory factor 5 | chr7 | c.579T>C | Heterozygous | Splice acceptor variant | 2.77e−5/1.76e−5 | Modulation of macrophages and neutrophil trafficking | Inflammatory bowel disease, inflammation | 13 | |
| IGSF9B | Immunoglobulin superfamily, member 9B | chr11 | c.3232C>T // R/* | Exon 18 | Heterozygous | Stop codon | NA/4.59e−6 | Inhibitory synaptic adhesion molecule | Disorders of the nervous system (schizophrenia, …) | 14,15 |
| PDIA2 | Protein disulfide isomerase family A member 2 | chr16 | c.1391_1392del // T/* | Exon 9 | Heterozygous | Frameshift variant | 6.45e−3/7.02e−3 | Protein folding catalyst and a molecular chaperone | Bicuspid aortic valve | 16,17 |
| TBC1D24 | TBC1 domain family, member 24 | chr16 | c.1008del // H/* | Exon 4 | Heterozygous | Frameshift variant | 6.61e−5/7.90e−5 | Regulator of neuronal differentiation | Disorders of the nervous system (epilepsy, deafness, …) | 18,19 |
| PSMB10 | Proteasome subunit, beta type, 10 | chr16 | c.56 + 1G>A | Exon 1 | Heterozygous | Splice donor variant | 3.62e−4/6.37e−4 | ubiquitin–proteasome system, e.g. regulation of cardiac fibrosis, inflammation | Neurological diseases, Cardiac diseases, … | 20,21 |
| MKS1 | Meckel syndrome, type 1 | chr17 | c.417 + 1G>A | Exon 4 | Heterozygous | splice donor variant | NA/4.06e−6 | Ciliary formation, epithelial morphogenesis | Meckel syndrome, ciliopathies, AV septum defects | 22,23 |
| ABCA8 | ATP-binding cassette, subfamily A (ABC1), member 8 | chr17 | c.2339_2340dup // -/* | Exon 17 | Heterozygous | Frameshift variant | 5.85e−4/5.81e−4 | Facilitator of cholesterol efflux, modulator of HDLc | low HDL cholesterol levels | 24 |
| FDX1L | Ferredoxin 1-like | chr19 | c.45C>T // Q/* | Exon 1 | Heterozygous | Stop codon | NA/NA | Heme A and Fe/S protein biosynthesis | Mitochondrial muscle myopathy | 25,26 |
| GNRH2 | Gonadotropin-releasing hormone 2 | chr20 | c.340_344del // EPR/E* | Exon 4 | Heterozygous | Frameshift variant | 6.15e−3/6.20e−3 | Stimulator of secretion of gonadotropins | 27 | |
| DCAF15 | DDB1 and CUL4 associated factor 15 | chr19 | c.1440 + 14_1440 + 33dup // E/EVGPGRA* | Exon 9 | Homozygous | Frameshift variant | NA/NA | Ubiquitination and degradation | – | 28 |
| SIGLEC12 | Sialic acid binding Ig-like lectin 12 (gene) | chr19 | c.193C>A; c.192_ 193insA // -/* | Exon 1 | Homozygous | Frameshift variant | NA/NA | CD33 signalling in immune cells | Prostate cancer | 29 |
| Gene | Chrom. | Mutation (DNA) // (protein) | Exon | Genotype | Impact of mutation | ExAC-MAF/ GnomAD | Gene Function | Associated phenotypes | References | |
|---|---|---|---|---|---|---|---|---|---|---|
| MITD1 | Microtubule interacting and trafficking domain containing 1 | chr2 | c.511C>T // Q/* | Exon 5 | Heterozygous | Stop codon | NA/NA | Regulation of abscission, final step of cytokinesis | – | 11 |
| CASD1 | CAS1 domain containing 1 | chr7 | c.693_696dup // A/AY* | Exon 8 | Heterozygous | Frameshift variant | NA/NA | 9-O-acetylation of sialic acids | – | 12 |
| IRF5 | Interferon regulatory factor 5 | chr7 | c.579T>C | Heterozygous | Splice acceptor variant | 2.77e−5/1.76e−5 | Modulation of macrophages and neutrophil trafficking | Inflammatory bowel disease, inflammation | 13 | |
| IGSF9B | Immunoglobulin superfamily, member 9B | chr11 | c.3232C>T // R/* | Exon 18 | Heterozygous | Stop codon | NA/4.59e−6 | Inhibitory synaptic adhesion molecule | Disorders of the nervous system (schizophrenia, …) | 14,15 |
| PDIA2 | Protein disulfide isomerase family A member 2 | chr16 | c.1391_1392del // T/* | Exon 9 | Heterozygous | Frameshift variant | 6.45e−3/7.02e−3 | Protein folding catalyst and a molecular chaperone | Bicuspid aortic valve | 16,17 |
| TBC1D24 | TBC1 domain family, member 24 | chr16 | c.1008del // H/* | Exon 4 | Heterozygous | Frameshift variant | 6.61e−5/7.90e−5 | Regulator of neuronal differentiation | Disorders of the nervous system (epilepsy, deafness, …) | 18,19 |
| PSMB10 | Proteasome subunit, beta type, 10 | chr16 | c.56 + 1G>A | Exon 1 | Heterozygous | Splice donor variant | 3.62e−4/6.37e−4 | ubiquitin–proteasome system, e.g. regulation of cardiac fibrosis, inflammation | Neurological diseases, Cardiac diseases, … | 20,21 |
| MKS1 | Meckel syndrome, type 1 | chr17 | c.417 + 1G>A | Exon 4 | Heterozygous | splice donor variant | NA/4.06e−6 | Ciliary formation, epithelial morphogenesis | Meckel syndrome, ciliopathies, AV septum defects | 22,23 |
| ABCA8 | ATP-binding cassette, subfamily A (ABC1), member 8 | chr17 | c.2339_2340dup // -/* | Exon 17 | Heterozygous | Frameshift variant | 5.85e−4/5.81e−4 | Facilitator of cholesterol efflux, modulator of HDLc | low HDL cholesterol levels | 24 |
| FDX1L | Ferredoxin 1-like | chr19 | c.45C>T // Q/* | Exon 1 | Heterozygous | Stop codon | NA/NA | Heme A and Fe/S protein biosynthesis | Mitochondrial muscle myopathy | 25,26 |
| GNRH2 | Gonadotropin-releasing hormone 2 | chr20 | c.340_344del // EPR/E* | Exon 4 | Heterozygous | Frameshift variant | 6.15e−3/6.20e−3 | Stimulator of secretion of gonadotropins | 27 | |
| DCAF15 | DDB1 and CUL4 associated factor 15 | chr19 | c.1440 + 14_1440 + 33dup // E/EVGPGRA* | Exon 9 | Homozygous | Frameshift variant | NA/NA | Ubiquitination and degradation | – | 28 |
| SIGLEC12 | Sialic acid binding Ig-like lectin 12 (gene) | chr19 | c.193C>A; c.192_ 193insA // -/* | Exon 1 | Homozygous | Frameshift variant | NA/NA | CD33 signalling in immune cells | Prostate cancer | 29 |
ATP, antitachycardia pacing; AV, atrioventricular; ExAC-MAF, Exome Aggregation Consortium-minor allele frequency; GnomAD, Genome Aggregation Database, indicated are the frequencies of the variants; HDL, high density lipoprotein; NA, not available, indicating that this (new) variant has not been found in the database.
