





Abstract
Risk stratification for sudden death in arrhythmogenic right ventricular cardiomyopathy (ARVC) is challenging in clinical practice. We lack recommendations for the risk stratification of exclusive left-sided phenotypes. The aim of this study was to investigate genotype–phenotype correlations in patients carrying a novel DSP c.1339C>T, and to review the literature on the clinical expression and the outcomes in patients with DSP truncating mutations.
Genetic screening of the DSP gene was performed in 47 consecutive patients with a phenotype of either an ARVC (n = 24) or an idiopathic dilated cardiomyopathy (DCM), who presented with ventricular arrhythmias or a family history of sudden death (n = 23) (aged 40 ± 19 years, 62% males). Three unrelated probands with DCM were found to be carriers of a novel mutation (c.1339C>T). Cascade family screening led to the identification of 15 relatives who are carriers. Penetrance in c.1339C>T carriers was 83%. Sustained ventricular tachycardia was the first clinical manifestation in six patients and nine patients were diagnosed with left ventricular impairment (two had overt severe disease and seven had a mild dysfunction). Cardiac magnetic resonance revealed left ventricular involvement in nine cases and biventricular disease in three patients. Extensive fibrotic patterns in six and non-compaction phenotype in five patients were the hallmark in imaging.
DSP c.1339C>T is associated with an aggressive clinical phenotype of left-dominant arrhythmogenic cardiomyopathy and left ventricular non-compaction. Truncating mutations in desmoplakin are consistently associated with aggressive phenotypes and must be considered as a risk factor of sudden death. Since ventricular tachycardia occurs even in the absence of severe systolic dysfunction, an implantable cardioverter-defibrillator should be indicated promptly.
We identified a new desmoplakin mutation (DSP c.1339C>T) associated with a severe phenotype of arrhythmogenic cardiomyopathy and a high burden of ventricular arrhythmia.
Left ventricular non-compaction with high personal and familiar arrhythmic burden should arouse suspicion towards desmosomal disease.
Vast majority of desmoplakin truncated mutations reported in the literature are associated with severe phenotypes.
Introduction
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a primary myocardial disorder clinically characterized by ventricular arrhythmias, sudden cardiac death (SCD), and end-stage heart failure. It is a common cause of SCD among young people and athletes,1 and fatal arrhythmia may even occur in the absence of significant ventricular remodelling.
Arrhythmogenic right ventricular cardiomyopathy was thought to primarily affect the right ventricle (RV) although in a proportion of cases, the disease has been shown to affect the left ventricle (LV) not only at the end stage but also as a primary target. In this scenario, isolated arrhythmogenic left ventricular cardiomyopathy (ALVC) is frequently misdiagnosed as dilated cardiomyopathy (DCM) or myocarditis.2,3
Mutations in genes encoding desmosomal proteins (desmoplakin, plakophilin-2, plakoglobin, desmocollin-2, and desmoglein-2)4 were first identified in classical ARVC and were later found in left dominant forms.5,6 Other non-desmosomal genes have recently been associated with arrhythmogenic cardiomyopathy (ACM).7
The aim of this study was to investigate genotype–phenotype correlations in patients carrying a novel DSP c.1339C>T, and to review the literature on the clinical expression and the outcomes in patients with DSP truncating mutations.
Methods
The study was approved by the Ethics Committee of our hospital (Virgen de la Arrixaca University Hospital, Murcia) and it complied with the ethical principles of the Declaration of Helsinki. All participants gave their written consent. A genealogical pedigree of each family was obtained. Clinical assessment consisted of a 12-lead digital electrocardiogram (ECG), a signal-averaged ECG (SA-ECG), a 2D-Doppler echocardiogram, cardiac magnetic resonance (CMR), an exercise test, and 24 h Holter monitoring. In addition, an electrophysiological study (EPS) was performed in three patients. Patients were followed up annually [median 26 (13–87) months].
Study population
The population studied included 47 unrelated patients [29 (62%) males, aged 40 ± 19 years]. Twenty three (49%) met the criteria for ARVC and 24 for DCM, showing either sustained ventricular tachycardia (S-VT) or a history of SCD in a first- or second-degree relative under 35 years of age. We chose a DCM population with such a particular aggressive arrhythmic burden that arouse the suspicion of arrhythmogenic left ventricular cardiomyopathy as the first diagnosis to be considered.
A genetic study of the five desmosomal genes (DSP, DSG2, DSC2, PKP2, PKG), PLN, and LDB3 was performed in the 47 patients. A novel DSP c.1339C>T mutation was identified in three unrelated probands. First-degree relatives from these three families were invited to participate in a clinical and genetic investigation. Fifteen relatives were found to carry DSP c.1339C>T. Two additional patients previously diagnosed with DCM, mild systolic dysfunction, and ventricular arrhythmia died before the study started and in light of their family history, they were considered to be obligatory carriers.
