





Abstract
Precision microbiome modulation as a novel treatment strategy is a rapidly evolving and sought goal. The aim of this study is to determine relationships among systemic gut microbial metabolite levels and incident cardiovascular disease risks to identify gut microbial pathways as possible targets for personalized therapeutic interventions.
Stable isotope dilution mass spectrometry methods to quantitatively measure aromatic amino acids and their metabolites were used to examine sequential subjects undergoing elective diagnostic cardiac evaluation in two independent cohorts with longitudinal outcome data [US (n = 4000) and EU (n = 833) cohorts]. It was also used in plasma from humans and mice before vs. after a cocktail of poorly absorbed antibiotics to suppress gut microbiota. Multiple aromatic amino acid-derived metabolites that originate, at least in part, from gut bacteria are associated with incident (3-year) major adverse cardiovascular event (MACE) risks (myocardial infarction, stroke, or death) and all-cause mortality independent of traditional risk factors. Key gut microbiota-derived metabolites associated with incident MACE and poorer survival risks include: (i) phenylacetyl glutamine and phenylacetyl glycine (from phenylalanine); (ii) p-cresol (from tyrosine) yielding p-cresol sulfate and p-cresol glucuronide; (iii) 4-OH-phenyllactic acid (from tyrosine) yielding 4-OH-benzoic acid and 4-OH-hippuric acid; (iv) indole (from tryptophan) yielding indole glucuronide and indoxyl sulfate; (v) indole-3-pyruvic acid (from tryptophan) yielding indole-3-lactic acid and indole-3-acetyl-glutamine, and (vi) 5-OH-indole-3-acetic acid (from tryptophan).
Key gut microbiota-generated metabolites derived from aromatic amino acids independently associated with incident adverse cardiovascular outcomes are identified, and thus will help focus future studies on gut-microbial metabolic outputs relevant to host cardiovascular health.
Gut microbiota-generated metabolites and pathways derived from aromatic amino acids that independently associate with incident major adverse cardiovascular event (MACE) and death risk are identified using a combination of large-scale clinical association studies (US and European cohorts with longitudinal outcome data), and both human and animal studies involving gut microbial suppression with oral ingestion of a cocktail of poorly absorbed antibiotics.
See the editorial comment for this article ‘A roadmap for gut microbiome-derived aromatic amino acids for improved cardiovascular risk stratification’, by B.E. Stähli et al., https://doi.org/10.1093/eurheartj/ehad367.
Introduction
Changes in both gut microbial composition and circulating levels of certain microbial metabolites have been associated with multiple human diseases, including cardiovascular disease (CVD).1–5 Mechanistic studies in both humans6 and animal models,7–11 have further linked microbial metabolites as participants in the development or exacerbation of host CVD phenotypes. These findings have served as a basis for developing new therapeutic approaches aimed at restoring microbiome health with the use of pre- and probiotics,12–14 and/or by modulating gut-microbial functional metabolic output. Illustrations of the latter approach include non-lethal inhibition (i.e. non-antibiotic) of gut microbial enzymes and sequestering or supplementing selected gut-microbial metabolites.15–18 For example, the gut microbial pathway involving the conversion of choline into trimethylamine (TMA), the precursor to trimethylamine-N-oxide, has previously been linked to the development of CVD. Animal model studies have shown that inhibiting the enzyme catalyzing choline into TMA can suppress atherosclerosis development,16 reduce both thrombosis potential17,19 and renal fibrosis,20,21 improve cardiac function,22 and lessen obesity.23 Inhibitors of gut-bacterial bile salt hydrolases have also been used to modulate the bile acid pool within the host,24,25 and non-lethal targeted inhibition of the gut bacterial β-glucuronidase has reportedly enhanced anticancer drug efficacy within hosts.26,27
Precision microbiome modulation—whether by using small molecule inhibitors of specific gut microbiota-driven pathways, dietary interventions, or by changing microbial compositions and function to alter microbial community metabolic output (e.g. shifting metabolic production from potentially detrimental to potentially beneficial products for the host)—requires comprehensive and quantitative analysis of microbial metabolites, as well as both an understanding of their associations with and effects on host adverse phenotypes.1,28–32 The vast majority of metabolomics studies report only qualitative relative levels (i.e. not quantitative absolute concentrations), and are not optimized for separation of structural isomers. Consequently, results reported can be difficult to replicate, and require validation with more quantitative and robust methods specifically designed to meet the rigor of in vitro clinical diagnostics use.
Numerous studies suggest that aromatic amino acid metabolism by gut microbiota gives rise to metabolites potentially linked to cardiometabolic diseases. For example, we recently identified a phenylalanine (Phe)-derived gut microbiota metabolite, phenylacetyl gultamine (PAGln), that is both clinically and mechanistically linked to CVD via adrenergic receptor signaling.11,33,34 Gut microbes also catabolize protein aromatic amino acids tyrosine (Tyr) and tryptophan (Trp) into precursors of the well-known uremic toxins p-cresol sulfate (pCS) and indoxyl sulfate (IS), respectively.35–37pCS and IS have been associated with cardiovascular mortality in subjects with impaired renal function,38–40 and have been reported to increase thrombosis potential in various in vitro and in vivo models.41–46 Further, while cross sectional studies have reported the association of these uremic toxins with heightened carotid atherosclerotic plaque burden, and in individuals with relatively preserved kidney function,47,48 the relationship of classic uremic toxins like IS and pCS to incident risk of adverse cardiovascular events among subjects without renal functional impairment is unclear. Conversely, alternative microbiota-associated metabolites of aromatic amino acids have been reported to be inversely associated with cardiometabolic disease risks [e.g. indole-3-propionic acid (I3PA) and reduced diabetes risk49], or positively associated with healthy eating habits and increased microbiome diversity (e.g. hippurate50,51).
