





Abstract
In one-third of patients with acute coronary syndrome (ACS), thrombosis occurs despite an intact fibrous cap (IFC) (IFC–ACS, ‘plaque erosion’). Recent studies emphasize neutrophils as the immediate inflammatory response in this pathology, but their exact molecular activation patterns are still poorly understood and may represent future therapeutic targets.
Thirty-two patients with IFC–ACS and matched patients with ACS with ruptured fibrous cap (RFC) (RFC–ACS) from the OPTICO–ACS study were included, and blood samples were collected from the local site of the culprit lesion and the systemic circulation. Neutrophil surface marker expression was quantified by flow cytometry. Neutrophil cytotoxicity towards endothelial cells was examined in an ex vivo co-culture assay. Secretion of active matrix metalloproteinase 9 (MMP9) by neutrophils was evaluated using zymography in supernatants and in plasma samples. Optical coherence tomography (OCT)–embedded thrombi were used for immunofluorescence analysis. Toll-like receptor 2 (TLR2) expression was higher on neutrophils from IFC–ACS than RFC–ACS patients. TLR2 stimulation increased the release of active MMP9 from local IFC–ACS–derived neutrophils, which also aggravated endothelial cell death independently of TLR2. Thrombi of IFC–ACS patients exhibited more hyaluronidase 2 with concomitant increase in local plasma levels of the TLR2 ligand: hyaluronic acid.
The current study provides first in-human evidence for distinct TLR2-mediated neutrophil activation in IFC–ACS, presumably triggered by elevated soluble hyaluronic acid. Together with disturbed flow conditions, neutrophil-released MMP9 might be promoting endothelial cell loss–triggered thrombosis and therefore providing a potential future target for a phenotype-specific secondary therapeutic approach in IFC–ACS.
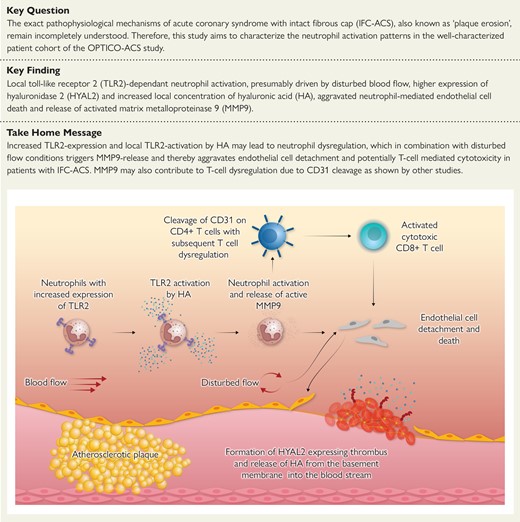
TLR2, Toll-like receptor 2; HA, hyaluronic acid; MMP9, matrix metalloproteinase 9; HYAL2, hyaluronidase 2.
See the editorial comment for this article ‘Unravelling the heart’s comic drama: can TLRs and hyaluronan metabolism stoke neutrophil rage in acute coronary syndrome?’, by G. Franck, https://doi.org/10.1093/eurheartj/ehad455.
The functional characterization of neutrophils from the coronary lesion site of patients with ACS provides mechanistic insights into the immuno-vascular processes in different ACS phenotypes. The enhanced TLR2 response of local IFC–ACS–derived neutrophils, resulting in MMP9 release and endothelial cell detachment, implicates possible future therapeutic targets and prevention strategies specifically in this ACS entity.
Introduction
Ischemic heart disease and its most dramatic manifestation acute coronary syndrome (ACS) remain the leading cause of death worldwide, despite effective lipid lowering therapies.1,2 Extensive effort has been made over the past decades to better characterize the molecular pathomechanisms of ACS, in order to predict and minimize cardiovascular event burden.2
These investigations have mainly focused on the mechanisms of rupture of the fibrous cap covering the plaque,3 the most frequent phenotype of ACS.4,5 However, autopsy studies have suggested another mechanism of ACS, i.e. coronary plaque erosion, characterized by thrombus formation on a plaque with intact fibrous cap (IFC) and, compared with ruptured fibrous cap (RFC) plaques, less lipid accumulation and local endothelial injury with loss of the luminal endothelial layer on the surface of the plaque.4 Contemporary intravascular imaging studies using optical coherence tomography (OCT) implicate IFC in up to one-third of ACS cases.6 However, the underlying pathophysiological mechanisms of IFC–ACS remain incompletely understood. Few ACS studies observed macrophage infiltrations at eroded ACS–causing sites and thereby suggest that inflammatory mechanisms may play a crucial role in this pathology.7,8,9 Notably, the neutrophils are shown to be the first immune cells to arrive at ACS–causing culprit sites and are thereby considered to be key initiators of the following inflammatory cascade after myocardial infarction.10 Neutrophil accumulation, disturbed flow, and engagement of the endothelial TLR2 receptor have been previously suggested to aggravate endothelial cell detachment in an in vitro model of plaque erosion.11 Therefore, this study aimed to characterize the mechanisms behind the neutrophil activation in patients with IFC–ACS with a special focus on neutrophil TLR2 activation.
Materials and methods
Study design
The current sub-study of the observational, prospective OPTICO–ACS study program enrolled patients with non-ST elevation myocardial infarction12 or ST elevation myocardial infarction13 and undergoing OCT imaging of the ACS–causing culprit lesion according to the inclusion and exclusion criteria.8 The OPTICO–ACS study was designed not only to assess the rates of major adverse cardiovascular events in patients with IFC–ACS vs. RFC–ACS as a primary endpoint but also to characterize these two populations by a combination of clinical description, OCT imaging of the culprit lesion, and immunophenotyping using flow cytometry.8
In the current nested case–control sub-study, 32 patients with IFC–ACS were matched by known factors to affect the neutrophil activation: age (±5 years),14–16 biological sex,17 and diabetes mellitus type 218 to patients with RFC–ACS (see Supplementary data online, Material S1). On the variables ‘sex’ and ‘diabetes mellitus type 2’, we performed an exact matching strategy (one to one), whereby on ‘age’, we performed an individual matching with a range of ±5 years. If more than one matched RFC–ACS control was available, the patient with the closest age to the matched IFC–ACS patient was randomly selected. The study was approved by the local ethics committee (No. EA1/270/16) and conducted in accordance with the Declaration of Helsinki.
Biosample collection
Systemic blood samples (SYS) were obtained during primary percutaneous coronary intervention (PCI) by aspiration of 30 ml blood from the arterial sheath, while local blood samples (LOC) were collected directly from the ACS–causing culprit lesion (LOC) by a the Export Advance™ aspiration catheter system (Medtronic, Minneapolis, MN, USA) (see Supplementary data online, Material S2). All experiments were performed in a blinded manner without revealing the underlying ACS–causing pathology to the operator.
Flow cytometry characterization of neutrophil and monocyte receptor repertoire
LOC and SYS whole EDTA–anticoagulated blood samples (100 µl) were stained with a cocktail of fluorochrome-labeled antibodies for a carefully pre-selected set of surface markers expressed by neutrophils and monocytes (see Supplementary data online, Material S3A), followed by a 1:10 dilution/fixation in 0.5% phosphate-buffered paraformaldehyde solution (final volume 1000 µl). Samples were acquired 16–24 h afterwards on an Attune NxT Acoustic Focusing Flow Cytometer (Thermo Fisher Scientific, USA), and data were analyzed by the Kaluza v1.5 software (Beckman Coulter Inc., USA) (see Supplementary data online, Material S3B, for the gating strategy). The receptor expression density per cell is described by the median fluorescence intensity of each marker. Due to the fact that neutrophils are the main, but not only immune cell population activated immediately after myocardial infarction,19 we also analyzed the distribution and TLR2 expression of the three main monocyte subtypes: classical (defined as CD14++ CD16−), intermediate (defined as CD14++ CD16+), and alternative monocytes (defined as CD14+ CD16+) using flow cytometry.
Polymorphonuclear granulocyte isolation
Polymorphonuclear granulocytes (PMNs) were isolated from both blood samples (LOC and SYS) using Polymorphprep density separation medium (Alere Technologies AS, Oslo, Norway) according to the manufacturer’s instructions.
Matrix metalloproteinase activity in supernatants and plasma samples from isolated PMNs
Isolated PMNs were either activated by a human recombinant TLR2/1 agonist (Pam3CSK4) or pre-treated with a selective inhibitory anti-TLR2 antibody. Fluorescence gelatine zymography was performed according to the manufacturer’s instructions (Invitrogen, Fisher Scientific, Göteborg, Sweden) to quantify gelatinase activity of the pre-treated PMNs in a time course. Furthermore, using the same technique the gelatinolytic activity in local and systemic citrate plasma samples was assessed, too.
Gelatine gel zymography of the above described supernatants was used for size discrimination between pro-matrix metalloproteinase (pro-MMP2)/MMP2 (80/72 kDa), pro-MMP9/MMP9 (92/84 kDa), and MMP9/neutrophil gelatinase–associated lipocalin (NGAL) complex (125 kDa) and performed as previously described by Toth et al.20
Immuno-enzymatic assay for detection of MMP9, NGAL, and hyaluronic acid plasma concentration
The concentrations of MMP9, NGAL, and hyaluronic acid (HA) in human plasma samples were assessed by commercially available ELISA kits (MMP 9 Human ELISA, Thermo Fischer, USA; NGAL ELISA Kit from Bioporto, Hamburg, and HA Test Kit, Corgenix, Broomfield, CO, USA, respectively). All tests were performed according to the manufacturer’s instructions.