A LV end-diastolic diameter (LVEDd) of >117% from normal and/or LV systolic impairment (<45%) was used for the diagnosis of DCM in probands. Familial criteria were applied in the remaining carriers.8 Arrhythmogenic right ventricular cardiomyopathy diagnosis was made according to the revised Task Force Criteria (TFC).9 Jenni et al.10 criteria were employed for the definition of left ventricular non-compaction (LVNC).
Genetic analysis
Deoxyribonucleic acid was extracted from peripheral blood samples using the Maxwell 16 Blood Purification kit. Previously published DSP, DSC2, DSG2, PKP2, PKG, PLN primer sequences were used for amplification. Results were compared with the sequences available in Gen Bank (RefSeq: NC_000006.11) and analysed by SeqScape software (AppliedBiosystem).11 MYBPC3 and MYH7 were studied in two selected individuals from families with DSP c.1339C>T with LVNC phenotype. DSP c.1339C>T variant was absent in 500 Spanish control samples and was not present in the 5000 exome project [Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP) (URL: http://evs.gs.washington.edu/EVS/) [1/12/2013].] Four published markers that flank the DSP gene were used to investigate the presence of a potential founder effect in carriers of DSP c.1339C>T. All of them are located within the DSP gene. Microsatellite 1 (Des.mic.1) is located in intron 1 and Microsatellite 2 (Des.mic.23) is located in intron 23 of the DSP gene. Both of them were indentified in a sequence of a BAC clone mapping to 6p23-p24 (GeneBank accession no. AL031058). The primer sequences for Des.mic 1 and 3 are as follows:
Des.mic.1 forward, 5′-CCCATCTATGCATAATGCAACC-3′; reverse, 5′-GTCCTCACGGATGTGCTACAAG-3′
Des.mic.23 forward, 5′-CGCTTTTGATCATGGCCCTAGTG-3′; reverse, 5′-CTCACCTGTTACAGCTAGATG-3′″
Immunohistochemistry
We analysed endomyocardial biopsy (EMB) samples from right ventricular (RV) septum in one case (C.III.12). They were formalin fixed, paraffin embedded, cut at a thickness of 4 mm, and mounted on slides. We incubated the slides with a primary antibody [monoclonal mouse anti-N-cadherin (1 : 400) (Sigma); monoclonal mouse anti-plakoglobin (1 : 1000) (Sigma); monoclonal rabbit anti-connexin 43 (1 : 400) (Sigma) and monoclonal rabbit anti-desmin (1 : 200)(Abcam)]. The slides were incubated with secondary goat anti-mouse or goat anti-rabbit (1 : 400) (Jackson ImmunoResearch). Stained slides were checked using a confocal microscope (LSM510, Meta Zeiss). Findings were compared with a control sample from a patient deceased for no cardiovascular reason in which necropsy did not show any evidence of cardiac disease. Case and control samples were fixed and embedded according to the same protocol and stained under similar conditions.
Electrocardiogram monitoring tests
One or more abnormal parameters in SA-ECG were considered as pathological (MAC 5500 ECG Diagnosis System—GE Healthcare). Patients with right bundle branch block (RBBB) or left bundle branch block (LBBB) were excluded from the SA-ECG analysis. Tracings from ambulatory 24 h Holter monitoring (Marquette Electronics and Synetec, ELA Medical) and treadmill exercise tests (Marquette Electronics, Inc. and General Electric T-2100) were reviewed. Non-sustained ventricular tachycardia (NS-VT) was defined as the presence of three or more ventricular beats at a rate of >100 b.p.m. Sustained ventricular tachycardia was defined as VT lasting >30 s or lasting less if it was poorly tolerated.
Echocardiogram
All participants underwent 2D-Doppler echocardiogram (Hewlett-Packard Sonos 7500, Hewlett-Packard). The standard analysis of the LV included the measurement of end systolic and end diastolic diameters and volumes, ejection fraction, wall motion abnormalities, and pathological valvular flows. Right ventricle analysis consisted of outflow tract diameter calculations (measured on both the long and short axis) and in order to assess RV performance, TAPSE, Doppler tissue imaging (DTI) and the fractional shortening ratios were both measured.
Cardiac magnetic resonance
Cardiac magnetic resonance was performed on 10 DSP c.1339C>T carriers using a 1.5T magnet (Achieva CV, Philips Medical Systems). SSFP end-expiratory breath-hold cine imaging was acquired. After a bolus injection of Gadobutrol (0.1 mmol/kg, Gadovist; Bayer Shering Pharma, Berlin, Germany) the T1 measurement was calculated using standard late gadolinium enhancement (LGE) sequences. The analysis was performed using a personal computer and semi-automated software (Philips, Software work space 2.6.3.2). Five patients were excluded from this analysis [three already carried an implantable cardioverter-defibrilator (ICD) and two rejected the test].