Results of whole exome sequencing
| Gene | Chrom. | Mutation (DNA) // (protein) | Exon | Genotype | Impact of mutation | ExAC-MAF/ GnomAD | Gene Function | Associated phenotypes | References | |
|---|---|---|---|---|---|---|---|---|---|---|
| MITD1 | Microtubule interacting and trafficking domain containing 1 | chr2 | c.511C>T // Q/* | Exon 5 | Heterozygous | Stop codon | NA/NA | Regulation of abscission, final step of cytokinesis | – | 11 |
| CASD1 | CAS1 domain containing 1 | chr7 | c.693_696dup // A/AY* | Exon 8 | Heterozygous | Frameshift variant | NA/NA | 9-O-acetylation of sialic acids | – | 12 |
| IRF5 | Interferon regulatory factor 5 | chr7 | c.579T>C | Heterozygous | Splice acceptor variant | 2.77e−5/1.76e−5 | Modulation of macrophages and neutrophil trafficking | Inflammatory bowel disease, inflammation | 13 | |
| IGSF9B | Immunoglobulin superfamily, member 9B | chr11 | c.3232C>T // R/* | Exon 18 | Heterozygous | Stop codon | NA/4.59e−6 | Inhibitory synaptic adhesion molecule | Disorders of the nervous system (schizophrenia, …) | 14,15 |
| PDIA2 | Protein disulfide isomerase family A member 2 | chr16 | c.1391_1392del // T/* | Exon 9 | Heterozygous | Frameshift variant | 6.45e−3/7.02e−3 | Protein folding catalyst and a molecular chaperone | Bicuspid aortic valve | 16,17 |
| TBC1D24 | TBC1 domain family, member 24 | chr16 | c.1008del // H/* | Exon 4 | Heterozygous | Frameshift variant | 6.61e−5/7.90e−5 | Regulator of neuronal differentiation | Disorders of the nervous system (epilepsy, deafness, …) | 18,19 |
| PSMB10 | Proteasome subunit, beta type, 10 | chr16 | c.56 + 1G>A | Exon 1 | Heterozygous | Splice donor variant | 3.62e−4/6.37e−4 | ubiquitin–proteasome system, e.g. regulation of cardiac fibrosis, inflammation | Neurological diseases, Cardiac diseases, … | 20,21 |
| MKS1 | Meckel syndrome, type 1 | chr17 | c.417 + 1G>A | Exon 4 | Heterozygous | splice donor variant | NA/4.06e−6 | Ciliary formation, epithelial morphogenesis | Meckel syndrome, ciliopathies, AV septum defects | 22,23 |
| ABCA8 | ATP-binding cassette, subfamily A (ABC1), member 8 | chr17 | c.2339_2340dup // -/* | Exon 17 | Heterozygous | Frameshift variant | 5.85e−4/5.81e−4 | Facilitator of cholesterol efflux, modulator of HDLc | low HDL cholesterol levels | 24 |
| FDX1L | Ferredoxin 1-like | chr19 | c.45C>T // Q/* | Exon 1 | Heterozygous | Stop codon | NA/NA | Heme A and Fe/S protein biosynthesis | Mitochondrial muscle myopathy | 25,26 |
| GNRH2 | Gonadotropin-releasing hormone 2 | chr20 | c.340_344del // EPR/E* | Exon 4 | Heterozygous | Frameshift variant | 6.15e−3/6.20e−3 | Stimulator of secretion of gonadotropins | 27 | |
| DCAF15 | DDB1 and CUL4 associated factor 15 | chr19 | c.1440 + 14_1440 + 33dup // E/EVGPGRA* | Exon 9 | Homozygous | Frameshift variant | NA/NA | Ubiquitination and degradation | – | 28 |
| SIGLEC12 | Sialic acid binding Ig-like lectin 12 (gene) | chr19 | c.193C>A; c.192_ 193insA // -/* | Exon 1 | Homozygous | Frameshift variant | NA/NA | CD33 signalling in immune cells | Prostate cancer | 29 |
| Gene | Chrom. | Mutation (DNA) // (protein) | Exon | Genotype | Impact of mutation | ExAC-MAF/ GnomAD | Gene Function | Associated phenotypes | References | |
|---|---|---|---|---|---|---|---|---|---|---|
| MITD1 | Microtubule interacting and trafficking domain containing 1 | chr2 | c.511C>T // Q/* | Exon 5 | Heterozygous | Stop codon | NA/NA | Regulation of abscission, final step of cytokinesis | – | 11 |
| CASD1 | CAS1 domain containing 1 | chr7 | c.693_696dup // A/AY* | Exon 8 | Heterozygous | Frameshift variant | NA/NA | 9-O-acetylation of sialic acids | – | 12 |
| IRF5 | Interferon regulatory factor 5 | chr7 | c.579T>C | Heterozygous | Splice acceptor variant | 2.77e−5/1.76e−5 | Modulation of macrophages and neutrophil trafficking | Inflammatory bowel disease, inflammation | 13 | |