Assessment of endothelial cell death and neutrophil survival in co-culture
1 × 106 PMNs, isolated and pre-treated as previously described, were co-cultured with 1 × 105 human aortic endothelial cells (HAECs) for 24 h in a humidified CO2 incubator (5% CO2, 37°C). HAECs without the addition of PMNs served as intra-experimental control. After the indicated exposure time, all cells were harvested by trypsinization and labelled with a cocktail of fluorochrome labeled antibodies, following measurement on an Attune Nxt Acoustic Focusing Flow Cytometer (Thermo Fisher Scientific, USA) and evaluation using Kaluza v1.5 (Beckman Coulter Inc., USA) analysis software. The neutrophil survival upon TLR2 activation was assessed directly by AnnexinV/Sytox Orange staining and indirectly by shedding of CD16.21 For the gating strategy, please refer to Supplementary data online, Material S4.
Effect of disturbed flow and recombinant MMP9 on endothelial detachment
HAECs were grown to 80% confluence on 1% gelatine pre-coated double-y-shaped microchannel slides (Ibidi, Gräfelfing, Germany) under laminar flow conditions (12 dyn/cm2 unidirectional flow). HAECs were either pre-treated with 500 ng/mL human recombinant MMP9 (Enzo Lifesciences, New York, USA) or control growth factor–deprived EBM medium. Endothelial cell detachment under different flow conditions: ‘laminar’ defined as unidirectional flow with 12 dyn/cm2 or ‘oscillatory’ defined as bidirectional flow with 12 dyn/cm2 was evaluated after 24 h using immunofluorescence stating for phalloidin for F-zctin and Hoechst for cell nuclei. Images showing the % detachment area were taken on a BZ-9000 inverted fluorescence phase contrast microscope (Keyence, USA) in 10× magnification and evaluated using ImageJ software version 1.48 (NIH/University of Wisconsin, USA).
Confocal microscopy immunofluorescence of intracoronary thrombus aspirates
Aspirated thrombus specimens from IFC–ACS and RFC–ACS patients (n = 20) were fixed in 4% paraformaldehyde solution, dehydrated in 30% sucrose solution, and embedded in OCT medium for long-term preservation at −80°C.
Assessment of HYAL2 expression in pre-flowed endothelial cell monolayers
Using reverse transcription polymerase chain reaction (RT-PCR), we assessed the gene expression of hyaluronidase 2 (HYAL2) in pre-flowed endothelial cell monolayers as described above. The relative protein expression of HYAL2 (Abcam, USA) to GAPDH (Cell Signaling, The Netherlands) was validated by western blot analysis.
Statistical analysis
Statistical analysis was based on a matched case–control study design. Distributions of variables were reported by standard descriptive statistics as described in the figure legends. Two group comparisons of continuously distributed variables were performed with Wilcoxon matched-pairs signed rank tests and categorical variables by Mc Nemar chi² test due to the case–control study design.22,23 Data with multiple readings within a patient were analyzed by a repeated measures analysis of variance including a factor (group, e.g. plaque phenotype). The overall effect of group, time, and interaction group × time was reported. Multiple post-hoc comparisons were performed in order to test for group differences (e.g. pathology) at each time point with a Bonferroni‘s correction for multiple comparisons in post-hoc tests. A two-sided P < 0.05 was considered to be statistically significant.
In addition, we reported the effect sizes Cohen’s d (standardized mean difference [SMD])24 for pairwise comparisons with rules of thumb to evaluate the magnitude of the effect size d reported in the figure legends.25 For ANOVA with repeated measures, we calculated the partial η² as an appropriate effect measure with a rule of thumb in the figure legends.25 Partial η² depicts the extent to which the total variance of the outcome (e.g. MMP9 activity) is explained by the covariates (e.g. time and pathology). Statistical analysis was performed with GraphPad Prism Software (La Jolla, CA) and SAS 9.3.
For detailed description of the methods, please refer to Supplementary data online, Material S5.
Results
Patients clinical characteristics
The baseline characteristics of the groups were similar, except for the expected higher proportion of dyslipidemia with higher total and LDL cholesterol (Table 1) in patients with RFC–ACS, which indeed is the pathophysiological hallmark of a RFC.8,26 We observed a higher pre-clinical use of statins in RFC–ACS vs. IFC–ACS (Table 1). Furthermore, the matched cohort was considered representative for the main study cohort of the OPTICO–ACS trial (see Supplementary data online, Material S6) and well balanced as shown by the SMDs of the continuous baseline characteristics (Table 2).
Clinical baseline characteristics of the study cohort. For comparison between categorical parameters, a McNemar test was used; for comparison between continuously distributed parameters, the Wilcoxon matched-pairs signed rank test was used. Values are expressed as a median with interquartile range (IQR); n = 32 per group
| RFC–ACS (n = 32) | IFC–ACS (n = 32) | P value | |
|---|---|---|---|
| Patient characteristics | |||
| Age (years) | 62.0 ± 16.75 | 61.0 ± 16.5 | 0.793 |
| Male (n;%) | 26 (81) | 26 (81) | 0.999 |
| Family history for CAD (n;%) | 14 (44) | 14 (44) | 0.999 |
| Active smoking (n;%) | 13 (40) | 17 (53) | 0.459 |
| Diabetes mellitus type 2 (n;%) | 7 (22) | 7 (22) | 0.999 |
| Arterial hypertension (n;%) | 29 (91) | 26 (81) | 0.214 |
| Dyslipidemiaa (n;%) | 30 (94) | 22 (68) | 0.002 |
| BMI (kg/m2) | 25.5 ± 6.1 | 25.6 ± 3.9 | 0.652 |
| Previous history of PCI (n;%) | 6 (19) | 1 (3) | 0.071 |
| Laboratory data | |||
| Total cholesterol (mg/l) | 204.0 ± 61.0 | 179.0 ± 50.2 | 0.040 |
| LDL cholesterol (mg/l) | 129.0 ± 53.0 | 113.0 ± 44.5 | 0.026 |
| HDL cholesterol (mg/l) | 40.0 ± 14.0 | 50.0 ± 20.0 | 0.417 |
| Serum creatinine (mg/l) | 0.94 ± 0.2 | 0.97 ± 0.2 | 0.256 |
| Hemoglobin (g/dl) | 14.7 ± 1.8 | 14.8 ± 1.6 | 0.681 |
| Leukocytes (per nl) | 10.5 ± 4.3 | 11.0 ± 6.0 | 0.367 |
| hsCRP (mg/l) | 2.1 ± 5.4 | 2.0 ± 3.2 | 0.721 |
| ACS characteristics | |||
| Presentation as STE–ACS (n;%) | 20 (63) | 18 (50) | 0.298 |
| CK peak (U/I) | 902.0 ± 720 | 569.0 ± 1.063 | 0.267 |
| LVEF at discharge (%) | 59.0 ± 11 | 55.0 ± 12.5 | 0.382 |
| Concomitant medication (n; %) | |||
| PCSK9 inhibitor | 0 (0) | 0 (0) | 0.241 |
| Ezetimibe | 0 (0) | 1 (3.1) | 0.191 |
| Statin | 3 (9.4) | 1 (3.1) | 0.010 |