Results
A novel heterozygous DSP variant (c.1339C>T) inherited in an autosomal dominant manner with high penetrance (83%) was identified in 3 patients. The mutation was a C to T transition at exon 11 in the DSP gene leading to a premature stop codon. This change occurred at the N-terminal region, and it is thought to generate a truncated peptide (85% in length). The affected amino acid at position c.1339C>T is located in one of the globular head domains of desmoplakin. This domain is shown to mediate the interaction of desmoplakin with two catenin proteins: plakoglobin and plakophilin-212 (Figure 1). The microsatellite study suggested a founder effect in the three families and the presence of a common ancestor.
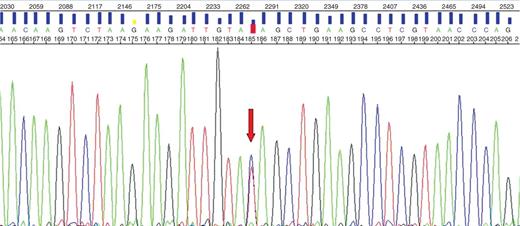
Electropherogram. The mutation was a C -> T transition at exon 11 in the DSP gene leading to a premature stop codon.
Family A
The 37-year-old proband (A.III.5) (Figure 2) was diagnosed after an episode of S-VT at rest. An echocardiogram demonstrated severe LV dilatation and systolic impairment. The patient had a history of longstanding alcohol drinking, smoking, and occasional cocaine use. The angiogram excluded coronary lesions and an ICD was implanted.
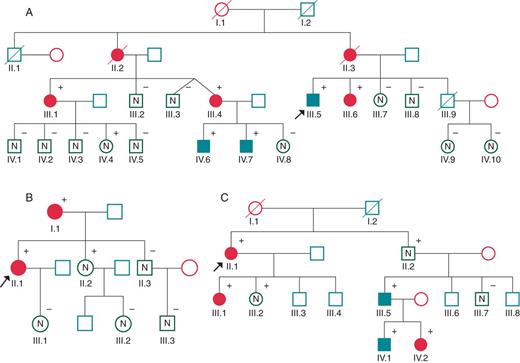
Family tree of families A, B, and C. Arrow points to probands. Black filled symbols stand for affected carriers.
His mother (A.II.3) had died suddenly at 67 years of age. She had been diagnosed with DCM after ruling out coronary disease. The maternal grandfather suffered from heart failure and died in his sleep at the age of 37 years (A.I.1). A maternal aunt had previously died of heart failure (A.II.2).
Prior to the proband's admission, a cousin aged 55 years (A.III.1) who presented with recurrent S-VT and a structurally normal heart underwent ICD implantation. Familial cascade screening led to a new diagnosis of four asymptomatic, though affected, individuals (A.III.4, A.III.6, A.IV.6, and A.IV.7). It should be noted that the diagnoses of A.III.4 and A.III.6 were LVNC. A severely increased LGE pattern was observed in A.IV.6 and A.IV.7 alongside mild systolic dysfunction. During the follow-up period, both siblings have suffered multiple episodes of chest pains with troponin rise. Coronary artery disease was ruled out in both cases. They had an ICD implanted for primary prevention in the view of progressive systolic dysfunction.
Family B
The proband (B.II.1), a 42-year-old woman, was admitted after a presyncopal S-VT episode. Echocardiography revealed global left ventricular hypokinesia and a moderately impaired systolic function.
The mother (B.I.1), aged 72 years, had a history of presyncopal episodes associated with S-VT. An echocardiogram showed severe systolic impairment and angiography ruled out coronary disease. She underwent an EPS where poorly tolerated S-VT of a different morphology was induced. Both patients received an ICD.
A maternal aunt had previously died suddenly at the age of 40 years. Family genetic screening led to diagnosis of an asymptomatic non-affected carrier: a sister (B.II.2) aged 50 years. The echocardiogram was normal and the patient rejected CMR due to claustrophobia. Four maternal aunts, one maternal uncle, and a nephew refused a cardiac and genetic study.
Family C
The 62-year-old female proband was admitted to hospital after a first episode of acute heart failure (C.II.1). Echocardiography demonstrated moderately impaired systolic function and MRI results led to the diagnosis of LVNC.
His daughter (C.III.1) had suffered an episode of acute myocarditis at the age of 32 years.
Six years after the diagnosis of the proband, a 45-year-old nephew (C.III.5) presented with cardiac arrest while walking. He was diagnosed with LVNC and moderate systolic dysfunction. One month after having an ICD implanted, he received four shocks due to S-VT during mild exertion (Figure5). Both his 13-year old son (C.IV.1) and his 17-year-old daughter (C.IV.2) were asymptomatic and they were diagnosed with LVNC in familial screening. The latter was found to have frequent ventricular ectopics and couplets on holter monitoring.