Mapping the pathways and products of gut microbiota-dependent metabolism of aromatic amino acids has not been comprehensively investigated in humans, and detailed exploration using quantitative stable isotope dilution methods of the numerous metabolites designed to provide absolute concentrations free of contaminating structural isomers has yet to be reported for most metabolites. As noted above, most studies that report metabolite associations with disease risks are cross sectional and/or qualitative (based on untargeted metabolomics), only reporting ‘relative levels’. The majority of such studies also do not unambiguously distinguish between structural isomers (i.e. compounds that possess the same elemental composition, physical properties, and often co-elute). Untargeted metabolomics analyses thus need to be separately validated using quantitative methods designed and verified to be both quantitative and specific for the target analytes desired.
Rational design of personalized therapeutic strategies that target gut microbiota driven pathways will require a comprehensive understanding of both gut microbial metabolism and the relationship between systemic levels of metabolites and incident CVD event risks. Thus, the aim of this study is to identify gut microbe-derived metabolites and microbial pathways associated with incident adverse CVD event risk. Gut microbial enzymes ferment essential aromatic amino acids into a plethora of small molecules via deamination, oxidation, and/or reduction reactions, to name a few. As the portal vein drains blood from most of the gastrointestinal tract into the liver, some molecules derived from gut microbes are further metabolized, either fully or in part, by host liver enzymes before they reach the systemic circulation. Accordingly, we herein develop a reverse phase, stable-isotope-dilution liquid chromatography tandem mass spectrometry (LC-MS/MS) method for the quantitative analysis of numerous metabolites potentially derived from dietary sources of the aromatic amino acids Phe, Tyr, and Trp via gut bacteria. We then examine the associations between quantitative measures of these circulating metabolites in two independent cohorts of stable individuals undergoing elective diagnostic cardiac evaluation with adjudicated longitudinal follow-up. In addition, we evaluate the contribution of gut microbiota to circulating metabolite levels in both human and mice intervention studies using poorly absorbed antibiotic cocktails to suppress gut microbiota. The collective data assembled, overlaid on metabolic pathways delineated, provide a map or ‘atlas’ of microbiota-dependent metabolic pathways of aromatic amino acid catabolism associated with incident CVD risks.
Methods
Study approvals
All clinical study protocols and informed consent for human subjects complied with the Declaration of Helsinki, and received ethical approval by the Cleveland Clinic Institutional Review Board or the ethics committee of Charité-Universitätsmedizin Berlin. Written informed consent was obtained from all subjects. All animal model studies were approved by the Institutional Animal Care and Use Committee at the Cleveland Clinic.
Human subjects
US cohort—GeneBank
All subjects gave written informed consent, and study protocols were approved by the Cleveland Clinic Institutional Review Board. The cohort (n = 4000) was obtained from sequential consenting subjects enrolled in the study GeneBank (ClinicalTrials.gov Identifier NCT00590200), a well-characterized cohort with broad geographic catchment from the quaternary referral center, the Cleveland Clinic. GeneBank subjects were comprised of sequential stable subjects 18 years of age or older without evidence of acute coronary syndrome (ACS) (cardiac troponin I <0.03 ng/mL) undergoing elective diagnostic coronary angiography (cardiac catheterization or coronary computed tomography) for evaluation of CVD risk.3,10 All patients were followed prospectively over the ensuing 3 years by telephone contact, mailing, and medical record review by designated research personnel independent of study investigators. All major adverse cardiovascular events (MACE), defined as death, nonfatal myocardial infarction (MI), or nonfatal stroke after enrollment, were adjudicated with source documentation. Significantly obstructive coronary artery disease (CAD) was defined as any clinical history of MI, percutaneous coronary intervention, coronary artery bypass graft surgery, or angiographic evidence of CAD (≥50% stenosis) in ≥1 major coronary artery. Subsequent MI was adjudicated from source documentation with supporting evidence including elevated cardiac enzymes, significant Q-wave definitive electrocardiographic evidence of new infarction, the presence of myocardial wall akinesis or scar on imaging, or treatment with thrombolytic agents or direct percutaneous intervention. Stroke was defined as documented loss of neurologic function caused by an ischemic event with residual symptoms continuing ≥24 h after onset (not to include transient ischemic attacks, microvascular infarcts, or amarosis fugax). High-sensitivity C-reactive protein (hsCRP), lipid profiles, fasting glucose, creatinine, troponin, and hemoglobin A1C were all measured on a Roche Cobas platform (Roche Diagnostics, Basel, Switzerland). Estimated glomerular filtration rate (eGFR) was calculated on the basis of the 2021 Chronic Kidney Disease Epidemiology Collaboration creatinine equation.52
EU cohort—LipidCardio
Plasma samples (n = 833) were also obtained from subjects enrolled in the LipidCardio study at the Charité-Universitätsmedizin, Berlin, which aims to study the role of lipoproteins in CVD.53 The study was approved by the local research ethics committee (approval number: EA1/135/16) and registered under German Clinical Trial Register (drks.de); Identifier: DRKS00020915. All participants provided written informed consent. Patients aged 18 years and older undergoing elective diagnostic cardiac catheterization at a single large academic center (Department of Cardiology, Campus Benjamin Franklin, Charité-Universitätsmedizin Berlin), except those with ACS, were eligible for inclusion. All patients were followed prospectively over the 3 years. In this cohort, acute ischemic stroke was diagnosed by a neurologist (according to current guidelines) and confirmed by cerebral imaging (CT or MRI). MI was defined as the detection of a rise and/or fall of troponin with at least one value above the 99th percentile URL and with at least one of the following: symptoms of acute myocardial injury, new ischemic electrocardiography changes, development of pathological Q waves, imaging evidence of new loss of viable myocardium or new regional wall motion abnormality in a pattern consistent with an ischemic etiology, and identification of a coronary thrombus by angiography.