| RFC–ACS, ruptured fibrous cap—acute coronary syndrome; IFC–ACS, intact fibrous cap acute coronary syndrome; SD, standard deviation; n, number; BMI, body mass index; LVEF, left ventricular ejection fraction; hsCRP, high-sensitivity C-reactive protein; CK, creatinine kinase; LDL, low-density lipoprotein; HDL, high-density lipoprotein; STE–ACS, ST-elevation myocardial infarction; PCI, percutaneous coronary intervention. aDefined as TC ≥ 220 mg/dl, triglycerides ≥ 150 mg/dl, LDL-C ≥ 140 mg/dl, HDL-C ≤ 40 mg/dl or taking medication for dyslipidemia. Bold values represent statistically significant results with a p value < 0.05. | |||
| RFC–ACS (n = 32) | IFC–ACS (n = 32) | P value | |
|---|---|---|---|
| Patient characteristics | |||
| Age (years) | 62.0 ± 16.75 | 61.0 ± 16.5 | 0.793 |
| Male (n;%) | 26 (81) | 26 (81) | 0.999 |
| Family history for CAD (n;%) | 14 (44) | 14 (44) | 0.999 |
| Active smoking (n;%) | 13 (40) | 17 (53) | 0.459 |
| Diabetes mellitus type 2 (n;%) | 7 (22) | 7 (22) | 0.999 |
| Arterial hypertension (n;%) | 29 (91) | 26 (81) | 0.214 |
| Dyslipidemiaa (n;%) | 30 (94) | 22 (68) | 0.002 |
| BMI (kg/m2) | 25.5 ± 6.1 | 25.6 ± 3.9 | 0.652 |
| Previous history of PCI (n;%) | 6 (19) | 1 (3) | 0.071 |
| Laboratory data | |||
| Total cholesterol (mg/l) | 204.0 ± 61.0 | 179.0 ± 50.2 | 0.040 |
| LDL cholesterol (mg/l) | 129.0 ± 53.0 | 113.0 ± 44.5 | 0.026 |
| HDL cholesterol (mg/l) | 40.0 ± 14.0 | 50.0 ± 20.0 | 0.417 |
| Serum creatinine (mg/l) | 0.94 ± 0.2 | 0.97 ± 0.2 | 0.256 |
| Hemoglobin (g/dl) | 14.7 ± 1.8 | 14.8 ± 1.6 | 0.681 |
| Leukocytes (per nl) | 10.5 ± 4.3 | 11.0 ± 6.0 | 0.367 |
| hsCRP (mg/l) | 2.1 ± 5.4 | 2.0 ± 3.2 | 0.721 |
| ACS characteristics | |||
| Presentation as STE–ACS (n;%) | 20 (63) | 18 (50) | 0.298 |
| CK peak (U/I) | 902.0 ± 720 | 569.0 ± 1.063 | 0.267 |
| LVEF at discharge (%) | 59.0 ± 11 | 55.0 ± 12.5 | 0.382 |
| Concomitant medication (n; %) | |||
| PCSK9 inhibitor | 0 (0) | 0 (0) | 0.241 |
| Ezetimibe | 0 (0) | 1 (3.1) | 0.191 |
| Statin | 3 (9.4) | 1 (3.1) | 0.010 |
| RFC–ACS, ruptured fibrous cap—acute coronary syndrome; IFC–ACS, intact fibrous cap acute coronary syndrome; SD, standard deviation; n, number; BMI, body mass index; LVEF, left ventricular ejection fraction; hsCRP, high-sensitivity C-reactive protein; CK, creatinine kinase; LDL, low-density lipoprotein; HDL, high-density lipoprotein; STE–ACS, ST-elevation myocardial infarction; PCI, percutaneous coronary intervention. aDefined as TC ≥ 220 mg/dl, triglycerides ≥ 150 mg/dl, LDL-C ≥ 140 mg/dl, HDL-C ≤ 40 mg/dl or taking medication for dyslipidemia. Bold values represent statistically significant results with a p value < 0.05. | |||
| RFC-ACS (n = 32) | IFC–ACS (n = 32) | P value | |
|---|---|---|---|
| Culprit segment TIMI flow < 1 (n; %) | 11 (34%) | 8 (25%) | 0.219 |
| Culprit vessel (n; %) | |||
| LM | 0 (0) | 1 (3) | 0.241 |
| LAD | 15 (47) | 16 (50) | |
| LCX | 4 (13) | 6 (19) | |
| RCA | 13 (41) | 9 (28) | |
| Involved culprit segment (n; %) | |||
| Proximal | 8 (25) | 18 (56) | 0.06 |
| Medial | 23 (72) | 12 (38) | |
| Distal | 1 (3) | 2 (6) | |
| Number of diseased vessels (n; %) | |||
| 1 | 16 (50) | 22 (69) | 0.284 |
| 2 | 9 (28) | 6 (19) | |
| 3 | 7 (22) | 4 (13) | |
| PCI characteristics | |||
| Total stented length (mm) | 26.0 ± 30.6 | 23.0 ± 11.5 | 0.362 |
| Largest stent diameter (mm) | 3.5 ± 0.50 | 3.4 ± 1.0 | 0.992 |
| RFC-ACS (n = 32) | IFC–ACS (n = 32) | P value | |
|---|---|---|---|
| Culprit segment TIMI flow < 1 (n; %) | 11 (34%) | 8 (25%) | 0.219 |
| Culprit vessel (n; %) | |||
| LM | 0 (0) | 1 (3) | 0.241 |
| LAD | 15 (47) | 16 (50) | |
| LCX | 4 (13) | 6 (19) | |
| RCA | 13 (41) | 9 (28) | |
| Involved culprit segment (n; %) | |||
| Proximal | 8 (25) | 18 (56) | 0.06 |
| Medial | 23 (72) | 12 (38) | |
| Distal | 1 (3) | 2 (6) | |
| Number of diseased vessels (n; %) | |||
| 1 | 16 (50) | 22 (69) | 0.284 |
| 2 | 9 (28) | 6 (19) | |
| 3 | 7 (22) | 4 (13) | |
| PCI characteristics | |||
| Total stented length (mm) | 26.0 ± 30.6 | 23.0 ± 11.5 | 0.362 |
| Largest stent diameter (mm) | 3.5 ± 0.50 | 3.4 ± 1.0 | 0.992 |
RFC–ACS, ruptured fibrous cap—acute coronary syndrome; IFC–ACS, intact fibrous cap acute coronary syndrome; SD, standard deviation; TIMI, thrombolysis in myocardial infarction; LM, left main; LAD, left anterior descending artery; LCX, left circumflex artery; RCA, right coronary artery.
Clinical baseline characteristics of the study cohort. For comparison between categorical parameters, a McNemar test was used; for comparison between continuously distributed parameters, the Wilcoxon matched-pairs signed rank test was used. Values are expressed as a median with interquartile range (IQR); n = 32 per group
| RFC–ACS (n = 32) | IFC–ACS (n = 32) | P value | |
|---|---|---|---|
| Patient characteristics | |||
| Age (years) | 62.0 ± 16.75 | 61.0 ± 16.5 | 0.793 |
| Male (n;%) | 26 (81) | 26 (81) | 0.999 |
| Family history for CAD (n;%) | 14 (44) | 14 (44) | 0.999 |
| Active smoking (n;%) | 13 (40) | 17 (53) | 0.459 |
| Diabetes mellitus type 2 (n;%) | 7 (22) | 7 (22) | 0.999 |
| Arterial hypertension (n;%) | 29 (91) | 26 (81) | 0.214 |
| Dyslipidemiaa (n;%) | 30 (94) | 22 (68) | 0.002 |
| BMI (kg/m2) | 25.5 ± 6.1 | 25.6 ± 3.9 | 0.652 |
| Previous history of PCI (n;%) | 6 (19) | 1 (3) | 0.071 |
| Laboratory data | |||
| Total cholesterol (mg/l) | 204.0 ± 61.0 | 179.0 ± 50.2 | 0.040 |
| LDL cholesterol (mg/l) | 129.0 ± 53.0 | 113.0 ± 44.5 | 0.026 |
| HDL cholesterol (mg/l) | 40.0 ± 14.0 | 50.0 ± 20.0 | 0.417 |
| Serum creatinine (mg/l) | 0.94 ± 0.2 | 0.97 ± 0.2 | 0.256 |
| Hemoglobin (g/dl) | 14.7 ± 1.8 | 14.8 ± 1.6 | 0.681 |
| Leukocytes (per nl) | 10.5 ± 4.3 | 11.0 ± 6.0 | 0.367 |
| hsCRP (mg/l) | 2.1 ± 5.4 | 2.0 ± 3.2 | 0.721 |
| ACS characteristics | |||
| Presentation as STE–ACS (n;%) | 20 (63) | 18 (50) | 0.298 |
| CK peak (U/I) | 902.0 ± 720 | 569.0 ± 1.063 | 0.267 |
| LVEF at discharge (%) | 59.0 ± 11 | 55.0 ± 12.5 | 0.382 |
| Concomitant medication (n; %) | |||
| PCSK9 inhibitor | 0 (0) | 0 (0) | 0.241 |
| Ezetimibe | 0 (0) | 1 (3.1) | 0.191 |
| Statin | 3 (9.4) | 1 (3.1) | 0.010 |
| RFC–ACS, ruptured fibrous cap—acute coronary syndrome; IFC–ACS, intact fibrous cap acute coronary syndrome; SD, standard deviation; n, number; BMI, body mass index; LVEF, left ventricular ejection fraction; hsCRP, high-sensitivity C-reactive protein; CK, creatinine kinase; LDL, low-density lipoprotein; HDL, high-density lipoprotein; STE–ACS, ST-elevation myocardial infarction; PCI, percutaneous coronary intervention. aDefined as TC ≥ 220 mg/dl, triglycerides ≥ 150 mg/dl, LDL-C ≥ 140 mg/dl, HDL-C ≤ 40 mg/dl or taking medication for dyslipidemia. Bold values represent statistically significant results with a p value < 0.05. | |||
| RFC–ACS (n = 32) | IFC–ACS (n = 32) | P value | |
|---|---|---|---|
| Patient characteristics | |||
| Age (years) | 62.0 ± 16.75 | 61.0 ± 16.5 | 0.793 |
| Male (n;%) | 26 (81) | 26 (81) | 0.999 |