Clinical presentation and therapy
The first clinical presentation was presyncopal S-VT in five patients, resuscitated SCD in one, and heart failure in two patients. Cardiac and genetic work-up led to the identification of seven asymptomatic patients, as well as three non-affected asymptomatic carriers. Eight carriers met the criteria for DCM, four for LVNC and seven carriers also fulfilled the diagnostic criteria for the definitive diagnosis of ARVC (Table 1).
Clinical characterization of DSP c.1339 C>T carriers
| Case | Family | Pedigree | Age/Sex | Syncope | Sustained VT | Morphology VT | CL (ms) | Non sustained VT | NYHA | ARVC diagnosis | LVEF | LGE distribution | Hypertrabeculation (CMR) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | A | II.2 | 59F | Yes | Yes | RBBB | 330 | Yes | I | Borderline | 40 | NA | NA |
| 2 | A | II.3 | 65F | No | No | NA | No | No | II | Possible | 40 | NA | NA |
| 3 | A | III.1 | 55F | No | Yes | NA | NA | No | I | Definitive | 64 | NA | NA |
| 4 | A | III.6 | 45F | No | No | No | No | No | I | Definitive | 40 | Inferoposterior and lateral | Yes |
| 5 | A | III.4 | 44F | No | No | NA | NA | Yes | I | Borderline | 50 | Global | Yes |
| 6 | A | III.5 | 36M | No | Yes | NA | NA | Yes | I | Definitive | 30 | NA | NA |
| 7 | A | IV.4 | 38F | No | No | No | No | No | I | Possible | 60 | No | NA |
| 8 | A | IV.6 | 14M | No | No | No | No | No | I | Possible | 45 | Lateral | No |
| 9 | A | IV.7 | 19M | No | No | No | No | No | I | Possible | 55 | Lateral | No |
| 10 | B | I.1 | 72F | No | Yes | LBBB. Axis −30° | 320 | Yes | II | Definitive | 30 | NA | NA |
| 11 | B | II.2 | 49F | No | No | No | No | No | I | Possible | 58 | NA | NA |
| 12 | B | II.1 | 42F | No | Yes | NA | NA | No | I | Definitive | 40 | NA | NA |
| 13 | C | II.1 | 62F | No | Yes | NA | NA | Yes | II | Definitive | 43 | Inferoposterior and lateral | No |
| 14 | C | III.5 | 45M | No | Yes | NA | 220 | No | I | Definitive | 42 | Global | Yes |
| 15 | C | II.3 | 73M | No | No | No | No | No | I | Borderline | 60 | No | No |
| 16 | C | III.1 | 38F | No | No | No | No | No | I | Borderline | 60 | No | No |
| 17 | C | IV.1 | 13M | No | No | No | No | No | I | Borderline | 66 | No | Yes |
| 18 | C | IV.2 | 17F | No | No | No | No | No | I | Borderline | 60 | No | Yes |
| Case | Family | Pedigree | Age/Sex | Syncope | Sustained VT | Morphology VT | CL (ms) | Non sustained VT | NYHA | ARVC diagnosis | LVEF | LGE distribution | Hypertrabeculation (CMR) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | A | II.2 | 59F | Yes | Yes | RBBB | 330 | Yes | I | Borderline | 40 | NA | NA |
| 2 | A | II.3 | 65F | No | No | NA | No | No | II | Possible | 40 | NA | NA |
| 3 | A | III.1 | 55F | No | Yes | NA | NA | No | I | Definitive | 64 | NA | NA |
| 4 | A | III.6 | 45F | No | No | No | No | No | I | Definitive | 40 | Inferoposterior and lateral | Yes |
| 5 | A | III.4 | 44F | No | No | NA | NA | Yes | I | Borderline | 50 | Global | Yes |
| 6 | A | III.5 | 36M | No | Yes | NA | NA | Yes | I | Definitive | 30 | NA | NA |
| 7 | A | IV.4 | 38F | No | No | No | No | No | I | Possible | 60 | No | NA |
| 8 | A | IV.6 | 14M | No | No | No | No | No | I | Possible | 45 | Lateral | No |
| 9 | A | IV.7 | 19M | No | No | No | No | No | I | Possible | 55 | Lateral | No |
| 10 | B | I.1 | 72F | No | Yes | LBBB. Axis −30° | 320 | Yes | II | Definitive | 30 | NA | NA |
| 11 | B | II.2 | 49F | No | No | No | No | No | I | Possible | 58 | NA | NA |
| 12 | B | II.1 | 42F | No | Yes | NA | NA | No | I | Definitive | 40 | NA | NA |
| 13 | C | II.1 | 62F | No | Yes | NA | NA | Yes | II | Definitive | 43 | Inferoposterior and lateral | No |
| 14 | C | III.5 | 45M | No | Yes | NA | 220 | No | I | Definitive | 42 | Global | Yes |
| 15 | C | II.3 | 73M | No | No | No | No | No | I | Borderline | 60 | No | No |
| 16 | C | III.1 | 38F | No | No | No | No | No | I | Borderline | 60 | No | No |
| 17 | C | IV.1 | 13M | No | No | No | No | No | I | Borderline | 66 | No | Yes |
| 18 | C | IV.2 | 17F | No | No | No | No | No | I | Borderline | 60 | No | Yes |
F, female; M, male; CL, cycle length; NA, not available. Probands are in bold.