The description of both cohort studies in this manuscript is in compliance with the Strenghtening the Reporting of Observational Studies in Epidemiology.54
Suppression of gut microbiota in subjects using oral antibiotics
In additional studies, healthy volunteers (n = 10) were subjected to a cocktail of oral poorly absorbed antibiotics for 7 days (ciprofloxacin, metronidazole and vancomycin; or ciprofloxacin, metronidazole, vancomycin, and neomycin), as previously described.3 Volunteers were not pregnant and did not have a chronic illness (including known history of heart failure, renal failure, pulmonary disease, gastrointestinal disorders, or hematologic diseases). Blood was collected after overnight fasting and then, following consumption of breakfast and eating ad libitum, serially at 2, 4, 6, 8, and 24 h later on three different days (relative to antibiotic use) as follows: at baseline (before antibiotics treatment; Pre-Abx), immediately following the 7-day antibiotics regimen (Abx), and after an antibiotics washout period (minimum 3 weeks) to permit repopulation of normal gut microorganisms (Post-Abx). Antibiotic suppression of gut microbial metabolite levels in humans were performed from samples collected in the pilot phase under an approved protocol registered under ClinicalTrials.gov Identifier: NCT01731236 (version Submitted Date: 16 November 2012).
Animal studies
All animal studies adhered to protocols approved by the Cleveland Clinic Institutional Animal Care and Use Committee. The impact of gut microbes on metabolite levels was studied in mice. Details of mouse studies are provided in the Supplementary material online (Supplementary Methods).
Targeted LC-MS/MS analysis of human and mouse plasma samples
Stable-isotope-dilution LC-MS/MS was used for quantification of metabolites in plasma using methods optimized for each of the indicated metabolites [including use of synthetic (as needed) or commercial isotope labeled internal standards]. Details of sample preparation, internal standards used, and LC-MS/MS conditions are provided in the Supplementary material online (Supplementary Methods) and Supplementary material online, Table S1 for original candidate metabolite list employed.
Statistics
The Wilcoxon rank sum test or Student’s t-test for continuous variables and chi-square test for categorical variables were used to examine the difference between groups. Cox proportional hazards analysis was used to determine hazard ratio (HR) and 95% confidence intervals (95% CI) for MACE and all-cause mortality stratified according to metabolite level quartiles [quartile 4 (Q4) vs. quartile 1 (Q1)]. Adjustments were made for age, sex, smoking status, systolic blood pressure and diastolic blood pressure or hypertension, low-density lipoprotein (LDL) cholesterol, high-density lipoprotein (HDL) cholesterol, triglycerides, hsCRP, and diabetes. Spearman's correlation was used to examine the associations among metabolites. Differences among metabolites levels before, during and after antibiotics (Abx) treatment were calculated by non-parametric one-way ANOVA (Kruskal Wallis). All analysis was performed with RStudio-R version 4.1.3. (2022–03–10) (Vienna, Austria), or GraphPad Prism 9. A P-value of <0.05 was considered statistically significant.
Results
Gut microbe-generated metabolites of aromatic amino acids and their association with incident adverse cardiac event risks
We began by assembling a comprehensive list of candidate metabolites, and subsequently developed stable isotope dilution LC-MS/MS methods for their quantitation, differentiating candidates from structural isomers where necessary (see Supplementary material online, Tables S1 and S2). Only metabolites that were detected in over three quarters of fasting plasma samples and had reproducible values in quality control samples (included at beginning, middle and end of every batch, typically n = 81 samples per batch, of samples analyzed) over the course of the study (CV < 20%) were included for further analysis.
To asses which metabolites were indeed linked to gut microbial metabolism, we next explored whether suppression of gut microbiota with poorly absorbed cocktail of oral antibiotics could reduce plasma levels of the metabolites in humans and in mice (see Supplementary material online, Figures S1–5). Briefly, healthy control subjects were placed on oral cocktail of poorly absorbed antibiotics. The impact of 7 days of antibiotic suppression on metabolite levels was assessed both at fasting and at multiple post prandial time points as described in Methods. Gut microbiota suppression studies were also performed on mice (see Supplementary material online, Figures S3 and S5). In parallel, levels of compounds potently resulting from gut microbial fermentation of aromatic amino acid were examined for their associations with incident adverse cardiovascular event risks and all-cause mortality. For these studies, we utilized two independent clinical cohorts composed of sequential individuals undergoing elective diagnostic cardiac evaluation with longitudinal follow-up [a US Cohort, GeneBank (n = 4000); and a European Cohort, LipidCardio (n = 833); Methods]. The overall associations observed between metabolite levels and adverse outcomes within each cohort were highly similar; consequently, analyses presented within the main manuscript are for the combined cohort, with results for the individual cohorts presented in Supplementary material online. Baseline clinical and laboratory characteristics of the combined cohort (n = 4833) are summarized in Table 1 (see Supplementary material online, Table S3 for the individual cohorts). The combined cohort had more males (65.4%), a median age of 64.9 years, and 14.7% of subjects experienced an incident adjudicated MACE (defined as non-fatal MI, non-fatal stroke or death) during 3 years of follow-up.