| Family history for CAD (n;%) | 14 (44) | 14 (44) | 0.999 |
| Active smoking (n;%) | 13 (40) | 17 (53) | 0.459 |
| Diabetes mellitus type 2 (n;%) | 7 (22) | 7 (22) | 0.999 |
| Arterial hypertension (n;%) | 29 (91) | 26 (81) | 0.214 |
| Dyslipidemiaa (n;%) | 30 (94) | 22 (68) | 0.002 |
| BMI (kg/m2) | 25.5 ± 6.1 | 25.6 ± 3.9 | 0.652 |
| Previous history of PCI (n;%) | 6 (19) | 1 (3) | 0.071 |
| Laboratory data | |||
| Total cholesterol (mg/l) | 204.0 ± 61.0 | 179.0 ± 50.2 | 0.040 |
| LDL cholesterol (mg/l) | 129.0 ± 53.0 | 113.0 ± 44.5 | 0.026 |
| HDL cholesterol (mg/l) | 40.0 ± 14.0 | 50.0 ± 20.0 | 0.417 |
| Serum creatinine (mg/l) | 0.94 ± 0.2 | 0.97 ± 0.2 | 0.256 |
| Hemoglobin (g/dl) | 14.7 ± 1.8 | 14.8 ± 1.6 | 0.681 |
| Leukocytes (per nl) | 10.5 ± 4.3 | 11.0 ± 6.0 | 0.367 |
| hsCRP (mg/l) | 2.1 ± 5.4 | 2.0 ± 3.2 | 0.721 |
| ACS characteristics | |||
| Presentation as STE–ACS (n;%) | 20 (63) | 18 (50) | 0.298 |
| CK peak (U/I) | 902.0 ± 720 | 569.0 ± 1.063 | 0.267 |
| LVEF at discharge (%) | 59.0 ± 11 | 55.0 ± 12.5 | 0.382 |
| Concomitant medication (n; %) | |||
| PCSK9 inhibitor | 0 (0) | 0 (0) | 0.241 |
| Ezetimibe | 0 (0) | 1 (3.1) | 0.191 |
| Statin | 3 (9.4) | 1 (3.1) | 0.010 |
| RFC–ACS, ruptured fibrous cap—acute coronary syndrome; IFC–ACS, intact fibrous cap acute coronary syndrome; SD, standard deviation; n, number; BMI, body mass index; LVEF, left ventricular ejection fraction; hsCRP, high-sensitivity C-reactive protein; CK, creatinine kinase; LDL, low-density lipoprotein; HDL, high-density lipoprotein; STE–ACS, ST-elevation myocardial infarction; PCI, percutaneous coronary intervention. aDefined as TC ≥ 220 mg/dl, triglycerides ≥ 150 mg/dl, LDL-C ≥ 140 mg/dl, HDL-C ≤ 40 mg/dl or taking medication for dyslipidemia. Bold values represent statistically significant results with a p value < 0.05. | |||
| RFC-ACS (n = 32) | IFC–ACS (n = 32) | P value | |
|---|---|---|---|
| Culprit segment TIMI flow < 1 (n; %) | 11 (34%) | 8 (25%) | 0.219 |
| Culprit vessel (n; %) | |||
| LM | 0 (0) | 1 (3) | 0.241 |
| LAD | 15 (47) | 16 (50) | |
| LCX | 4 (13) | 6 (19) | |
| RCA | 13 (41) | 9 (28) | |
| Involved culprit segment (n; %) | |||
| Proximal | 8 (25) | 18 (56) | 0.06 |
| Medial | 23 (72) | 12 (38) | |
| Distal | 1 (3) | 2 (6) | |
| Number of diseased vessels (n; %) | |||
| 1 | 16 (50) | 22 (69) | 0.284 |
| 2 | 9 (28) | 6 (19) | |
| 3 | 7 (22) | 4 (13) | |
| PCI characteristics | |||
| Total stented length (mm) | 26.0 ± 30.6 | 23.0 ± 11.5 | 0.362 |
| Largest stent diameter (mm) | 3.5 ± 0.50 | 3.4 ± 1.0 | 0.992 |
| RFC-ACS (n = 32) | IFC–ACS (n = 32) | P value | |
|---|---|---|---|
| Culprit segment TIMI flow < 1 (n; %) | 11 (34%) | 8 (25%) | 0.219 |
| Culprit vessel (n; %) | |||
| LM | 0 (0) | 1 (3) | 0.241 |
| LAD | 15 (47) | 16 (50) | |
| LCX | 4 (13) | 6 (19) | |
| RCA | 13 (41) | 9 (28) | |
| Involved culprit segment (n; %) | |||
| Proximal | 8 (25) | 18 (56) | 0.06 |
| Medial | 23 (72) | 12 (38) | |
| Distal | 1 (3) | 2 (6) | |
| Number of diseased vessels (n; %) | |||
| 1 | 16 (50) | 22 (69) | 0.284 |
| 2 | 9 (28) | 6 (19) | |
| 3 | 7 (22) | 4 (13) | |
| PCI characteristics | |||
| Total stented length (mm) | 26.0 ± 30.6 | 23.0 ± 11.5 | 0.362 |
| Largest stent diameter (mm) | 3.5 ± 0.50 | 3.4 ± 1.0 | 0.992 |
RFC–ACS, ruptured fibrous cap—acute coronary syndrome; IFC–ACS, intact fibrous cap acute coronary syndrome; SD, standard deviation; TIMI, thrombolysis in myocardial infarction; LM, left main; LAD, left anterior descending artery; LCX, left circumflex artery; RCA, right coronary artery.
Standardized mean differences (SMDs, d) reported for continuous baseline characteristics show well-balanced matched populations; effect size d (absolute values of d: small effect d < 0.2, medium effect d between 0.2 and 0.8, large effect d > 0.8). Values are expressed as a median with interquartile range (IQR); n = 32 per group
| RFC–ACS | IFC–ACS | d | |
|---|---|---|---|
| PCI characteristics | |||
| Total stented length (mm) | 26.0 ± 30.6 | 23.0 ± 11.5 | 0.08 |
| Largest stent diameter (mm) | 3.5 ± 0.50 | 3.4 ± 1.0 | 0.05 |
| Patient characteristics | |||
| Age (years) | 62.0 ± 16.75 | 61.0 ± 16.5 | 0.04 |
| BMI (kg/m2) | 25.5 ± 6.1 | 25.6 ± 3.9 | 0.08 |
| Laboratory data | |||
| Total cholesterol (mg/l) | 204.0 ± 61.0 | 179.0 ± 50.2 | 0.49 |
| LDL cholesterol (mg/l) | 129.0 ± 53.0 | 113.0 ± 44.5 | 0.56 |
| HDL cholesterol (mg/l) | 40.0 ± 14.0 | 50.0 ± 20.0 | 0.23 |
| Serum creatinine (mg/l) | 0.94 ± 0.2 | 0.97 ± 0.2 | 0.17 |
| Hemoglobin (g/dl) | 14.7 ± 1.8 | 14.8 ± 1.6 | 0.09 |
| Leukocytes (per nl) | 10.5 ± 4.3 | 11.0 ± 6.0 | 0.16 |
| hsCRP (mg/l) | 2.1 ± 5.4 | 2.0 ± 3.2 | 0.19 |
| ACS characteristics | |||
| CK peak (U/I) | 902.0 ± 720 | 569.0 ± 1.063 | 0.13 |
| RFC–ACS | IFC–ACS | d | |
|---|---|---|---|
| PCI characteristics | |||
| Total stented length (mm) | 26.0 ± 30.6 | 23.0 ± 11.5 | 0.08 |
| Largest stent diameter (mm) | 3.5 ± 0.50 | 3.4 ± 1.0 | 0.05 |
| Patient characteristics | |||
| Age (years) | 62.0 ± 16.75 | 61.0 ± 16.5 | 0.04 |
| BMI (kg/m2) | 25.5 ± 6.1 | 25.6 ± 3.9 | 0.08 |
| Laboratory data | |||
| Total cholesterol (mg/l) | 204.0 ± 61.0 | 179.0 ± 50.2 | 0.49 |
| LDL cholesterol (mg/l) | 129.0 ± 53.0 | 113.0 ± 44.5 | 0.56 |
| HDL cholesterol (mg/l) | 40.0 ± 14.0 | 50.0 ± 20.0 | 0.23 |
| Serum creatinine (mg/l) | 0.94 ± 0.2 | 0.97 ± 0.2 | 0.17 |
| Hemoglobin (g/dl) | 14.7 ± 1.8 | 14.8 ± 1.6 | 0.09 |
| Leukocytes (per nl) | 10.5 ± 4.3 | 11.0 ± 6.0 | 0.16 |
| hsCRP (mg/l) | 2.1 ± 5.4 | 2.0 ± 3.2 | 0.19 |
| ACS characteristics | |||
| CK peak (U/I) | 902.0 ± 720 | 569.0 ± 1.063 | 0.13 |
Standardized mean differences (SMDs, d) reported for continuous baseline characteristics show well-balanced matched populations; effect size d (absolute values of d: small effect d < 0.2, medium effect d between 0.2 and 0.8, large effect d > 0.8). Values are expressed as a median with interquartile range (IQR); n = 32 per group
| RFC–ACS | IFC–ACS | d | |
|---|---|---|---|
| PCI characteristics | |||
| Total stented length (mm) | 26.0 ± 30.6 | 23.0 ± 11.5 | 0.08 |
| Largest stent diameter (mm) | 3.5 ± 0.50 | 3.4 ± 1.0 | 0.05 |
| Patient characteristics | |||
| Age (years) | 62.0 ± 16.75 | 61.0 ± 16.5 | 0.04 |
| BMI (kg/m2) | 25.5 ± 6.1 | 25.6 ± 3.9 | 0.08 |
| Laboratory data | |||
| Total cholesterol (mg/l) | 204.0 ± 61.0 | 179.0 ± 50.2 | 0.49 |
| LDL cholesterol (mg/l) | 129.0 ± 53.0 | 113.0 ± 44.5 | 0.56 |
| HDL cholesterol (mg/l) | 40.0 ± 14.0 | 50.0 ± 20.0 | 0.23 |
| Serum creatinine (mg/l) | 0.94 ± 0.2 | 0.97 ± 0.2 | 0.17 |
| Hemoglobin (g/dl) | 14.7 ± 1.8 | 14.8 ± 1.6 | 0.09 |
| Leukocytes (per nl) | 10.5 ± 4.3 | 11.0 ± 6.0 | 0.16 |
| hsCRP (mg/l) | 2.1 ± 5.4 | 2.0 ± 3.2 | 0.19 |
| ACS characteristics | |||