Clinical characterization of DSP c.1339 C>T carriers
| Case | Family | Pedigree | Age/Sex | Syncope | Sustained VT | Morphology VT | CL (ms) | Non sustained VT | NYHA | ARVC diagnosis | LVEF | LGE distribution | Hypertrabeculation (CMR) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | A | II.2 | 59F | Yes | Yes | RBBB | 330 | Yes | I | Borderline | 40 | NA | NA |
| 2 | A | II.3 | 65F | No | No | NA | No | No | II | Possible | 40 | NA | NA |
| 3 | A | III.1 | 55F | No | Yes | NA | NA | No | I | Definitive | 64 | NA | NA |
| 4 | A | III.6 | 45F | No | No | No | No | No | I | Definitive | 40 | Inferoposterior and lateral | Yes |
| 5 | A | III.4 | 44F | No | No | NA | NA | Yes | I | Borderline | 50 | Global | Yes |
| 6 | A | III.5 | 36M | No | Yes | NA | NA | Yes | I | Definitive | 30 | NA | NA |
| 7 | A | IV.4 | 38F | No | No | No | No | No | I | Possible | 60 | No | NA |
| 8 | A | IV.6 | 14M | No | No | No | No | No | I | Possible | 45 | Lateral | No |
| 9 | A | IV.7 | 19M | No | No | No | No | No | I | Possible | 55 | Lateral | No |
| 10 | B | I.1 | 72F | No | Yes | LBBB. Axis −30° | 320 | Yes | II | Definitive | 30 | NA | NA |
| 11 | B | II.2 | 49F | No | No | No | No | No | I | Possible | 58 | NA | NA |
| 12 | B | II.1 | 42F | No | Yes | NA | NA | No | I | Definitive | 40 | NA | NA |
| 13 | C | II.1 | 62F | No | Yes | NA | NA | Yes | II | Definitive | 43 | Inferoposterior and lateral | No |
| 14 | C | III.5 | 45M | No | Yes | NA | 220 | No | I | Definitive | 42 | Global | Yes |
| 15 | C | II.3 | 73M | No | No | No | No | No | I | Borderline | 60 | No | No |
| 16 | C | III.1 | 38F | No | No | No | No | No | I | Borderline | 60 | No | No |
| 17 | C | IV.1 | 13M | No | No | No | No | No | I | Borderline | 66 | No | Yes |
| 18 | C | IV.2 | 17F | No | No | No | No | No | I | Borderline | 60 | No | Yes |
| Case | Family | Pedigree | Age/Sex | Syncope | Sustained VT | Morphology VT | CL (ms) | Non sustained VT | NYHA | ARVC diagnosis | LVEF | LGE distribution | Hypertrabeculation (CMR) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | A | II.2 | 59F | Yes | Yes | RBBB | 330 | Yes | I | Borderline | 40 | NA | NA |
| 2 | A | II.3 | 65F | No | No | NA | No | No | II | Possible | 40 | NA | NA |
| 3 | A | III.1 | 55F | No | Yes | NA | NA | No | I | Definitive | 64 | NA | NA |
| 4 | A | III.6 | 45F | No | No | No | No | No | I | Definitive | 40 | Inferoposterior and lateral | Yes |
| 5 | A | III.4 | 44F | No | No | NA | NA | Yes | I | Borderline | 50 | Global | Yes |
| 6 | A | III.5 | 36M | No | Yes | NA | NA | Yes | I | Definitive | 30 | NA | NA |
| 7 | A | IV.4 | 38F | No | No | No | No | No | I | Possible | 60 | No | NA |
| 8 | A | IV.6 | 14M | No | No | No | No | No | I | Possible | 45 | Lateral | No |
| 9 | A | IV.7 | 19M | No | No | No | No | No | I | Possible | 55 | Lateral | No |
| 10 | B | I.1 | 72F | No | Yes | LBBB. Axis −30° | 320 | Yes | II | Definitive | 30 | NA | NA |
| 11 | B | II.2 | 49F | No | No | No | No | No | I | Possible | 58 | NA | NA |
| 12 | B | II.1 | 42F | No | Yes | NA | NA | No | I | Definitive | 40 | NA | NA |
| 13 | C | II.1 | 62F | No | Yes | NA | NA | Yes | II | Definitive | 43 | Inferoposterior and lateral | No |
| 14 | C | III.5 | 45M | No | Yes | NA | 220 | No | I | Definitive | 42 | Global | Yes |
| 15 | C | II.3 | 73M | No | No | No | No | No | I | Borderline | 60 | No | No |
| 16 | C | III.1 | 38F | No | No | No | No | No | I | Borderline | 60 | No | No |
| 17 | C | IV.1 | 13M | No | No | No | No | No | I | Borderline | 66 | No | Yes |
| 18 | C | IV.2 | 17F | No | No | No | No | No | I | Borderline | 60 | No | Yes |
F, female; M, male; CL, cycle length; NA, not available. Probands are in bold.