| IGSF9B | Immunoglobulin superfamily, member 9B | chr11 | c.3232C>T // R/* | Exon 18 | Heterozygous | Stop codon | NA/4.59e−6 | Inhibitory synaptic adhesion molecule | Disorders of the nervous system (schizophrenia, …) | 14,15 |
| PDIA2 | Protein disulfide isomerase family A member 2 | chr16 | c.1391_1392del // T/* | Exon 9 | Heterozygous | Frameshift variant | 6.45e−3/7.02e−3 | Protein folding catalyst and a molecular chaperone | Bicuspid aortic valve | 16,17 |
| TBC1D24 | TBC1 domain family, member 24 | chr16 | c.1008del // H/* | Exon 4 | Heterozygous | Frameshift variant | 6.61e−5/7.90e−5 | Regulator of neuronal differentiation | Disorders of the nervous system (epilepsy, deafness, …) | 18,19 |
| PSMB10 | Proteasome subunit, beta type, 10 | chr16 | c.56 + 1G>A | Exon 1 | Heterozygous | Splice donor variant | 3.62e−4/6.37e−4 | ubiquitin–proteasome system, e.g. regulation of cardiac fibrosis, inflammation | Neurological diseases, Cardiac diseases, … | 20,21 |
| MKS1 | Meckel syndrome, type 1 | chr17 | c.417 + 1G>A | Exon 4 | Heterozygous | splice donor variant | NA/4.06e−6 | Ciliary formation, epithelial morphogenesis | Meckel syndrome, ciliopathies, AV septum defects | 22,23 |
| ABCA8 | ATP-binding cassette, subfamily A (ABC1), member 8 | chr17 | c.2339_2340dup // -/* | Exon 17 | Heterozygous | Frameshift variant | 5.85e−4/5.81e−4 | Facilitator of cholesterol efflux, modulator of HDLc | low HDL cholesterol levels | 24 |
| FDX1L | Ferredoxin 1-like | chr19 | c.45C>T // Q/* | Exon 1 | Heterozygous | Stop codon | NA/NA | Heme A and Fe/S protein biosynthesis | Mitochondrial muscle myopathy | 25,26 |
| GNRH2 | Gonadotropin-releasing hormone 2 | chr20 | c.340_344del // EPR/E* | Exon 4 | Heterozygous | Frameshift variant | 6.15e−3/6.20e−3 | Stimulator of secretion of gonadotropins | 27 | |
| DCAF15 | DDB1 and CUL4 associated factor 15 | chr19 | c.1440 + 14_1440 + 33dup // E/EVGPGRA* | Exon 9 | Homozygous | Frameshift variant | NA/NA | Ubiquitination and degradation | – | 28 |
| SIGLEC12 | Sialic acid binding Ig-like lectin 12 (gene) | chr19 | c.193C>A; c.192_ 193insA // -/* | Exon 1 | Homozygous | Frameshift variant | NA/NA | CD33 signalling in immune cells | Prostate cancer | 29 |
ATP, antitachycardia pacing; AV, atrioventricular; ExAC-MAF, Exome Aggregation Consortium-minor allele frequency; GnomAD, Genome Aggregation Database, indicated are the frequencies of the variants; HDL, high density lipoprotein; NA, not available, indicating that this (new) variant has not been found in the database.
Statistical analyses
Analyses of survival and cumulative incidences of arrhythmia were performed with the Kaplan–Meier method; survival proportions were compared with the log-rank test. All analyses were performed with Prism 5 for Windows (Graph-Pad), and data is presented as means ± SD; n indicates the number of patients. A P-value of <0.05 was considered significant.
Results
Clinical presentation of index patient (IV:7)
The index patient first consulted a cardiologist at 48 years of age, because of limited exercise capacity and known muscular dystrophy. Her ECG showed sinus bradycardia and atrioventricular (AV)-block I° without any other abnormality (Figure 1A and B). An exercise ECG test was normal. A 24 h Holter-ECG revealed nocturnal bradycardia (34 b.p.m.) and frequent supraventricular and polymorphic ventricular extrasystoles. Echocardiography showed normal systolic and diastolic left ventricular function and normal dimensions. The patient’s family history was noteworthy for a high incidence of sudden death at early age and cases of muscular dystrophy, which made a genetic cause likely. The patient was, therefore, screened for a LMNA mutation. A novel mutation c.544C>T in exon 3 of the LMNA gene (mutation absent in databases ExAC/gnomAD) was found, which leads to the substitution of glutamine with a premature stop at codon 182 (p.Q182*), confirming the suspected diagnosis ‘laminopathy’. Dilated cardiomyopathy multi-gene-panel diagnostic excluded additional mutations in other DCM-related genes.