| CK peak (U/I) | 902.0 ± 720 | 569.0 ± 1.063 | 0.13 |
| RFC–ACS | IFC–ACS | d | |
|---|---|---|---|
| PCI characteristics | |||
| Total stented length (mm) | 26.0 ± 30.6 | 23.0 ± 11.5 | 0.08 |
| Largest stent diameter (mm) | 3.5 ± 0.50 | 3.4 ± 1.0 | 0.05 |
| Patient characteristics | |||
| Age (years) | 62.0 ± 16.75 | 61.0 ± 16.5 | 0.04 |
| BMI (kg/m2) | 25.5 ± 6.1 | 25.6 ± 3.9 | 0.08 |
| Laboratory data | |||
| Total cholesterol (mg/l) | 204.0 ± 61.0 | 179.0 ± 50.2 | 0.49 |
| LDL cholesterol (mg/l) | 129.0 ± 53.0 | 113.0 ± 44.5 | 0.56 |
| HDL cholesterol (mg/l) | 40.0 ± 14.0 | 50.0 ± 20.0 | 0.23 |
| Serum creatinine (mg/l) | 0.94 ± 0.2 | 0.97 ± 0.2 | 0.17 |
| Hemoglobin (g/dl) | 14.7 ± 1.8 | 14.8 ± 1.6 | 0.09 |
| Leukocytes (per nl) | 10.5 ± 4.3 | 11.0 ± 6.0 | 0.16 |
| hsCRP (mg/l) | 2.1 ± 5.4 | 2.0 ± 3.2 | 0.19 |
| ACS characteristics | |||
| CK peak (U/I) | 902.0 ± 720 | 569.0 ± 1.063 | 0.13 |
Differences in culprit plaque morphology assessed by optical coherence tomography
In line with the higher proportion of dyslipidemia and culprit features presented recently for the entire OPTICO–ACS study cohort8 as well as other studies,27 RFC–ACS–causing culprit plaques were predominantly thin-cap fibroatheromas and were characterized by larger maximum lipid arcs when compared with IFC–ACS culprit lesions (see Supplementary data online, Material S7).
Upregulation of TLR2 on neutrophils in IFC–ACS
Absolute neutrophil cell counts and the relative distribution as a ratio of LOC vs. SYS neutrophils were not different between patients with IFC–ACS and RFC–ACS (Figure 1A, Supplementary data online, Material S8). However, higher expression levels of TLR2 were detected on neutrophils from IFC–ACS patients compared with RFC–ACS patients (Figure 1B). The expression levels of the other neutrophil receptors from our pre-defined panel between IFC–ACS and RFC–ACS were similar (see Supplementary data online, Material S9), as were the distribution of the monocyte subpopulations and their TLR2 expression between both disease groups (see Supplementary data online, Material S10 and S11). Consequently, in subsequent experiments, we focused on the effect of an in vitro TLR2 stimulation of the neutrophils.
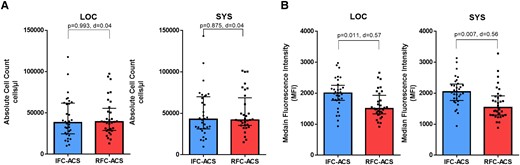
Flow cytometry analysis of neutrophil surface receptors shows equal neutrophil cell count in IFC–ACS vs. RFC–ACS (A), but higher TLR2 expression on local and systemic neutrophils from IFC–ACS patients vs. RFC–ACS patients (B). Wilcoxon matched-pairs signed rank test; effect size d (absolute values of d: small effect d < 0.2, medium effect d between 0.2 and 0.8, large effect d > 0.8); data are shown as median with interquartile range (IQR); n = 32 per each group. MFI, median fluorescence intensity; RFC–ACS, acute coronary syndrome with ruptured fibrous cap; IFC–ACS, acute coronary syndrome with intact fibrous cap; LOC, local; SYS, systemic; d, Cohen’s.
Increased local and systemic MMP9 concentration in IFC–ACS vs. RFC–ACS
Another surrogate marker for higher neutrophil-driven inflammatory activity in IFC–ACS was the higher local and systemic MMP9 concentration in the plasma samples (Figure 2A and B), as neutrophils represent not the only, but the largest immediate source of MMP9 after a myocardial infarction.28,29 Furthermore, local plasma samples from IFC–ACS patients showed higher gelatinolytic activity in comparison to RFC–ACS patients (Figure 2C). This effect was not observed in systemic samples (Figure 2D).
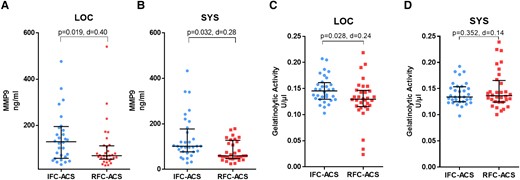
MMP9 concentration in local (A) and systemic (B) plasma samples from matched patients with IFC–ACS and RFC–ACS, measured in ng/mL; gelatinolytic activity in local (C) and systemic (D) plasma samples from matched IFC–ACS and RFC–ACS patients, measured in U/µl. Wilcoxon matched-pairs signed rank test; effect size d (absolute values of d: small effect d < 0.2, medium effect d between 0.2 and 0.8, large effect d > 0.8); data are presented as median with interquartile range (IQR); n = 32 per group. MMP9, matrix metalloproteinase 9; LOC, local; SYS, systemic; d = Cohen’s.
TLR2 activation specifically enhances release and activation of MMP9 from local IFC–ACS neutrophils
Upon TLR2 activation with Pam3CSK4, LOC IFC–ACS–derived neutrophils increased their gelatinolytic activity as compared with lesion-site RFC–ACS neutrophils (Figure 3A) and SYS IFC–ACS neutrophils (Figure 3B). This effect was observed neither in SYS neutrophils (Figure 3C) nor in RFC–ACS neutrophils in general (Figure 3D). Exemplary gelatine zymography supported MMP9 to be the predominant gelatinase in the supernatants of the stimulated neutrophils (Figure 3E). Furthermore, the local plasma samples from IFC–ACS patients showed an increased concentration of NGAL (Figure 3F), which prevents MMP9 inactivation.30
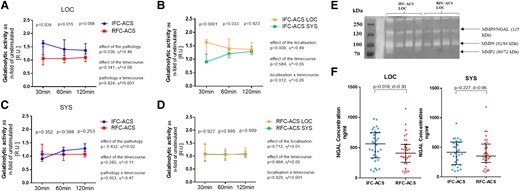
Higher gelatinolytic activity independent of the time course in local IFC–ACS (n = 8) vs. RFC–ACS TLR2 pre-treated neutrophils (n = 8) (A) and in local vs. systemic IFC–ACS neutrophils (B). TLR2 activation has no significant effect on the gelatinolytic activity in systemic IFC–ACS vs. RFC–ACS neutrophils (C) and in local vs. systemic RFC–ACS neutrophils (D); gelatinolytic activity measured in RU and expressed as n-fold of the unstimulated control group; representative graphic of gelatinolytic activity of supernatants of TLR2 pre-stimulated neutrophils assessed by gel zymography showing the pre-dominant MMP9 (E); local plasma samples from IFC–ACS patients show higher NGAL concentration than RFC–ACS patients as estimated by a Wilcoxon matched-pairs signed rank test, n = 32 per group (F); for all of the other comparisons, a repeated measures ANOVA with Bonferroni‘s multiple comparisons test has been used; data are shown as median ± interquartile range (IQR); effect size η² after repeated measures ANOVA (small effect η² < 0.01, medium effect η² between 0.01 and 0.14, large effect η² > 0.14), effect size d (absolute values of d: small effect d < 0.2, medium effect d between 0.2 and 0.8, large effect d > 0.8). RFC–ACS, acute coronary syndrome with ruptured fibrous cap; IFC–ACS, acute coronary syndrome with intact fibrous cap; RU, relative units; LOC, local; SYS, systemic; NGAL, neutrophil gelatinase-associated lipocalin; d = Cohen’s.
Upon TLR2 blockade by an anti-TLR2 antibody, MMP9 activity was not enhanced by Pam3CSK4 stimulation anymore, underlining the specific relation between the TLR2 activation and release of active MMP9 by neutrophils at the culprit lesion site of patients with IFC–ACS (Figure 4A). This effect was not observed in RFC–ACS–derived neutrophils in general and SYS IFC–ACS–derived neutrophils (Figure 4B, Supplementary data online, Material S12).