Beta-blockers were started in all patients. Nine patients with systolic impairment were on angiotensin-converting enzyme inhibitors or ARB-II blockers. Spironolactone was started in four patients. Five patients received an ICD for secondary prevention of sudden death (A.III.1; A.III.5; B I.1; B II.1; and C.III.5) and four for its primary prevention (A III.4; AIII.6; A.IV.6, and A.IV.7). Primary prevention was considered in the presence of any of the following events: moderate systolic impairment; frequent ventricular ectopic beats; and/or couplets on Holter monitoring despite beta-blocker administration. Two siblings (A.IV.6 and A.IV.7) had the ICD implanted for recurrent chest pains and troponin rise anlongside with worsening of the ejection fraction. During follow-up, two patients, B III.1 and C.III.5, had an appropriate ICD shock due to S-VT.
Twelve-lead electrocardiogram and signal-averaged electrocardiogram
Electrocardiograms from 16 mutation carriers were available for analysis. Nine (56%) showed T wave inversion in the precordial and/or frontal leads. Intraventricular conduction abnormalities were present in 4 patients (LBBB in two patients, bi-fascicular block in one, and non-specific conduction abnormalities in another case). A SA-ECG was performed in 14 patients (two had died before the study and two were excluded due to the existence of baseline wide QRS). Six showed late potentials (43%) (Figure 3).
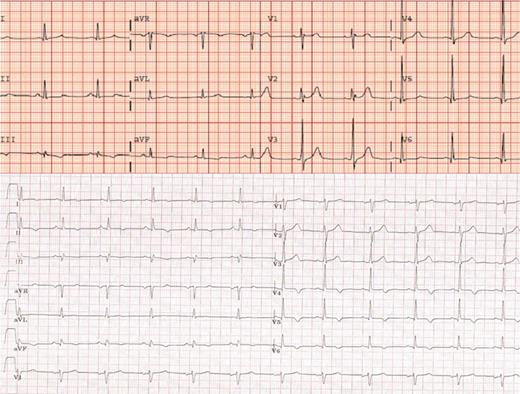
ECG from a LVNC patient (top) (C.III.5) and a DCM patient (bottom) (A.IV.7) T wave inversion in inferolateral leads was the most common abnormality in ECG.
Twenty-four hour electrocardiogram-Holter monitoring, an exercise test and ICD recordings
NS-VT was evidenced in four patients using 24 h Holter monitoring. Two additional patients had a NS-VT episode on ICD recordings, one of which had an episode of NS-VT while undergoing an exercise treadmill test. One patient suffered SVT one month after ICD implantation requiring four shocks to restore sinus rhythm. Frequent ventricular ectopics occurred during the treadmill test in two cases. Another young carrier without ECG or MRI abnormalities presented frequent ventricular ectopics and couplets.
Electrophysiological study
An EPS was performed on 3 patients, 2 of which (B.I.1 and B.II.1) were prior to ICD implantation. During the procedure the proband (B.I.1) developed S-VT different from the one previously recorded during hospitalization. Her daughter (B.II.1) developed S-VT during the EPS similar to the one originally recorded. Successful ablation was achieved. However, she did suffer a relapse 4 years later and further VT ablation was required. Endocardial voltage mapping using the CARTO navigator system in this later case showed no low voltage areas.
Echocardiogram
An echocardiographic study showed global or inferoposterior hypokinesia in 11 of 18 (61%) carriers. Two patients had severe systolic dysfunction and five had moderate systolic impairment. In three cases, LV apical trabeculae fulfilling the diagnostic criteria for LVNC were present. Neither RV enlargement nor dysfunction was detected in any case.
Cardiac magnetic resonance
Cardiac magnetic resonance was performed in 10 patients. Pathological findings were found in eight of them. Biventricular disease was observed in three patients and isolated LV involvement was present in six. Sub-epicardial myocardial fibrosis at the LV was the hallmark and it was detected in six patients. Fibrosis was distributed in the lateral wall in three cases, the inferoposterior wall in two, and it was globally distributed two more patients. Five patients were diagnosed with LVNC (Figure 4).