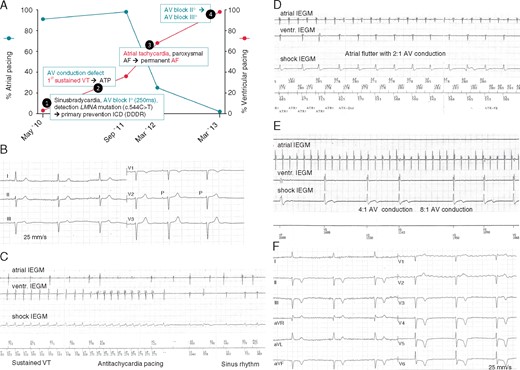
Clinical presentation of index patient. (A) Sequence of atrial and ventricular arrhythmia development in the index patient as exemplary time course in this LMNA family. Pacemaker pacing rates in the atrium (%, blue line, y-axis on the left) and in the ventricle (red line, y-axis on the right) are given over 3 years. Initially, sinus bradycardia with mainly atrial stimulation (91%) is observed, but no need for ventricular pacing (3%). Over time, AF burden increases and AV conduction worsens, leading to a decrease in atrial pacing (25%) and an increase in ventricular pacing (68%). Only 3 years after initial presentation, the patient is in permanent AF and complete AV-block (atrial pacing: 0%, ventricular pacing: 98%). (B) Initial ECG in 2010 showed sinus bradycardia (45 b.p.m.) and AV-block I° (PQ 250 ms) (indicated in time course as (1)). (C) IEGM obtained from ICD memory. A sustained VT (185 b.p.m.) is terminated by ATP 6 months after ICD implantation (indicated in time course as (2)). (D) Atrial flutter (atrial rate approx. 215–220 b.p.m.) with 2:1 AV conduction (indicated in time course as (3)). (E) Atrial flutter with 4:1 and 8:1 AV conduction. (F) Spontaneous ECG with pacemaker inhibition during regular pacemaker-check 5 years after initial diagnosis. The patient is in permanent AF and complete AV-block with junctional AV-escape rhythm (36 b.p.m.) (indicated in time course as (4)). Negative precordial T-waves are caused by permanent ventricular pacing (cardiac memory). AF, atrial fibrillation; ATP, antitachycardia pacing; AV, atrioventricular; DDDR, dual-chamber rate adaptive pacemaker; ECG, electrocardiogram; ICD, implantable cardioverter-defibrillator; IEGM, intra-cardiac electrocardiogram; VT, ventricular tachycardia.
A dual-chamber ICD (DDDR, Boston Scientific) was implanted for primary prevention. Only 6 months later, the patient experienced a sustained VT (185 b.p.m.), which was successfully terminated by anti-tachycardia pacing (ATP) (Figure 1C). Concurrently, paroxysmal AF rapidly progressed to permanent AF, and AV-block I° worsened to complete AV-block, both reflected by a decrease in atrial pacing and an increase in ventricular pacing (Figure 1A–F). Six years after diagnosis, the patient developed heart failure with exertional dyspnoea [New York Heart Association (NYHA) II–III]. Echocardiography demonstrated a slightly dilated left ventricle (left ventricular end diastolic diameter 57 mm) and a borderline systolic ejection fraction (EF) of 50%. An increase of NT-proBNP to 2760 pg/mL (reference value: <120 pg/mL) was noticed. After heart failure therapy had been intensified by adding a loop diuretic and increasing the dosage of angiotensin-converting enzyme-inhibitor, the patient improved clinically and NT-proBNP levels decreased. During the 6 years after ICD implantation, the patient received one ATP to terminate a sustained VT, but no ICD-shock.
Genetic and clinical characteristics of family members with LMNA c.544C>T mutation
The constructed family tree included 67 family members over five generations. In total, 17 already-deceased and presumed mutation-carriers, and 8 genetically verified mutation-carriers (out of 23 living tested family members) were identified (Figure 2). Of note, similar to other characterizations of large multi-generation LMNA-cardiomyopathy families,30 blood, or other genetic materials were only available for family members of the younger generations; particularly as the SCD in the 1st and 2nd generations date back to 1920 and the 1950’s, respectively.
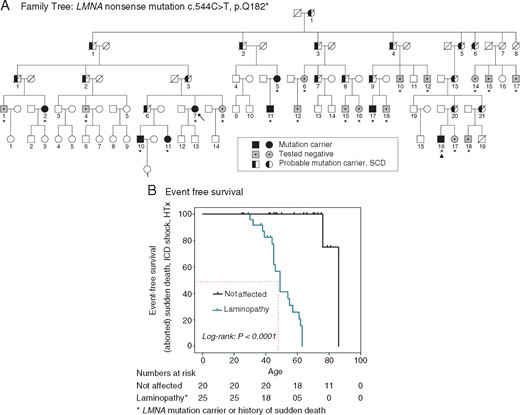
Family tree. DNA of 23 family members was analysed (indicated with an asterisk (*)): 8 mutation-carriers (35%; filled symbols) and 15 healthy individuals (65%; striated symbols with a dot) were identified. Furthermore, we identified 17 (already deceased) putative carriers (half-filled symbols). The index patient is marked with an arrow (→), the recipient of heart transplantation is marked with a triangle (▲). In four generations (n = 65), 17 SCDs occurred. SCD, sudden cardiac death.
In gel electrophoresis of LMNA exon 3 cDNA performed after the digestion, one band was visible in non-carrier controls, while mutation-carriers showed two bands (Figure 3A). Sequencing confirmed identified mutation-carriers with C to T transition at position 544 in exon 3 (Figure 3B).
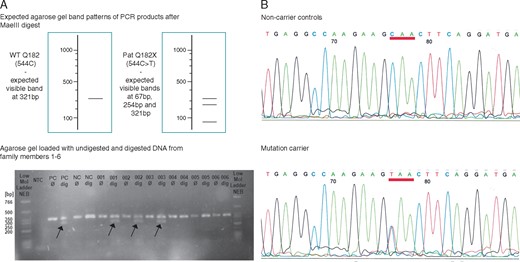
Genetic analysis in LMNA exon 3 (A) The upper image shows the expected agarose gel band patterns after digestion of PCR products with restriction enzyme MaeIII. The lower image is a representative agarose gel with non-digested (Ø) and digested (dig) samples from patients and non-carrier controls. After digestion, one band was seen in non-carriers, while two bands were seen in mutation-carriers due to the small size of the 3rd band. (B) Sequencing results of the PCR product in non-carrier control (upper panel) and mutation-carrier (lower panel). Different bases are indicated by letters and colour-coded. NC, negative control; NTC, no template control; PC, positive control; PCR, polymerase chain reaction; WT, wild type.