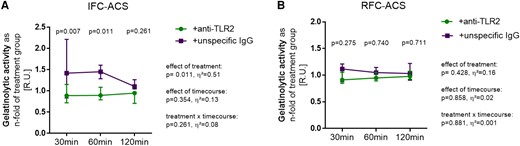
TLR2 inhibition by a monoclonal antibody against TLR2 leads to markedly decreased MMP9 activity in local neutrophils from IFC–ACS (A) but not RFC–ACS patients (B) measured in RU and expressed as n-fold of the TLR2-stimulated group (+Pam3CSK4); n = 6 per group; repeated measures ANOVA with Bonferroni‘s multiple comparisons test; data are shown as median ± interquartile range (IQR); effect size η² after repeated measures ANOVA (small effect η² < 0.01, medium effect η² between 0.01 and 0.14, large effect η² > 0.14).
Aggravated endothelial cell detachment by MMP9 and oscillatory flow
Turbulent flow conditions aggravated endothelial cell detachment exclusively in combination with MMP9 (Figure 5).
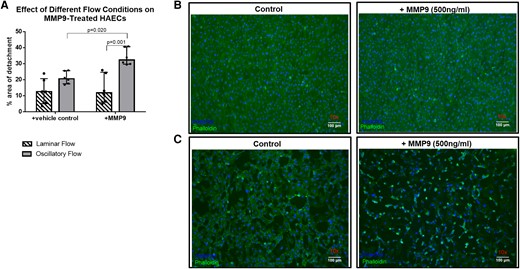
The combination of MMP9 and turbulent (oscillatory) flow leads to increased detachment of HAECs in vitro (A). Histological sections of HAECs showing normal detachment under laminar flow conditions (B) and increased detachment under proatherogenic oscillatory conditions, especially when combined with recombinant MMP9 (C); n = 6 per group; repeated measures ANOVA with Bonferroni‘s multiple comparisons test; data are shown as median with interquartile range (IQR).
TLR2 activation prevents neutrophil apoptosis in local IFC–ACS–derived neutrophils
High CD16b expression as an indirect marker of neutrophil survival21,31,32 was maintained in LOC IFC–ACS–derived neutrophils upon TLR2 activation by Pam3CSK4 (Figure 6A). Moreover, in SYS IFC–ACS and RFC–ACS neutrophils in general (Figure 6A and B), TLR2 activation did not prevent neutrophil apoptosis. This data was supported by a flow cytometry assessment of neutrophil survival, where we observed a decrease in the apoptotic rate of overall and CD16lo IFC–ACS–derived neutrophils upon TLR2 activation in comparison to RFC–ACS neutrophils (see Supplementary data online, Material S13 and S14).
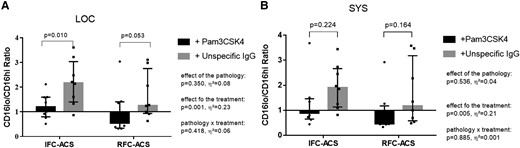
TLR2 activation with Pam3CSK4 increases survival in local IFC–ACS–derived neutrophils (A), but neither in systemic IFC–ACS neutrophils (B), nor in RFC–ACS–derived neutrophils in general (A for local, B for systemic); data are expressed as a ratio of CD16lo vs. CD16hi neutrophils. Repeated measures ANOVA with Bonferroni’s multiple comparisons test; data are presented as a median with interquartile range (IQR); n = 8 per group; effect size η² after repeated measures ANOVA (small effect η² < 0.01, medium effect η² between 0.01 and 0.14, large effect η² > 0.14). Pam3CSK4, Toll-like receptor 2 ligand; IFC–ACS, acute coronary syndrome with intact fibrous cap; RFC–ACS, acute coronary syndrome with ruptured fibrous cap.
Aggravated endothelial cell death in co-culture with local IFC–ACS neutrophils
Furthermore, LOC IFC–ACS, but not RFC–ACS–derived neutrophils, enhanced the rate of endothelial cell death, independently of TLR2 (Figure 7). Neutrophils from the systemic withdrawal site of IFC–ACS as compared with RFC–ACS did not affect endothelial cell survival (Figure 8).
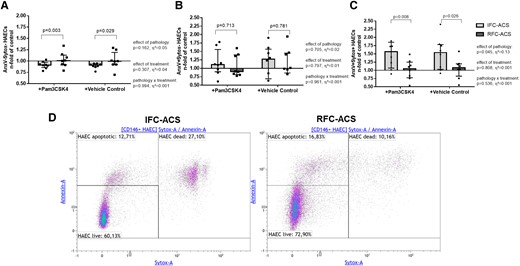
Increased rate of dead AnxV+Sytox + endothelial cells (C) in co-culture with either Pam3CSK4 pre-treated or non-treated (vehicle control) local neutrophils from IFC–ACS patients, but not RFC–ACS patients; no effect on live AnxV-Sytox- HAECs (A) and apoptotic AnxV + Sytox- HAECs (B); representative images of the applied gating startegy (D); repeated measures ANOVA with Bonferroni’s multiple comparisons test; data are presented as a median with interquartile range (IQR); n = 8 per group; effect size η² after repeated measures ANOVA (small effect η² < 0.01, medium effect η² between 0.01 and 0.14, large effect η² > 0.14). Pam3CSK4, Toll-like receptor 2 ligand; AnxV, Annexin V; HAECs, human aortic endothelial cells.
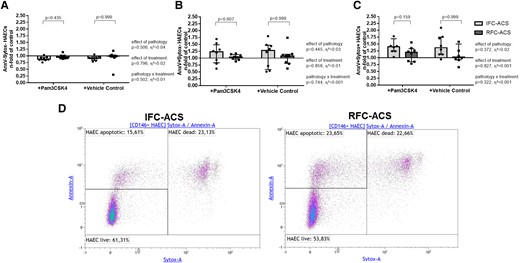
Constant rate of live (AnxV- Sytox-, A), apoptotic (AnxV+ Sytox-, B) and dead (AnxV + Sytox +, C) endothelial cells in co-culture with either Pam3CSK4 pre-treated or non-treated (vehicle control) systemic neutrophils from IFC–ACS and RFC–ACS patients; representative images of the applied gating strategy (D); repeated measures ANOVA with Bonferroni‘s multiple comparisons test; data are presented as a median with interquartile range (IQR); n = 8 per group; effect size η² after repeated measures ANOVA (small effect η² < 0.01, medium effect η² between 0.01 and 0.14, large effect η² > 0.14). Pam3CSK4, Toll-like receptor 2 ligand; AnxV, annexin V; HAECs, human aortic endothelial cells; IFC–ACS, acute coronary syndrome with intact fibrous cap; RFC–ACS, acute coronary syndrome with ruptured fibrous cap.
Elevated local hyaluronic acid concentration in IFC–ACS as endogenous activator of TLR2
In line with the observed selective TLR2 activation of LOC neutrophils from IFC–ACS patients, the immuno-enzymatic assays showed that the plasma concentration of HA, a well-known endogenous TLR2 ligand,33 was similar at SYS levels between patients presenting with IFC–ACS and RFC–ACS, while its LOC plasma levels were slightly higher in the IFC–ACS group (Figure 9).
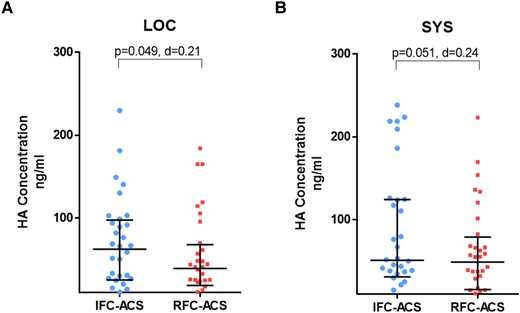
Scatter plots with bars showing HA concentrations (ng/mL) in local (A) and systemic (B) plasma samples from matched IFC–ACS vs. RFC–ACS patients. Wilcoxon matched-pairs signed rank test; effect size d (absolute values of d: small effect d < 0.2, medium effect d between 0.2 and 0.8, large effect d > 0.8); data are presented as median with interquartile range (IQR); n = 32 per group. HA, hyaluronan; LOC, local; SYS, systemic; d = Cohen’s.
Thrombus aspirates from IFC–ACS patients display high HYAL2 levels and a characteristic HYAL2 distribution
Confocal imaging of thrombus aspirates revealed that specimen collected from patients with IFC–ACS show higher HYAL2 expression than that from RFC–ACS patients, observed mainly at the outer surface of the thrombus, which was exposed to the blood stream (Figure 10). Exemplary thrombus sections for IFC–ACS and RFC–ACS are shown in Supplementary data online, Material S15. Furthermore, a trend towards higher local presence of neutrophils in IFC–ACS thrombi was observed by co-staining of neutrophil elastase (NE) and 4′,6-diamidino-2-phenylindole (DAPI) (see Supplementary data online, Material S16). Exemplary images of IFC–ACS thrombi implicate co-localization of HYAL2 and neutrophils in the border zones of the thrombi (white arrows in Supplementary data online, Material S16).