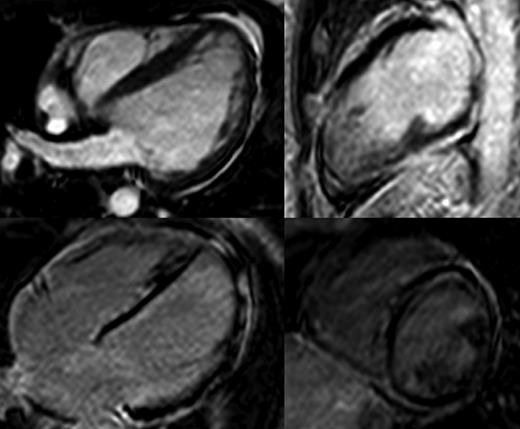
Magnetic resonance of a LVNC (C.III.5) (top) and DCM patient (A.IV.7)(bottom).
Immunohistochemistry
An EMB was analysed from C.III.5 during ICD implantation after cardiac arrest. Histology showed normal cardiomyocytes and no trace of either fibrofatty replacement, disarray, granuloma, inflammatory infiltrates or apoptosis (Figure 5). Staining for N-cadherin, desmoplakin, plakoglobin, connexin 43, and desmin was normal and no differences when compared the case with a control were observed (Figure 6).
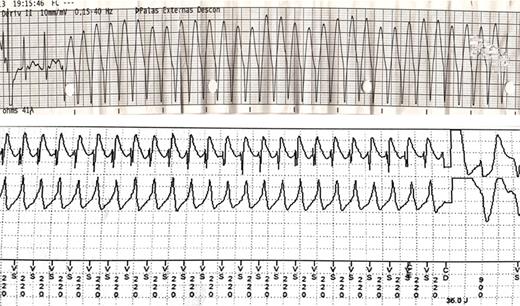
Sustained ventricular tachycardia (S-VT) recorded at the emergency department during resuscitation manoeuvres (top). Implantable cardioverter-defibrilator recordings showing S-VT and the beginning of another S-VT of a different morphology after shock delivery (bottom). Four shocks were necessary to restore sinus rhythm (C.III.5).
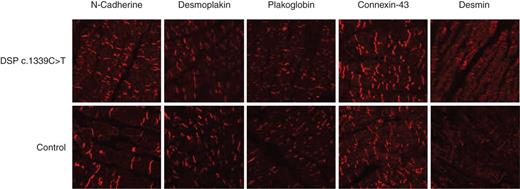
Immunohistochemistry from an EMB. No difference in the distribution of desmoplakin present in DSP c.1339C>T compared with the control. Plakoglobin, connexin-43 and desmin also show an identical distribution.
Discussion
To date, 42 DSP mutations leading to premature termination of translation have been reported. There is clinical information available on 120 carriers from 44 families. Of these, 86 (72%) are affected, with 18 (15%) SCD cases reported in carriers of one of these mutations. In the 14 families where clinical cascade screening was possible, disease penetrance was >50% in ten families (71%) and >90% in seven (50%) (Figure 7).
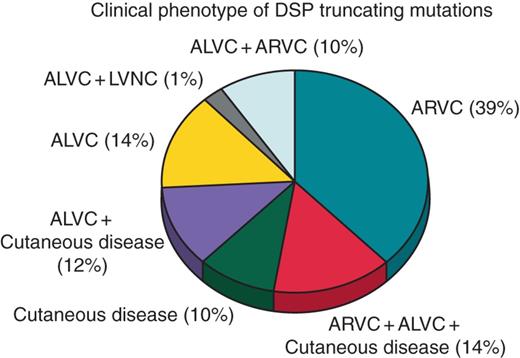
Clinical phenotypes of the 42 DSP truncating mutations reported to date. Plus indicates overlap syndrome.
Herein, we present the largest series of carriers of the same DSP truncating mutation reported to date. The microsatellite study suggests DSP c.1339C>T to have a founder effect. Several ARVC and/or ALVC families bearing truncating DSP mutations have been published, some of them, suffering SCD as a first clinical manifestation.13–15 These studies have reported highly penetrant disease phenotypes, with predominant LV involvement, a high incidence of malignant arrhythmias, and a poor prognosis. In keeping with these studies, the novel mutation herein presented is also associated with high disease penetrance (83%), LV dysfunction, extensive fibrosis, mild/no LV enlargement, left/anterior precordial lead repolarization abnormalities, and high prevalence of ventricular arrhythmias. It is notable that myocardial fibrosis was present even in young carriers (14 and 19 year olds). Such extensive fibrosis at an early age is an uncommon finding in cardiomyopathies and particularly rare in heterozygous carriers of autosomal dominant mutations underlying ACM.