Baseline characteristics of the participants in the US cohort (GeneBank) and the European cohort (LipidCardio) (n = 4833)
| Characteristics | All participants (n = 4833) |
|---|---|
| Age (years) | 64.9 (56.1–73.5) |
| Male sex (%) | 65.4 |
| Smoking (%) | 13.4 |
| Hypertension (%) | 46.0 |
| Diabetes (%) | 30.9 |
| CAD (%) | 76.2 |
| MACE at 3 years, (%) | 14.7 |
| HDL (mg/dL) | 35.7 (29.3–44.3) |
| LDL (mg/dL) | 95.0 (76.0–117.0) |
| TG (mg/dL) | 119.0 (87.0–166.0) |
| hsCRP (mg/L) | 2.34 (1.00–5.73) |
| eGFR (mL/min/1.73 m²) | 88.76 (71.97–99.13) |
| Baseline medications (%) | |
| Aspirin (%) | 70.7 |
| ACE inhibitors (%) | 53.6 |
| Beta blocker (%) | 62.3 |
| Statin (%) | 60.1 |
| Characteristics | All participants (n = 4833) |
|---|---|
| Age (years) | 64.9 (56.1–73.5) |
| Male sex (%) | 65.4 |
| Smoking (%) | 13.4 |
| Hypertension (%) | 46.0 |
| Diabetes (%) | 30.9 |
| CAD (%) | 76.2 |
| MACE at 3 years, (%) | 14.7 |
| HDL (mg/dL) | 35.7 (29.3–44.3) |
| LDL (mg/dL) | 95.0 (76.0–117.0) |
| TG (mg/dL) | 119.0 (87.0–166.0) |
| hsCRP (mg/L) | 2.34 (1.00–5.73) |
| eGFR (mL/min/1.73 m²) | 88.76 (71.97–99.13) |
| Baseline medications (%) | |
| Aspirin (%) | 70.7 |
| ACE inhibitors (%) | 53.6 |
| Beta blocker (%) | 62.3 |
| Statin (%) | 60.1 |
The cohort was made by merging individuals from the GeneBank [US cohort (n = 4000)] and LipidCardio [European cohort (n = 833)], which consists of sequential stable subjects without evidence of acute coronary syndrome (cardiac troponin I <0.03 ng/mL) who underwent elective diagnostic coronary angiography (cardiac catheterization or coronary computed tomography) for evaluation of coronary artery disease (CAD). MACE was defined as death, nonfatal myocardial infarction (MI), or nonfatal cerebrovascular accident (stroke) following enrollment.
Continuous data are presented as median (interquartile range), categorical variables are presented as %; ACE angiotensin-converting enzyme, eGFR = estimated glomerular filtration rate. HDL high-density lipoprotein, hsCRP high-sensitivity C-reactive protein, LDL low-density lipoprotein.
Baseline characteristics of the participants in the US cohort (GeneBank) and the European cohort (LipidCardio) (n = 4833)
| Characteristics | All participants (n = 4833) |
|---|---|
| Age (years) | 64.9 (56.1–73.5) |
| Male sex (%) | 65.4 |
| Smoking (%) | 13.4 |
| Hypertension (%) | 46.0 |
| Diabetes (%) | 30.9 |
| CAD (%) | 76.2 |
| MACE at 3 years, (%) | 14.7 |
| HDL (mg/dL) | 35.7 (29.3–44.3) |
| LDL (mg/dL) | 95.0 (76.0–117.0) |
| TG (mg/dL) | 119.0 (87.0–166.0) |
| hsCRP (mg/L) | 2.34 (1.00–5.73) |
| eGFR (mL/min/1.73 m²) | 88.76 (71.97–99.13) |
| Baseline medications (%) | |
| Aspirin (%) | 70.7 |
| ACE inhibitors (%) | 53.6 |
| Beta blocker (%) | 62.3 |
| Statin (%) | 60.1 |
| Characteristics | All participants (n = 4833) |
|---|---|
| Age (years) | 64.9 (56.1–73.5) |
| Male sex (%) | 65.4 |
| Smoking (%) | 13.4 |
| Hypertension (%) | 46.0 |
| Diabetes (%) | 30.9 |
| CAD (%) | 76.2 |
| MACE at 3 years, (%) | 14.7 |
| HDL (mg/dL) | 35.7 (29.3–44.3) |
| LDL (mg/dL) | 95.0 (76.0–117.0) |
| TG (mg/dL) | 119.0 (87.0–166.0) |
| hsCRP (mg/L) | 2.34 (1.00–5.73) |
| eGFR (mL/min/1.73 m²) | 88.76 (71.97–99.13) |
| Baseline medications (%) | |
| Aspirin (%) | 70.7 |
| ACE inhibitors (%) | 53.6 |
| Beta blocker (%) | 62.3 |
| Statin (%) | 60.1 |
The cohort was made by merging individuals from the GeneBank [US cohort (n = 4000)] and LipidCardio [European cohort (n = 833)], which consists of sequential stable subjects without evidence of acute coronary syndrome (cardiac troponin I <0.03 ng/mL) who underwent elective diagnostic coronary angiography (cardiac catheterization or coronary computed tomography) for evaluation of coronary artery disease (CAD). MACE was defined as death, nonfatal myocardial infarction (MI), or nonfatal cerebrovascular accident (stroke) following enrollment.
Continuous data are presented as median (interquartile range), categorical variables are presented as %; ACE angiotensin-converting enzyme, eGFR = estimated glomerular filtration rate. HDL high-density lipoprotein, hsCRP high-sensitivity C-reactive protein, LDL low-density lipoprotein.
When examining each analyte for its association with incident MACE risk, the distribution of each analyte quantified in both the merged and individual cohorts are shown in Supplementary material online (Supplementary material online, Tables S4-S6), with results (HR and 95% CI) stratified by whether the subjects experienced an incident MACE event during follow-up (3 years) (Figure 1; See Supplementary material online, Figures S6 and S7 for HR of each compound in each of the individual cohorts). Notably, all three aromatic amino acid nutrient precursors (Phe, Tyr, and Trp) had lower circulating levels among individuals with MACE vs. those without, but only Tyr and Trp levels reached significance (see Supplementary material online, Table S4). Tyr and Trp showed inverse associations with incident MACE risk, while in each case only a subset of the down-stream metabolites were associated with heightened risk, including following adjustments for comorbidities and CVD risk factors (Figure 1, See Supplementary material online, Figures S6 and S7). The results of analyses (HR of Q4 vs. Q1 and 95% CI) are overlaid on proposed pathway maps of each aromatic amino acid in Figures 2 and 3. Also shown are currently reported microbial enzyme sources, including insights into gut microbiota contribution based on results of the antibiotic suppression intervention studies performed in humans and rodents.