In four generations, 17 SCDs occurred (thus far no lethal arrhythmia has been documented in the 5th generation). Furthermore, one patient was successfully defibrillated and resuscitated (aged 63 years), and another patient (aged 49 years) received adequate ICD-therapies for VT. Mean age at time of SCD was 49.3 ± 10.0 years (Figure 4A). No significant sex difference in survival of mutation-carriers was observed (Figure 4B).
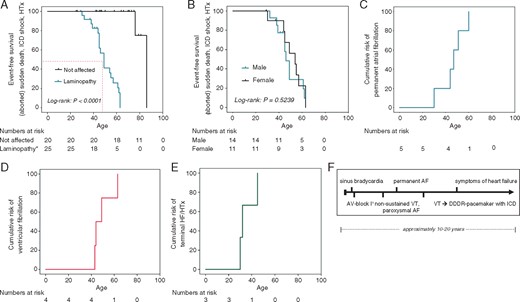
Clinical phenotype: Kaplan–Meier survival, cumulative risk for arrhythmia, and HF. (A) The Kaplan–Meier survival analysis demonstrates a significantly shorter life expectancy in LMNA mutation-carriers with early death due to SCD (VT/VF) and HF as compared to non-carrier family members. Average age of death in LMNA mutation-carriers is 49.3±10.0 years. (B) The Kaplan–Meier survival curves of male and female LMNA mutation-carriers indicate no significant sex differences in event-free survival. (C) Cumulative risk plot of patients to develop permanent AF: permanent AF occurred at a median age of 46 ± 10 years. (D) Cumulative risk plot of patients to develop ventricular fibrillation: ventricular fibrillation occurred at a median age of 43 ± 8 years. (E) Cumulative risk plot of patients in the ‘heart failure’ subfamily to develop decompensated HF and/or need for heart transplantations. Two patients died prior to HTX (one pregnancy-related cardiac decompensation); one patient received high-urgency HTX. (F) Typical clinical course of arrhythmia and HF development. *LMNA mutation carrier or history of sudden death. AF, atrial fibrillation; AV, atrioventricular; DDDR, dual-chamber rate adaptive pacemaker; HF, heart failure; HTX, heart transplantation; ICD, implantable cardioverter-defibrillator; SCD, sudden cardiac death; VT, ventricular tachycardia.
While five of eight living mutation-carriers were asymptomatic at initial screening, 4 years later 7/8 demonstrated signs of the disease. This is consistent with observations by Hasselberg et al.,31 who found a 61% cardiac penetrance over 4.4 years of follow-up in asymptomatic LMNA mutation-carriers.
Two distinct clinical phenotypes were identified within this family: ‘arrhythmogenic’ and ‘heart failure’.
Arrhythmogenic phenotype
The majority of patients presented an ‘arrhythmogenic’ subtype that closely resembles the previously described ‘classical’ phenotype: All symptomatic patients first developed arrhythmia-related symptoms such as palpitations (due to supraventricular and ventricular extrasystoles) at 35.5 ± 9.4 years (Table 2). Most patients initially presented sinus bradycardia and AV-block I° (36.5 ± 8.1 years). AV conduction disorder progressed to AV-block II° (42.6 ± 6.1 years) and AV-block III° (46.2 ± 9.8 years), leading to pacemaker dependence (Table 2). Most patients developed paroxysmal AF (41.8 ± 5.7 years), which progressed to permanent AF (46.2 ± 9.8 years) (Table 2, Figure 4C and D). Subsequently, non-sustained VT (43.3 ± 8.2 years) and syncope followed, eventually progressing to sustained VT, VF and SCD. Only patients protected from SCD by an ICD developed a ‘DCM phenotype’ with severe, rapidly-progressive heart failure (45.2 ± 10.6 years) (Figure 4E and F). Furthermore, mild skeletal muscular dystrophy and subjective ‘muscular weakness’ were reported in various patients and confirmed in two cases by neurological examinations and muscle biopsies.
Clinical symptoms
| Clinical symptoms (initial cause of presentation) | n |
| Palpitations | 8/9 (89%) |
| Dyspnoea | 5/9 (56%) |
| Syncope | 1/9 (11%) |
| Arrhythmia findings | Age (initial diagnosis) |
| AV conduction defect | |
| AV-block I° (n = 4) | 37 ± 8 years |
| AV-block II° (n = 3) | 43 ± 6 years |
| AV-block III° (n = 2) | 45 ± 10 years |
| AF | |
| Paroxysmal AF (n = 4) | 42 ± 6 years |
| Permanent AF (n = 5) | 46 ± 10 years |
| Non-sustained VT on Holter-ECG (n = 4) | 43 ± 8 years |
| Pacemaker (n = 2) | 48 ± 12 years |
| ICD (n = 5) | 48 ± 9 years |
| Time to 1st appropriate ICD therapy (n = 3) | 2 ± 2 years |
| Time to death despite ICD (n = 2) | 3 ± 2 years |
| Death from heart failure/HTX (n = 3, 1 subfamily) | 36 ± 7 years |
| Clinical symptoms (initial cause of presentation) | n |
| Palpitations | 8/9 (89%) |
| Dyspnoea | 5/9 (56%) |
| Syncope | 1/9 (11%) |
| Arrhythmia findings | Age (initial diagnosis) |
| AV conduction defect | |
| AV-block I° (n = 4) | 37 ± 8 years |
| AV-block II° (n = 3) | 43 ± 6 years |
| AV-block III° (n = 2) | 45 ± 10 years |
| AF | |
| Paroxysmal AF (n = 4) | 42 ± 6 years |
| Permanent AF (n = 5) | 46 ± 10 years |
| Non-sustained VT on Holter-ECG (n = 4) | 43 ± 8 years |
| Pacemaker (n = 2) | 48 ± 12 years |
| ICD (n = 5) | 48 ± 9 years |
| Time to 1st appropriate ICD therapy (n = 3) | 2 ± 2 years |
| Time to death despite ICD (n = 2) | 3 ± 2 years |
| Death from heart failure/HTX (n = 3, 1 subfamily) | 36 ± 7 years |
AF, atrial fibrillation; AV, atrioventricular; ECG, electrocardiogram; HTX, heart transplantation; ICD, implantable cardioverter-defibrillator; VT, ventricular tachycardia.