Characterizing a phenotype using an electrocardiogram
Six percent of the patients showed ECG abnormalities, even in the absence of overt structural disease. New diagnostic criteria for the diagnosis of ARVC include T wave inversion in V4–V6 as a marker of LV involvement. In the light of our results and those previously reported in other series13 we would like to point out that T-wave inversion in inferior leads may be sensitive markers of ALVC even in the absence of ventricular remodelling. Signal-averaged ECG findings failed to correlate with ECG or imaging abnormalities.
Characterizing a phenotype with cardiac magnetic resonance
Heterogeneous myocardial fibrosis pattern evidenced by means of gadolinium on CMR has been associated with progression to heart failure in patients with hypertrophic cardiomyopathy, and, additionally, ventricular arrhythmias have been reported to be more frequent in patients with LGE in ischemic and idiopathic DCM.16 The vast majority of the DSP c.1339C>T carriers showed extensive fibrosis in the LV, particularly at the sub-epicardium, similar to that seen in localized myocarditis. During a ‘hot phase’, ACM may mimic myocarditis in its clinical presentation or CMR appearance.2,17 In our series, CMR was performed on clinically stable patients. Despite morphological findings being compatible with the diagnosis of LVNC in 5 patients, the presence of a truncating mutation in DSP, arrhythmic burden being out of proportion with the degree of systolic dysfunction and the family history led us to the diagnosis of ACM.
Implications for prevention of malignant arrhythmias and sudden death
Identifying individuals at high risk of SCD is of undisputed importance. Whether ALVC is associated with a poorer prognosis than ARVC or DCM remains a matter of debate. In terms of risk stratification in ARVC, several factors have been proposed over the past few years, the presence of which would prompt ICD implantation. These include resuscitated cardiac arrest, syncope, recurrent or syncopal S-VT, and severe RV or LV involvement.18,19 Other markers such as family history of sudden death, induction of VT upon EPS, extensive bipolar low voltage area in RV,20 fragmented potentials within the scar,21 T wave inversion in three or more leads22 and dispersion of depolarization and repolarization have been also reported.23,24 On the other hand, primary ICD implantation in DCM is based almost exclusively on the severity of impairment of LV ejection fraction and functional status.25 The decision on ICD implantation in ALVC poses a real challenge. To date, there is no general consensus in support of the use of genetic studies for ICD implantation. There is, however, no doubt that some genetic conditions such as laminopathies are associated with a high incidence of arrhythmic events and SCD, which may precede severe myocardial remodelling and dysfunction.
In recent years, the classic concept or ARVC has been broadened to the general concept of ACM. We have found a high prevalence of a LVNC-like phenotype in our families, leading us to recommend screening for DSP mutations in LVNC patients affected by a high burden of personal and familial arrhythmia. In this particular scenario, LVNC may be another phenotype to be considered in the broad spectrum of ACM.
Desmoplakin c.1339C>T: an unexpected immunohistochemistry pattern
Through its N-terminus, desmoplakin associates with armadillo proteins at the cell membrane. Through its C-terminus, it anchors intermediate filaments tethering them to junctional sites. Herein, we describe a novel truncating mutation in the N-terminal domain, anticipated to disrupt the synthesis of desmoplakin.
Confocal microscopy revealed no difference in the expression of desmoplakin, plakoglobin, connexine-43 and desmin compared with the control (Figure 6). It is notable that the hallmark in ARVC immunohistochemistry; plakoglobin shifting from junctions26 was not present. This finding is in keeping with a series of phospholamban mutation carriers where the plakoglobin distribution significantly differed in the DCM phenotype compared with the ARVC phenotype.7
This difference in plakoglobin remodelling leads us to rise the question whether the signalling pathways differs in ALVC and ARVC. Further studies will be required to shed light on the pathophysiology of this particular phenotype.
Conclusion
We have reported on a novel mutation in desmoplakin (c.1339C>T) associated with a phenotype of ALVC and LVNC. Truncating mutations in desmoplakin seem to consistently cause extensive LV fibrosis with near normal RV performance. Ventricular arrhythmias and SCD occur in the absence of overt LV dysfunction or dilatation, and as a consequence, ICD implantation must be considered promptly. Genetic information seems to be of paramount prognostic value in this setting.
Funding
Juan Ramón Gimeno received support from the RIC from the Carlos III Health Institute (C03/01,RD06/0014/0017). Lorenzo Monserrat received a Carlos III Health Institute grant (PI11/0260).
Acknowledgements
The authors thank Dr Angeliki Asimaki and Dr Jeffrey E. Saffitz (Boston, USA, BIDMC and Harvard Medical School) for their essential help in the understanding of the pathophysiology of ACM; Dr FJ Nicolas from the University of Murcia, for his lessons on confocal microscopy; They also thank Mrs María López for her help with the clinical examinations. Last but not least, they thank the families that agreed to participate in this study.
Conflict of interest: Dr Lorenzo Monserrat is shareholder of Health in Code S.L.
None of the authors have any conflict of interest to declare.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}