![Association between MACE and plasma metabolites derived from phenylalanine, tyrosine and tryptophan via gut microbes. Hazard ratio (95% CI) for incident (3-year) risks for major adverse cardiovascular events (MACE) to gut microbial-derived metabolites from aromatic amino acids in a cohort of stable cardiac patients from combined two independent cohorts [US cohort (n = 4000) and European cohort (n = 833)] undergoing elective diagnostic coronary evaluation. Hazard ratio (unadjusted, filled circles) and multivariable Cox model adjusted (open circles; adjusted for age, sex, smoking, HDL, LDL, TG, hypertension, diabetes mellitus, and hsCRP). Red, green, and black color is for compounds positively, negatively, and not associated with MACE, respectively. The 5%–95% confidence interval is indicated by line length.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/eurheartj/44/32/10.1093_eurheartj_ehad333/1/m_ehad333f1.jpeg?Expires=1749806187&Signature=j~sE4bZ6tE6flM8R6dCzxXs~Bvr3G7FlyDJIhUEt--HlKG3aKtBnVhXHhR6XDK5fBQmcMYV3pnVozAQbVWzRrPwtnNSnaacQQINglyitQIIY0biHteCbaDvBQla1SuJcWh-ekVXSZTT~EiW0McL8nqNX7XgLdJvfF2eeYpeQtgkL43Ka-q2fDSujtrnO-yuwpuVGocAelmtagcD~4NVg~KSlHzHe~nNsfVV2Bul7mcot-tuCTJ4INhVZfHWIgCk4jGqv-eWm6e1-kpQkb-x~vTiCS34bYcJmDBnRBREn9KjtHGy61mM6gwQIBsaSPC~cWidYS-jfzy8Yjh7jj3R-Cw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Association between MACE and plasma metabolites derived from phenylalanine, tyrosine and tryptophan via gut microbes. Hazard ratio (95% CI) for incident (3-year) risks for major adverse cardiovascular events (MACE) to gut microbial-derived metabolites from aromatic amino acids in a cohort of stable cardiac patients from combined two independent cohorts [US cohort (n = 4000) and European cohort (n = 833)] undergoing elective diagnostic coronary evaluation. Hazard ratio (unadjusted, filled circles) and multivariable Cox model adjusted (open circles; adjusted for age, sex, smoking, HDL, LDL, TG, hypertension, diabetes mellitus, and hsCRP). Red, green, and black color is for compounds positively, negatively, and not associated with MACE, respectively. The 5%–95% confidence interval is indicated by line length.
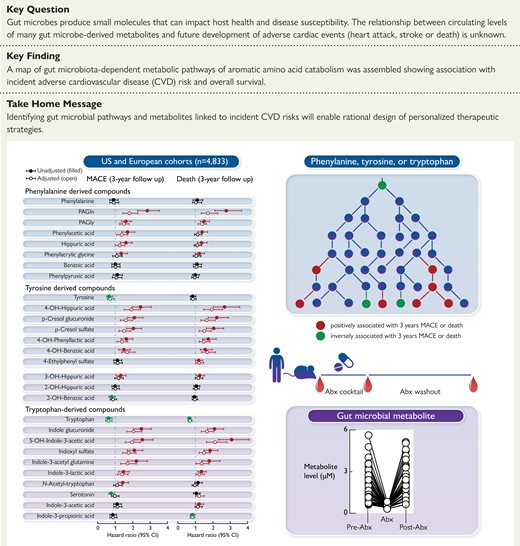
An atlas of microbial phenylalanine and tyrosine pathways associated with MACE. Metabolic pathways for aromatic acid fermentation together with their association with MACE (red chemical structures for compounds positively associated with MACE while green structures for compounds negatively associated with MACE after adjusting for traditional risk factors and hsCRP) and effect of gut microbe depletion on their circulation levels both in humans (blue (left) arrow) and mice (red (right) arrow); (−) no effect. Abx = antibiotics.
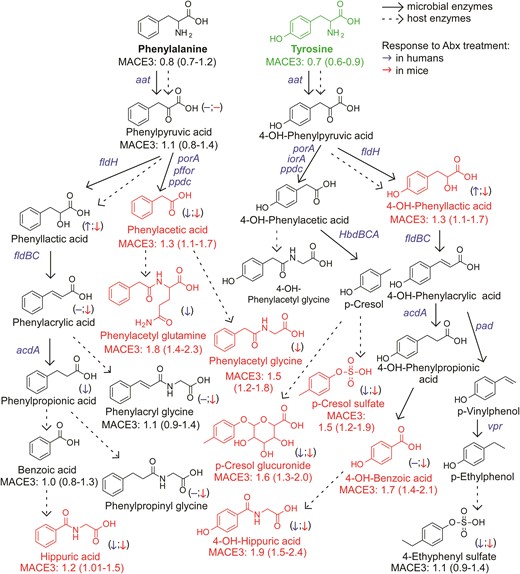
An atlas of microbial tryptophan pathways associated with MACE. Metabolic pathways for aromatic acid fermentation together with their association with MACE (red chemical structures for compounds positively associated with MACE while green structures for compounds negatively associated with MACE after adjusting for traditional risk factors and hsCRP) and effect of gut microbe depletion on their circulation levels both in humans (blue (left) arrow) and mice (red (right) arrow); (−) no effect. Abx = antibiotics.