Clinical symptoms
| Clinical symptoms (initial cause of presentation) | n |
| Palpitations | 8/9 (89%) |
| Dyspnoea | 5/9 (56%) |
| Syncope | 1/9 (11%) |
| Arrhythmia findings | Age (initial diagnosis) |
| AV conduction defect | |
| AV-block I° (n = 4) | 37 ± 8 years |
| AV-block II° (n = 3) | 43 ± 6 years |
| AV-block III° (n = 2) | 45 ± 10 years |
| AF | |
| Paroxysmal AF (n = 4) | 42 ± 6 years |
| Permanent AF (n = 5) | 46 ± 10 years |
| Non-sustained VT on Holter-ECG (n = 4) | 43 ± 8 years |
| Pacemaker (n = 2) | 48 ± 12 years |
| ICD (n = 5) | 48 ± 9 years |
| Time to 1st appropriate ICD therapy (n = 3) | 2 ± 2 years |
| Time to death despite ICD (n = 2) | 3 ± 2 years |
| Death from heart failure/HTX (n = 3, 1 subfamily) | 36 ± 7 years |
| Clinical symptoms (initial cause of presentation) | n |
| Palpitations | 8/9 (89%) |
| Dyspnoea | 5/9 (56%) |
| Syncope | 1/9 (11%) |
| Arrhythmia findings | Age (initial diagnosis) |
| AV conduction defect | |
| AV-block I° (n = 4) | 37 ± 8 years |
| AV-block II° (n = 3) | 43 ± 6 years |
| AV-block III° (n = 2) | 45 ± 10 years |
| AF | |
| Paroxysmal AF (n = 4) | 42 ± 6 years |
| Permanent AF (n = 5) | 46 ± 10 years |
| Non-sustained VT on Holter-ECG (n = 4) | 43 ± 8 years |
| Pacemaker (n = 2) | 48 ± 12 years |
| ICD (n = 5) | 48 ± 9 years |
| Time to 1st appropriate ICD therapy (n = 3) | 2 ± 2 years |
| Time to death despite ICD (n = 2) | 3 ± 2 years |
| Death from heart failure/HTX (n = 3, 1 subfamily) | 36 ± 7 years |
AF, atrial fibrillation; AV, atrioventricular; ECG, electrocardiogram; HTX, heart transplantation; ICD, implantable cardioverter-defibrillator; VT, ventricular tachycardia.
Heart failure phenotype
In one sub-family [two sisters (IV:20 and IV:21) and one son (V:16)], we noticed a different phenotype. These confirmed (V:16) and presumed (IV:20 and IV:21) mutation-carriers presented rapidly decompensating heart failure already at young age (terminal heart failure at 35.6 ± 6.6 years) (Figure 4E). Prior to symptomatic heart failure, these patients showed only minor or no arrhythmias; once symptoms occurred, heart failure progressed rapidly leading to listing for heart transplantation (HTX). Unfortunately, two patients died while on the waiting list (aged 30 and 45 years). One of the women (IV:21) exhibited a peripartum-associated aggravation during her second pregnancy. Before her pregnancy, she had only sinus bradycardia and ventricular extrasystoles; at 28–30 weeks of gestation, however, incipient DCM with severe heart failure occurred, which led to death three weeks after Caesarean section in week 32 of gestation. Her son (V:19) suffered from severe cerebral hypoxia at birth and developed mental and physical disability. He died 15 years later.
Since no genetic material was stored, we were not able to prove the presence of the LMNA mutation in this patient. Although mortality is a known risk in peri-partum cardiomyopathy per se, the course of the disease with minor rhythm abnormalities such as sinus bradycardia and ventricular extrasystoles prior to her pregnancy followed by rapid progression of heart failure as in her sister and her nephew—a verified mutation-carrier—however, strongly suggest that she was a LMNA mutation-carrier.
The son (V:16) of patient IV:20 was identified as mutation-carrier at the age of 28 years. At that time, he did not show symptoms and declined to be informed about the test result. Only 4 years later, he was admitted to the hospital with dyspnoea, massive pulmonary, and peripheral oedema. Echocardiography showed dilated ventricles and atria (Figure 5A) and a severely impaired left ventricular ejection fraction (LVEF) of 16%. Chest radiography revealed a globally enlarged heart (Figure 5C). Electrocardiogram showed atrial flutter, AV-block I° and non-sustained VTs (Figure 5B). As conservative treatment failed, he was listed for high-urgency HTX, which he successfully received 3 weeks later. Histology of the explanted heart showed cardiomyocyte cell-size heterogeneity, cytoplasmic vacuoles, and absence of inflammation (Figure 5D).