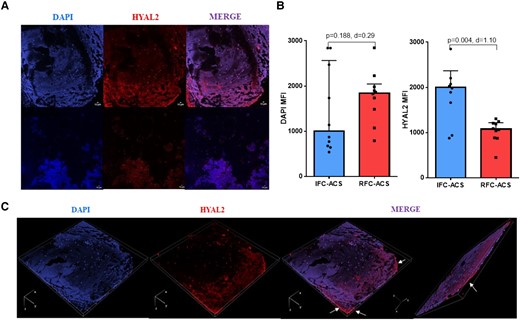
(A) Immunofluorescence analyses assessed by confocal microscopy showing DAPI and HYAL2 levels. Samples were stained with DAPI and HYAL2 antibody . Merge of the two labels is showed as composite image (co-expression of DAPI and HYAL2). Bars represent 10 μm. Images were taken at 20× magnification and at resolutions of 512 × 512. (B) Scatter plot with bars showing the DAPI and HYAL2 expressed as MFI (C) 3D representative projection of a sample of a coronary thrombus aspirated from a patient with plaque erosion created through a multiple stack series acquisition (dimension order: XYCZT). White arrows indicate the area of HYAL2 intense immunoreactivity; Wilcoxon matched-pairs signed rank test; effect size d (absolute values of d: small effect d < 0.2, medium effect d between 0.2 and 0.8, large effect d > 0.8); data are presented as median with interquartile range (IQR). DAPI, 4′,6-diamidino-2-phenylindole; HYAL2, hyaluronidase-2; mm, micrometer; MFI, mean fluorescence intensity; d = Cohen’s.
Endothelial cells do not upregulate HYAL2 under oscillatory shear stress
Under oscillatory flow conditions, endothelial cells interestingly reduce HYAL2 mRNA expression but do not downregulate the protein expression of HYAL2 in comparison to laminar flow conditions (see Supplementary data online, Material S17), thus pointing to another cell source of HYAL2 in the context of plaque erosion.
Discussion
In contrast to the well-characterized pathophysiology of RFC–ACS with the main target at the optimization of lipid accumulation, IFC–ACS represents an ACS–causing entity with rising prevalence, but still poorly understood underlying mechanisms, involving genetic/epigenetic pre-disposition, disturbed flow, and particularly distinct inflammatory activation patterns.8,10,34 With the development of intracoronary imaging, it has become feasible to characterize the morphology of the culprit lesion in vivo and thereby to distinguish between different ACS–causing pathologies in real time and not only post-mortem.35 This allows for development of precise-targeted treatment approaches for different ACS patient populations, which itself requires better understanding of the underlying pathophysiological mechanisms.
Since neutrophils have been proposed to be a key initiator of the progression and destabilization of the ACS–causing culprit plaques,10 our study focuses on the differences in the neutrophil activation between the two main ACS–causing pathophysiologies (IFC- and RFC–ACS). The study implicates an increased TLR2-dependent neutrophil activation and MMP9 release, presumably triggered by elevated HA and enhanced by disturbed flow conditions in patients with IFC–ACS (Structured Graphical Abstract). The provided in-patient functional data sharpen our mechanistic understanding of IFC–ACS and propose future prevention and therapeutic targets for this patient population.
Neutrophil activation in IFC–ACS: a question of functionality and not cell count
Neutrophil infiltration is both observed in atherosclerotic culprit lesions from RFC–ACS and IFC–ACS patients,36 but their exact functional properties in the context of these pathologies are still incompletely understood. Therefore, this study aimed to better characterize the activation patterns of human neutrophils from patients with IFC–ACS as compared with RFC–ACS.
While previous studies show mainly quantitative alterations, i.e. of neutrophil count in ACS,37 the current study highlights a different functionality with an increased TLR2 expression of the neutrophils in IFC–ACS, despite similar neutrophil abundance. Previous murine and in vitro data converge on a ‘two-hits’ model of plaque erosion: injured endothelium at localizations with high shear stress expresses higher levels of TLR2 and thereby acts as a trigger for the second hit: the neutrophil attraction and activation, which aggravates the endothelial cell detachment and thereby leads to plaque erosion.11 The current study advances this concept by providing complementary first-in-human data on IFC–ACS–derived neutrophils in a more disease-centered approach, showing specific TLR2-dependent mechanisms of neutrophil activation and MMP9 release and thereby suggesting potential therapeutic targets for this patient population.
TLR2-dependent neutrophil activation in IFC–ACS
The involvement of TLRs in sterile inflammatory diseases such as autoimmune diseases and atherosclerosis was postulated in the beginning of the 21st century by showing interactions with endogenous molecules like HA and other damage-associated molecular patterns.38 Intriguingly, histological evidence shows that especially IFC–ACS lesions contain high amounts of HA,33,39 suggesting that local exposure of HA and in particular low-molecular-weight hyaluronic acid (LMW-HA) may trigger innate immune response in plaque erosion via TLR2.33,39–41 LMW-HA is capable of inducing quick recruitment and activation of classical innate immune cells such as monocytes42 and neutrophils.33 In correspondence, we observed not only higher expression of TLR2 exclusively on local neutrophils and not monocytes from IFC–ACS patients but also higher abundance of HYAL2, the key enzyme responsible for the HA catabolism and release into the bloodstream,43 in the thrombus material from patients with plaque erosion. This finding may account for the observed increased concentration of HA in the local plasma samples of the IFC–ACS patients. In this context, our study provides a plausible association between the higher HYAL2 expression with correspondingly higher HA local concentrations and the subsequent activation of the TLR2 on neutrophils as one of the pathophysiological mechanisms in IFC–ACS.
In order to study the functional impact of the TLR2 activation in neutrophils, we mimic the endogenous TLR2 activation of isolated neutrophils from IFC–ACS and RFC–ACS patients by stimulating them ex vivo and studying their differential inflammatory response. Nevertheless, for this purpose, we used an agonist of the TLR2/TLR1 heterodimer: Pam3CSK4 and not HA in order to study the TLR2-specific response of neutrophils and not the general HA-driven activation of also other HA-binding receptors. As demonstrated by the current study, IFC–ACS–derived neutrophils do not only express higher amounts of TLR2 compared with RFC–ACS neutrophils, but the TLR2 pathway appears to be generally more susceptible to activation as shown by an increased release of MMP9 at the local site of IFC–ACS and not systemically, suggesting a disease-specific pattern. In this context, we consider the increased expression of HYAL2 in the coronary thrombi and thereby the increased local concentration of cleaved HA as one of the main amplifier of the TLR2-mediated neutrophil activation in IFC–ACS, determining their reaction specificity at the lesional site and not elsewhere in the artery system. The reaction specificity appears also to be determined by the fact that high shear stress, which is normally ubiquitous in the artery system, does not increase the HYAL2 expression in endothelial cells, supporting previous studies showing that lesional monocytes, but not the endothelium itself, seem to be the main source of HYAL2.39 In this context, our immunofluorescence analysis shows a tendency towards a co-localization of HYAL2 and neutrophils in IFC–ACS thrombi, thereby proposing neutrophils as possible HYAL2 sources themselves and thereby also supporting the hypothesis of HYAL2 originating rather from immune cells than from endothelial cells in IFC–ACS.
TLR2-dependent increase in MMP9 secretion by IFC–ACS–derived neutrophils
Obviously, more factors are simultaneously required for the TLR2-dependent neutrophil activation: not only the higher TLR2 expression by the LOC IFC–ACS–derived neutrophils but also the distinct local environment of the IFC–ACS causing culprit lesion, rich in endogenous TLR2 ligands like LMW-HA39 and characterized by a distinct inflammatory signature as depicted in the Structured Graphical Abstract.8
Upon TLR2 stimulation of LOC IFC–ACS neutrophils, we observe an increase in the secretion of active MMP9. This effect was not seen in SYS IFC–ACS, nor in RFC–ACS neutrophils, and was shown to be reversible under TLR2 blockade, supporting a specific TLR2-dependent neutrophil activation pattern at the site of the culprit lesion in plaque erosion. Correspondingly, we observe higher local MMP9 concentrations in plasma samples obtained from the culprit site of IFC–ACS as compared with RFC–ACS, which might originate from LOC neutrophils from IFC–ACS patients, with greater sensitivity towards locally available TLR2 substrates, e.g. HA.40
In contrast to the well-established role of MMP9 in plaque rupture and fissure,44 where increased MMP9 concentration is associated with plaque instability due to collagen breakdown inside the coronary vessel wall,45,46 little is known about its contribution to the pathology of plaque erosion due to a different localization of action: the blood stream.47 The different localization of the immune cells and their secretory mediators seems to be of a great importance for the better understanding of the pathology of RFC–ACS and IFC–ACS. RFC–ACS–causing lesions are characterized by more prone infiltration of immune cells into the vessel wall,48,49 whereby in IFC–ACS, the inflammatory cascade is mainly taking place intraluminal;8,10 therefore, our study design focuses on the local intraluminal environment at the culprit lesion.