When mapping the metabolism of each of the dietary aromatic amino acids by gut microbiota, the initial step typically involves removal of the alpha amino group by deaminases, an enzymatic activity that is promiscuous among gut microbiota.55 Of note, circulating levels of the proximal deamination product of Phe, phenylpyruvic acid, were not appreciably different among individuals who experienced MACE vs. those that did not (Figures 1, 2). Phenylpyruvic acid is further metabolized by either oxidative (forming phenylacetic acid) or reductive (forming phenyllactic acid) metabolism.11,34,56 Notably, PAGln and PAGly, downstream metabolites from phenylactetic acid, were associated with incident MACE risk, including following adjustments for traditional CVD risk factors (Figures 1, 2). Products of reductive degradation of phenylpyruvic acid, which terminate in hippuric acid, showed a trend toward increased levels with MACE, and remained significantly associated with incident MACE risk following adjustments (Figures 1, 2).
A map of gut microbiota-dependent catabolism of dietary Tyr metabolites with overlaid associations with incident MACE risks in the combined US and European cohort is also shown in Figure 2. The Tyr-derived metabolites that remained associated with MACE risks after adjustments for traditional CVD risk factors were derived either from p-cresol (both pCS and p-cresol glucuronide), 4-OH-phenyllactic acid, 4-OH-benzoic acid and 4-OH-hippuric acid. Notably, while structural isomers of 4-OH-hippuric acid (2-OH and 3-OH isomers) were also included in the analyses, only 4-OH-hippuric acid remained significantly associated with MACE following adjustments (Figures 1, 2).
Dietary Trp gives rise to a large array of gut microbe-generated metabolites (Figure 3). Circulating levels of this essential amino acid were inversely associated with incident MACE risk (Figures 1, 3). Upon further examination of Trp metabolites, indole glucuronide, IS, 5-OH-indole-3-acetic acid, indole-3-acetyl glutamine and indole-3-lactic acid remained significantly associated with incident MACE risk following adjustments.
We next examined the relationship between fasting plasma levels of each analyte and all-cause mortality (3-year). The distribution of each analyte quantified in both the merged and individual cohorts are shown in Supplementary material online (Supplementary material online, Tables S7-S9), with mortality risk shown in Figure 4, and overlaid onto proposed metabolic pathways in Figures 5 and 6. Associations among metabolites and mortality risk followed trends notably similar to those observed for incident MACE risk, with some modest exceptions. Notably, circulating levels of the aromatic amino acid Tyr were not negatively associated with over-all mortality risk, but its gut microbial metabolite 4-ethylphenyl sulfate, a metabolite reportedly linked to neurocognitive impairment,57 did show positive association with incident death risk. Additionally, hippuric acid showed an attenuated association with poorer survival following adjustments, while the Trp metabolite I3PA remained negatively associated with all-cause mortality following adjustments (Figures 4–6).
![Association between all-cause mortality and plasma metabolites derived from phenylalanine, tyrosine and tryptophan via gut microbes. Hazard ratio (95% CI) for incident (3-year) risks for all-cause-mortality) to gut microbial-derived metabolites from aromatic amino acids in a cohort of stable cardiac patients from combined two independent cohorts [US cohort (n = 4000) and European cohort (n = 833)] undergoing elective diagnostic coronary evaluation. Hazard ratio (unadjusted, filled circles) and multivariable Cox model adjusted (open circles; adjusted for age, sex, smoking, HDL, LDL, TG, hypertension, diabetes mellitus, and hsCRP). Red, green, and black color is for compounds positively, negatively and not associated with MACE, respectively. The 5%–95% confidence interval is indicated by line length.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/eurheartj/44/32/10.1093_eurheartj_ehad333/1/m_ehad333f4.jpeg?Expires=1749806187&Signature=Ow55UBekFuOwLphLK-4T6sAambk8gq7EJl9UTWW77XQ2hFxKtLI9gD0JbdpV5SDL6VD~0~mMGU7-urdEpwRRi0y7z~hY3nUuMQcvLfNezMVcZZoqcSapC~sl8H1ZLzRi-Fvh0Ctx9yb9Wb~DGTkKjVlgRHtNgo5bsywqC9htrJI~tX-cFBOIDNsd1sBjm4CLy7xcWNBnbsChrB5gDKp3uDIOSmXdqGwfvnntctTStI~xM5BQrq0EdCfGR74pwSN15GZ-r~0NgIULp1DEPvJgLaCl~e0mx6e1al7o7YQM9-HMVOkP-cT1w~-gewzkANAkjqrGpADAZcv5w~4KG3Ut2Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Association between all-cause mortality and plasma metabolites derived from phenylalanine, tyrosine and tryptophan via gut microbes. Hazard ratio (95% CI) for incident (3-year) risks for all-cause-mortality) to gut microbial-derived metabolites from aromatic amino acids in a cohort of stable cardiac patients from combined two independent cohorts [US cohort (n = 4000) and European cohort (n = 833)] undergoing elective diagnostic coronary evaluation. Hazard ratio (unadjusted, filled circles) and multivariable Cox model adjusted (open circles; adjusted for age, sex, smoking, HDL, LDL, TG, hypertension, diabetes mellitus, and hsCRP). Red, green, and black color is for compounds positively, negatively and not associated with MACE, respectively. The 5%–95% confidence interval is indicated by line length.
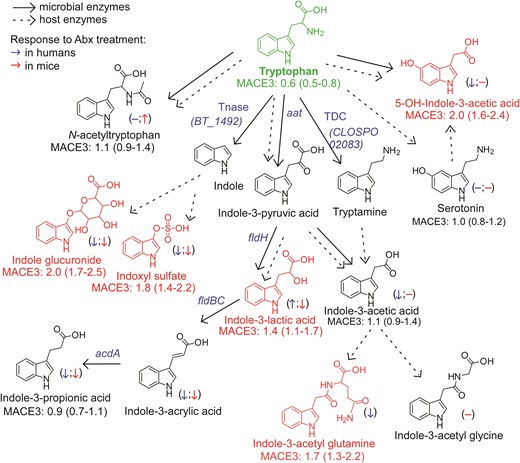
An atlas of microbial phenylalanine and tyrosine pathways associated with 3-year all-cause mortality. Metabolic pathways for aromatic acid fermentation together with their association with all-cause mortality (death) (red chemical structures for compounds positively associated with death while green structures for compounds negatively associated with death after adjusting for traditional risk factors and hsCRP) and effect of gut microbe depletion on their circulation levels both in humans (blue (left) arrow) and mice (red (right) arrow); (−) no effect. Abx = antibiotics.