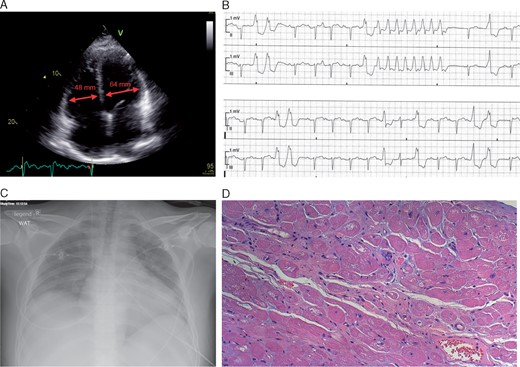
Clinical features in subtype with early severe heart failure. (A) Transthoracic echocardiogram with dilated cardiac cavities of patient V:16. left ventricular end diastolic diameter = 64 mm, right ventricular end diastolic diameter = 48 mm. (B) Non-sustained ventricular tachycardia on electrocardiogram telemetry. (C) Globally enlarged heart in chest radiography. (D) Histology of the explanted heart showed increased extracellular matrix, increased cardiomyocyte cell-size heterogeneity, cytoplasmic vacuoles, and absence of an inflammatory infiltrate. Hematoxylin and eosin stain.
In this patient, further genetic testing was performed to identify additional mutations or potential disease-modifying variants. Dilated cardiomyopathy multi-gene-panel testing excluded additional known DCM-related mutations and only revealed variants of unknown significance in SCN5A and ABCC9 (see Supplementary material online, Table S1), some of which have previously been described as ‘disease-modifiers’.32 Whole exome sequencing identified several heterozygous and homozygous variants exclusively in this patient. Due to the large amount of variants detected, we focused primarily on truncating variants, i.e. nonsense, frameshift and splice donor/acceptor variants with probable high-impact severity, to identify potential ‘disease-modifiers’ (Table 1). No variants were found in TTN or BAG3, known DCM-related genes not covered by the DCM multi-gene-panel. Of all the genes carrying variants with ‘high-impact severity’, PSMB10 and FDX1L have been described as being linked to cardiomyopathy or mitochondrial muscle dystrophy, respectively (Table 1).
Discussion
We identified a large family, spanning five generations, with the novel heterozygous LMNA nonsense mutation c.544C>T in exon 3 that is predicted to lead to premature termination of translation of Lamin A/C (p.Q182*). This mutation causes familial cardiomyopathy with a pronounced arrhythmogenic phenotype with 17 cases of SCD in four generations and successfully terminated VT/VF in two patients by external defibrillator and ICD, respectively.
This mutation is one of 27 known LMNA nonsense-mutations to date, and within those, it encompasses, along with one other report, the largest family described so far. The other large family was reported in the first publication linking mutations in LMNA with cardiomyopathy,33 which was later characterized in detail.30 In their study, the authors reported 29 affected individuals either as carriers of LMNA mutation c.16C>T or presumed mutation-carriers with a history of SCD. In eight of these individuals SCD occurred at 48.4 ± 9.5 years, while another five died of decompensated heart failure at 51.8 ± 6.6 years.30 In the here-described family, we identified 25 affected family members, 8 of which were genetically verified and 17 presumed mutation-carriers with SCD. Within four generations, 17 cases of SCD occurred at 49.3 ± 10.0 years and in another two mutation-carriers sustained VT/VF was successfully terminated.
Nonsense mutations make up only 5.3% of all 488 known LMNA mutations, none of which lies within exon 3 (based on Human Gene Mutation Database; http://www.hgmd.cf.ac.uk/ac; research performed in April 2017)—except for the mutation here-identified. It is noteworthy that 20 out of these 27 nonsense mutations lead to DCM and/or heart rhythm abnormalities,34,35 while the remaining 7 lead to musculoskeletal diseases (see Supplementary material online, Table S2).
In a multi-centre study conducted in 2012, several risk factors for malignant ventricular arrhythmias in LMNA mutation-carriers were identified, with the most predictive being the occurrence of non-sustained VT, left-ventricular EF of <45%, male sex, and ‘non-missense’ mutations.8 This last observation is of particular interest since a nonsense—i.e. a non-missense—mutation underlies the highly arrhythmogenic phenotype in our family as well.
Furthermore, a prolonged PR-interval was identified as early indicator for increased arrhythmogenic risk36—a finding observed in nearly all mutation-carriers in our family. Several multi-centre studies found a male predominance of 65% in symptomatic patients with LMNA mutations with a particularly elevated risk for malignant VT, end-stage heart failure, and SCD in male patients37,38; while conduction disorder, atrial tachyarrhythmia and non-sustained VT showed no sex differences.38 With 14 affected male (56%) and 11 female (44%) patients in this family, the same trend was observed in this study. However, since there was no sex difference in survival, we could not confirm male sex as a negative predictor for survival in LMNA c.544C>T.
Different phenotypes in patients carrying LMNA nonsense mutation c.544C>T
Two distinct, highly malignant clinical phenotypes were observed: most family members initially exhibited arrhythmia-related symptoms and early onset of malignant ventricular arrhythmia/SCD followed by late onset of heart failure-related symptoms at older age, consistent with previous reports demonstrating high arrhythmogenic risk even with preserved systolic and diastolic left ventricular function.39 One sub-family, in contrast, developed early onset, rapidly progressing heart failure at young age preceded by only minor rhythm-abnormalities such as sinus bradycardia. This pronounced clinical variability has been previously described in other LMNA mutations7 and may be partially caused by genetic modifiers that determine the disease phenotype in the individual patient. While some of the phenotypic variability might also be caused by the mutation itself, the fact that a very distinct ‘early onset heart failure’ phenotype was observed in one sub-family without preceding rhythm-abnormalities contrasting with the predominant ‘arrhythmic phenotype’ in all other family members prompted us to investigate, whether we might identify potential genetic modifiers.