One hypothesis suggests that intraluminal cleavage of non-fibrillar collagen, such as collagen IV, by gelatinases (MMP2 and MMP9)50 may lead to degradation of the basal membrane to which endothelial cells attach and thereby trigger endothelial cell desquamation as a hallmark of plaque erosion.10,34 In a proof-of-concept in vitro experiment, we demonstrate that recombinant human MMP9 aggravates endothelial cell detachment in a flow chamber mimicking coronary artery bifurcations, which might point to the requirement of both factors, i.e. flow conditions and active MMP9, for the initiation of IFC–ACS. This seems to be in line with our previous observation that the culprit lesion is more frequently located close to bifurcations in IFC than in RFC–ACS.8
Furthermore, MMP9 may function as an important positive feedback loop for neutrophil chemotaxis and activation by cleaving pro-interleukin (IL)1β and IL8 into their active forms.51
As shown by some studies, one of the major sources of active MMP9 after myocardial infarction is the neutrophils themselves52 and their ability to quickly release MMP9 from their storage in secondary granulae designates them to be the immediate source of MMP9 after acute myocardial infarction.28,53 However, the impact of MMP9 on the post-infarct remodeling of the myocardium is controversially discussed due to the two-sided role of MMP9 depending on the timing after myocardial infarction. On the one hand, high MMP9 levels in the early phase are associated with higher neutrophil counts and seem to have detrimental effects on left ventricular remodeling; on the other hand, increased MMP9 concentrations in the late phase of wound healing are associated with preservation of left ventricular function.54 Therefore, further investigation into the impact of MMP9 on post-infarct remodeling, specifically in the context of IFC–ACS, is needed in order to develop precise therapeutic targets with negligible off-side effects, which however extends beyond the scope of the current hypothesis-generating study.
TLR2-independent neutrophil cytotoxicity in IFC–ACS
Furthermore, our study points toward another possible cellular mechanism of IFC–ACS: direct cytotoxic effects of activated neutrophils on endothelial cells.10 We have previously postulated T-cell–mediated cytotoxicity as one of the main contributors to endothelial cell death in plaque erosion.8 However, MMP9 may function as a cross-link between the innate and adaptive immunity by cleaving the functional domain of CD31 on CD4+ T-cells and thereby inducing T-cell dysregulation, as exemplary shown in non-ST-elevation myocardial infarction patients.55 However, not only T-cell–mediated cytotoxicity but also neutrophil cytotoxicity towards endothelial cells seems to play an important role in IFC–ACS, as it was suggested to be higher than of neutrophils isolated from RFC–ACS patients and was independent of the TLR2 activation, thereby proposing a complementary mechanism of endothelial cell loss in IFC–ACS. This might involve NETosis56,57 or cytotoxic effects of the neutrophil secretome as previously shown,58 which have not been the focus of the current study and may represent promising neutrophil activation pathways to be further examined in future studies.
TLR2 activation prolongs neutrophil survival in IFC–ACS
The whole process may be further boosted by a decreased apoptosis rate of local IFC–ACS–derived neutrophils upon TLR2 engagement, which may lead to prolonged neutrophil activation at the culprit site of IFC–ACS as suggested by the current study. The importance of maintaining a healthy neutrophil turnover is shown in infectious diseases such as sepsis, where neutrophil survival, infiltration, and damage of vital organs such as the lungs, kidneys, and heart are aggravated in a TLR2-dependent manner.59,60
In this context, the presented study emphasizes the associative involvement of many different local factors in the neutrophil activation at the lesion site of IFC–ACS with TLR2 activation being one of the main drivers of neutrophil MMP9 release and neutrophil survival, which may presumably lead to detachment of endothelial cells, especially under turbulent flow conditions, and thereby thrombus formation at the culprit site of plaque erosion (Structured Graphical Abstract).
Anti-TLR2 treatment in IFC–ACS: translational opportunities
In this context, a protective role of temporary anti-TLR2 treatment seems to be plausible as shown for other diseases like early sepsis.61 The beneficial effects of anti-TLR2 treatment in rats with transient focal ischemia-induced brain damage through inhibition of neuroinflammation were previously demonstrated by Ziegler et al.62 In the context of cardiovascular inflammation, experimental data suggest that inhibition of the TLR2 pathway either by monoclonal antibodies or in a TLR2 knockout mouse model may reduce cardiac fibrosis by attenuating macrophage-mediated cardiac infiltration and subsequent inflammation.63 Furthermore, a post-infarct treatment with a humanized anti-Toll-like receptor 2 antibody (OPN-305) seems to reduce the infarct size by 45% and enhance cardiac function in a porcine model of ischemia/reperfusion.64 Yet, only experimental data on anti-TLR2 treatment exist so far, as some limitations like increased mortality due to infections61 of this therapeutic approach has come to light.
Another potential therapeutic target could be the modulation of the HYAL2 activity in IFC–ACS patients and thereby reducing the amount of HA which may lead to lower TLR2 activation in neutrophils. Hyaluronidase-triggered anticancer treatment has shown promising results in drug-resistant breast cancer.65 Furthermore, there is some evidence for the anti-thrombotic potential of an anti-HYAL2 treatment through prevention of platelet–monocyte aggregates in ACS.42 The effect of a similar therapeutic intervention on the TLR2-dependent activation of the neutrophils specifically in IFC–ACS remains to be studied and may represent a therapeutic approach with limited side effects due to careful timing and pre-selection of the patient population. In this regard, developing a profound understanding of the multiple complex events involved in IFC–ACS (Structured Graphical Abstract) and pre-selecting the target population seem to be one of the crucial steps in the development of a successful immunotherapy in patients with IFC–ACS.66
Limitations of the study
As typical for studies of acute clinical events, we lack information about the inflammatory activation status at a ‘baseline’ time point before the acute coronary event. Therefore, we may only speculate about the inflammatory activation status and the underlying pathophysiological mechanisms before the collection time point. Correspondingly, we can mimic the involvement/connection of some activation factors ex vivo, but we are limited by default in complete restoration of the exact timing and sequence of occurrence of different factors as it is happening in vivo, which of course allows mainly for associative assumptions of the whole pathophysiological process.
Furthermore, the matching strategy may control for some known covariates of neutrophil activation but still leaves some other possible factors unconsidered. Therefore, residual confounding cannot be ruled out completely, which should be taken into consideration when analyzing the data. Due to the small sample size, multivariable analysis was not performed.
We used Pam3CSK4 as a synthetic TLR2/TLR1 agonist, which does not inform on the activation of TLR2/6 in the current context. Even though the same intracellular pathway is being demonstrated for both heterodimers by previous studies,67 we may still see some different biological effects due to off-target activation of other pathways depending on the ligand being used,68 which should be taken into account when analyzing the results.
In the current study, we focused on HA as a known endogenous ligand of TLR2 based on previous knowledge about the HA distinct abundance4,10,39 and metabolism39 in plaque erosion, but the use of broad proteomics approaches in order to identify possible new TLR2 endogenous ligands is of future interest to our group. We measured total HA concentration and not specifically LMW-HA as methods available for HA size differentiation have significant constraints and require many purification steps with the removal of plasma proteins, use of filters, etc. and thereby may create artefacts and affect the reliability of the results.69–71 Nevertheless, in the blood stream in contrast to synovial fluids,72,73 HA polymers are naturally degraded by hyaluronidase to its LMW counterpart.72–74
Conclusion
In conclusion, the current study suggests a multiple-hit hypothesis of plaque erosion with distinct TLR2-dependent neutrophil activation, presumably perpetuated by a higher local expression of HYAL2 and thereby increased local cleavage of HA at the coronary culprit lesion of IFC–ACS, leading to enhanced release of active MMP9 by neutrophils and thereby aggravated detachment of luminal endothelial cells in combination with disturbed flow conditions (Structured Graphical Abstract). These data emphasize not only the importance of the TLR2 pathway in patients with IFC–ACS but also the generally aggravated TLR2-independent neutrophil cytotoxicity towards endothelial cells as a hallmark of plaque erosion. MMP9 might further function as a connection between the innate and adaptive immunity, triggering a complementary T-cell–mediated response in IFC–ACS.8 Thereby, the current study sharpens our understanding of the complex pathology of IFC–ACS, showing that many different factors seem to be simultaneously involved in the inflammatory cascade of IFC–ACS. Further studies need to address the potential therapeutic value of these findings with focus on a possible specific inflammation-targeted treatment of IFC–ACS in the era of personalized medicine.
Acknowledgements
Thanks are due to S. Knüppel and N. Rösener for the technical assistance.
Supplementary data
Supplementary data are available at European Heart Journal online.
Declarations
Disclosure of Interest
Abbott, Amgen, AstraZeneca and Daiichi Sankyo (UL, FC, GL, DL), NovoNordisk, Sanofi, Amarin, Berlin-Chemie, Novartis, Pfizer, CRISPR Therapeutics, Bayer, Boeringer Ingelheim and Cardiac Dimensions (UL), Menarini and Chiesi (FC). American Heart Association, Italian National Health Service and Italian Minister of Education (GL).
Data Availability
The data underlying this article will be shared on reasonable request to the corresponding author.
Funding
German Centre for Cardiovascular Research (DZHK) Partner site Berlin, Berlin Institute of Health (BIH) and Italian National Project Grant PRIN 2017, Protocol 2017WJBKKW_001.
Ethical Approval
The study was approved by the local ethics committee (No. EA1/270/16) and conducted in accordance with the Declaration of Helsinki.
Pre-registered Clinical Trial Number
The pre-registered clinical trial number is NCT03129503.
References
Author notes
Ulf Landmesser, David M Leistner and Nicolle Kränkel contributed equally to this work.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}