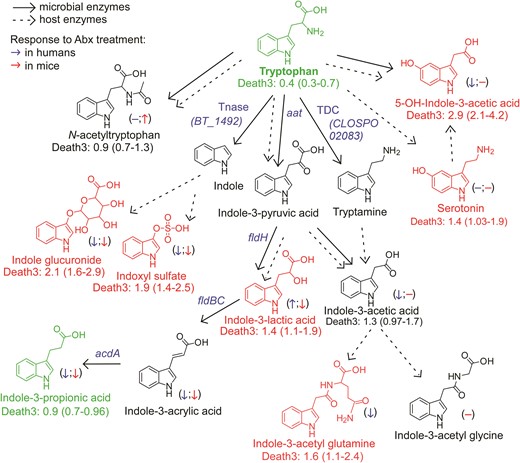
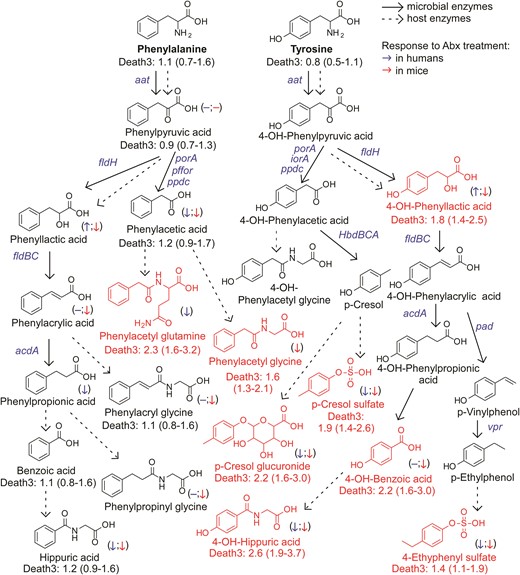
An atlas of microbial tryptophan pathways associated with 3-year all-cause mortality. Metabolic pathways for aromatic acid fermentation together with their association with all-cause mortality (death) (red chemical structures for compounds positively associated with death while green structures for compounds negatively associated with death after adjusting for traditional risk factors and hsCRP) and effect of gut microbe depletion on their circulation levels both in humans (blue (left) arrow) and mice (red (right) arrow); (−) no effect. Abx = antibiotics.
Discussion
As more and more microbial metabolites are shown to contribute to disease-relevant phenotypes, enhanced interest in modifying the functional metabolic output of the gut-microbial community is emerging as a potential therapeutic approach for treating multiple diseases.17,58 Moreover, providing gut microbiome-relevant supplements as therapeutic agents has had beneficial effects on cholesterol levels in animal models, and in randomized, double-blinded, placebo-controlled human intervention trials.59 But in order to design personalized therapeutic strategies that target gut microbiota driven pathways, either as a non-lethal bacterial enzyme inhibitors16,17,20,23–27,60 or newly emerging phage therapies,61 we first need a comprehensive understanding of both gut microbial metabolism and the relationship between systemic levels of metabolites and incident CVD risks. If we inhibit a specific gut microbial pathway, the ‘flow of carbon’ through the microbial community will be shifted and diverted to alternative end metabolites. Whether these alternative metabolites are associated with benign or harmful outcomes, is critical information for informed therapeutics development. Thus, for precision medicine targeting of the gut microbiome, both overall metabolic pathways, and whether a given structurally specific metabolite is associated with incident CVD risks, and thus appropriate for further mechanistic studies, is needed. The present studies provide a metabolic roadmap of aromatic amino acid metabolites, emphasizing direct experimentally-observed gut microbial contribution to their circulating levels, and their association with incident CVD adverse event risks in both US and European cohorts (Structured Graphical Abstract).
Some major conclusions noted by the present studies and pathway analyses are that multiple final products of microbial fermentation of Phe and Tyr via oxidative pathways (PAGln, PAGly, pCS, and p-cresol glucuronide) are associated with MACE and mortality risk in human subjects. It is of interest that each of these metabolites have previously been shown to affect platelet function and thrombosis potential both in vitro and in vivo.11,42,43 Additionally, the strong correlations observed between PAGln vs. pCS and p-cresol glucuronide in the present studies supports a commonality in their metabolic pathway origins (see Supplementary material online, Figure S8). Also, it is notable that all of these metabolites are almost exclusively of microbial origin, with dramatic suppression of their circulating levels with poorly absorbed antibiotics. The existence of shared microbial metabolic origins suggests the potential for development of therapeutic interventions to inhibit a single pathway, preventing downstream production of numerous MACE-associated metabolites (i.e. diversion of carbon flow in the microbial community toward more ‘benign’ metabolites).
Trp products associated with MACE (indole glucuronide and IS) are derived from indole, the microbiota-generated metabolite produced by the enzyme trpytophanase.35 IS has been reported to enhance platelet reactivity, including increased expression of P-selectin, formation of platelet microparticles, platelet-monocyte aggregates45 and increases in thrombus formation in animal models of vascular injury.41 PAGln, pCS, and IS have all been associated with heightened mortality in individuals with renal functional impairment.38–40,62 The present studies show these so-called ‘uremic toxins’ are also associated with incident adverse cardiac event risks among subjects with primarily preserved kidney function (eGFR for the combined cohort, 89 mL/min/1.73 m2).