Other DCM-related genes were excluded in this sub-family, but some ‘variants of unknown significance’ in ABCC9 and SCN5A that were previously described as ‘disease-modifiers’ (HGDM) were identified in the ‘heart failure’ sub-family patient (V:16). In addition, WES revealed ‘high-impact severity’ mutations in PSMB10 and FDX1L in the ‘heart failure’ patient (Table 1); genes that have been previously linked to cardiomyopathy or mitochondrial muscle dystrophy in case of homozygous mutations or in knock-out models (Table 1). Whether these identified mutations may act as LMNA disease-modifiers needs to be assessed in future research.
Pregnancy-associated aggravation of LMNA cardiomyopathy
In one patient (IV:21) of the ‘cardiomyopathy sub-family’, pregnancy-associated fast-progressing heart failure with lethal outcome was observed. While classical peripartal cardiomyopathy (PPCM) is a diagnosis by exclusion, defined as a non-familial, idiopathic form of cardiomyopathy inducing systolic dysfunction (LVEF <45%) and heart failure in the peripartal period,40 some studies identified a history of familial DCM and links to DCM-mutations in some PPCM patients.41–43 The patient in our family, however, is the first presumed LMNA mutation-carrier with pregnancy-induced severe heart-failure described to date. Since SCN5A mutations have been associated with PPCM, one might speculate that the SCN5A variants identified in the ‘heart failure’ sub-family might also have contributed to the pregnancy-associated decompensation.41 Identifying genetic predisposition for familial cardiomyopathy (including LMNA mutations) may thus help to identify patients at risk of pregnancy-associated heart failure.
Comparison of clinical phenotype of nonsense mutation c.544C>T and missense mutations in exon 3
To better assess the impact of the genotype on the clinical phenotype, we compared our family to another family within our clinical reach with LMNA missense mutation c.568C>T in exon 3, which is located in very close proximity to the novel nonsense mutation and leads to substitution of arginine into tryptophan (p.Arg190Trp).44 This family (see Supplementary material online, Results and Figure S1) demonstrated a completely different clinical phenotype with early DCM and heart failure requiring HTX in two patients (III: 3 and III: 4, at 45 and 43 years), and prompting their father’s (II: 2) early cardiac death due to heart failure (see Supplementary material online, Results).
These two families exemplify how two mutations, despite being closely located to each other, may cause diseases with very different clinical phenotypes: one displaying mostly arrhythmia and conduction-disorder, whilst the other shows mainly DCM and heart failure, likely due to their different impact on the Lamin A/C protein,34 e.g. premature termination of translation in the nonsense mutation vs. amino acid change in the missense mutation. We, therefore, compared all 18 missense mutations hitherto identified in exon 3 (according to HGMD®Professional).7,9,34,35,44–57 Of these, 15 present very similar clinical phenotypes with fast-progressing heart failure at young age requiring HTX. Differences were mainly apparent in the extent of arrhythmia (ranging from no arrhythmia to persistent AF), while no complex VT were described7,34,35,44,45,47–49,51,53–57 (see Supplementary material online, Table S3). These findings highlight a significant correlation between genotype/type of mutation and phenotype: DCM with almost no arrhythmias and heart failure-related death in most missense mutations, contrasting with pronounced arrhythmogenic phenotype and SCD due to VT/VF in nonsense mutations such as the novel mutation c.544C>T.
Clinical implications
Many LMNA nonsense mutations such as the here-presented mutation c.544C>T display a severe arrhythmogenic phenotype with high risk of sudden cardiac death. In addition, once heart failure develops in laminopathy, there is often a very fast progression to end-stage heart failure; with a similarly high mortality. Therefore, a careful and timely follow-up is required in patients with laminopathy with early consideration of ICD implantation and listing for HTX—particularly as usual parameters for the assessment of arrhythmic risk and for the inclusion in the heart-transplantation line may not be applicable to laminopathy patients. Novel therapeutic approaches targeting nonsense mutations have been developed and may offer mechanism-oriented therapies in addition to standard symptomatic heart failure therapy and ICD implantation. Ataluren (PTC-124) is such a drug that enables skipping the premature termination codon (PTC) leading to regular translation of the complete protein.58 This drug has already been approved (and proven efficient) for various genetic disorders caused by nonsense mutations.58 It would be intriguing to investigate whether it is similarly effective in laminopathy with nonsense mutations such as LMNA c.544C>T.
Limitations and future directions
In this study, we identified potential ‘disease-modifiers’ in one patient of the heart failure sub-family. Ideally, WES analyses had been performed in all affected members of this sub-family, as this would allow identification not only of truncating variants, but also of potentially relevant missense or other ‘lower-impact severity’ variants. As the other two patients had both died prior to HTX and no blood samples were available, however, this was impossible.
The relevance of the identified potential LMNA ‘disease-modifiers’ in conferring the heart-failure subtype stills needs to be assessed in future research. One potential approach would comprise a comparison of electrophysiological and mechanical parameters in iPS-derived cardiomyocytes from patients of the different phenotypic sub-families to investigate whether an intrinsic functional difference in the cardiomyocytes can be attributed to the genetic variants although there are some limitations to this method such as that these iPSC-CM represent ‘immature’ cardiomyocytes and that complex cell–cell interactions may not be assessed with cellular systems.
Conclusion
We identified a large family, spanning five generations, with the novel LMNA nonsense mutation c.544C>T in exon 3. This mutation causes familial cardiomyopathy with a severe arrhythmogenic phenotype, with 17 SCDs in four generations and successfully terminated VT/VF in another two patients. On average, patients not protected by an ICD died (SCD) approximately 15 years after first (arrhythmia-related) symptoms. Importantly, a second distinct clinical phenotype was identified in this family, leading to a similarly malignant phenotype but related to early onset of rapidly decompensating heart failure at young age with only minor prior arrhythmia-related symptoms, indicating a potential role of genetic modifiers in determining the disease phenotype in the individual patient.
Acknowledgements
The authors would like to thank Anton Glöcklhofer (Austria) for identifying and treating the index patient.
Conflict of interest: none declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}