Another conclusion noted from the present studies is that the reductive fermentation (fldH pathway) of Trp produces a metabolite (I3PA) that showed negative association with all-cause mortality, and similar tendency toward inverse association with MACE risk. In previous studies, I3PA has been negatively associated with diet-induced obesity63 and both coronary artery calcification and advanced atherosclerosis.64,65 In our study, the end product from Phe catalyzed by the same microbial pathway, hippurate, was positively associated with MACE, but the positive association with all-cause mortality was attenuated following adjustments for CVD risk factors. Urinary levels of the same metabolite has in cross sectional studies been associated with increased microbial diversity, higher intake of fruit and whole grains, and reduced odds of having metabolic syndrome.50,51 Further, studies have reported that urine hippurate levels are lower in obese individuals,66 in type 2 diabetes patients,67 among those with Crohn’s disease,68 and are inversely associated with blood pressure.69 Some studies have also reported a positive correlation between plasma hippurate and MACE during follow-up in advanced atherosclerosis cohorts,65 as well as increased hippurate excretion in individuals with type 2 diabetes.70,71
Terminal metabolites generated from Tyr via the reductive gut microbial catabolism branch (4-OH-benzoic acid and 4-OH-hippurate) showed positive associations with MACE risk and death, unlike metabolites derived from Trp and in part from Phe. While other mechanisms can account for this (e.g. differences in their excretion), our interpretation of these results suggests that the metabolites do not share a metabolic origin (lack of correlation between Phe- vs. Tyr-derived metabolites; Supplementary material online, Figure S9), and that additional or different gut microbes, microbial enzymes, and/or nutrient precursors (like polyphenols72), may be involved in the production of 4-OH-metabolites.
Two intermediate products of the Trp and Tyr reductive pathways—indole-3-lactic acid and 4-OH-phenyllactic acid—were strongly associated with MACE risk and poorer survival. While it has yet to be determined whether these metabolites directly affect cardiovascular health, our experiments using antibiotics to deplete gut bacteria suggest that circulating levels of aromatic lactates are produced primarily via endogenous enzymatic processes (e.g. aromatic α-keto acid reductase and lactic dehydrogenase) within the host (humans), since their levels are not significantly reduced. Alternatively, these metabolites could be produced mainly by antibiotic resistant bacteria and/or fungi in the gut that ‘bloom’ during antibiotic exposure, possibly explaining the modest increases observed in some cases (we also note that increases in levels with antibiotics could signify reduced competition of nutrient precursors by suppression of gut microbiota, leaving more precursor for host absorption). In addition, the strong correlation between circulating levels of 4-OH-phenyllactic acid and indole-3-lactic acid, and the poor correlation between indole-3-lactic acid and I3PA, further support our notion that these lactic acids originate from distinct metabolic pathways.
The present studies have numerous strengths and limitations that deserve discussion. A major strength is that our analyses draw from two geographically distinct cohorts with adjudicated outcome data (and each quaternary referral centers with wide catchment on different continents). Further, analyses were performed using quantitative isotope dilution LC-MS/MS methods optimized to distinguish between structural isomers, and where absolute quantification was correlated to reference standards—unlike untargeted MS analyses, which are only semi-quantitative, and thus threshold levels for clinically relevant risk associated with these metabolites remains elusive. Untargeted metabolic studies also only putatively identify candidate metabolites, and are not typically optimized to distinguish structural isomers, and thus require separate validation studies. We also report median and interquartile range levels for each metabolite stratified by MACE, and mortality, which could further serve as a resource for risk stratification efforts and is much more informative than relative values obtained by untargeted metabolomics approaches. Further, buy using unique sample sets obtained from humans and rodent studies with suppressed gut microbiota, we were able to both identify and confirm metabolites of microbial origin in humans and mice. In the current studies, the associations of circulating fasting levels of gut microbial metabolites derived from aromatic amino acids, and incident adverse CVD event risks, were examined among stable patients undergoing elective diagnostic cardiac evaluations. Whether such associations extend to alternative patient cohorts will require further studies. Two limitations of the present studies are a lack of fecal microbial composition data, and as well as lack of available dietary records with protein intake73,74. We also note that like any clinical observational study, in contrast to a randomized clinical outcome trial, a limitation is that there may be unknown confounders that are not adequately adjusted for. However, the inclusion of multiple independent cohorts enhances the legitimacy of the reported findings. While the US and European cohorts were run independently and possess subtle differences in age, definitions and adjudication of endpoints, the overall similar findings, for both MACE and all-cause mortality as endpoints observed in distinct cohorts with broad geographic catchment from two different contents, argues for the results observed to be robust and reproducible.
Novel therapies for cardiovascular and metabolic disease treatments aimed at shifting nutrient fermentation toward metabolites that are more beneficial to the host will require comprehensive analyses of microbial metabolites, their associations with disease relevant phenotypes, and their effects on the host. The present results provide a ‘roadmap’ and meaningful starting point for the development of such therapeutic treatments.
Supplementary data
Supplementary data is available at European Heart Journal online.
Data availability
Where permissible, the datasets generated and/or analyzed during the present studies are available from the corresponding author on request. There are restrictions to the availability of some of the clinical data generated in the present study, because we do not have permission in our informed consent from research subjects to share data outside our institution without their authorizations. Under these situations, data shared will be in summary format.
Funding
This work is supported by grants from the National Institutes of Health (NIH) and the Office of Dietary Supplements (P01 HL147823, R01HL103866 and R01HL160747) and the Foundation Leducq (17CVD01). K.A.R. was supported in part by training grant T32HL134622 from the National Heart, Lung, and Blood Institute (NHLBI) of the NIH. M.W. was supported in part by an award from the Deutsche Forschungsgemeinschaft (WI 5229/1-1). Mass spectrometry studies were performed on instrumentation housed in a facility supported in part through a Shimadzu Center of Excellence award. A.H. is participant in the BIH-Charité Advanced Clinician Scientist Pilotprogram funded by the Charité—Universitätsmedizin Berlin and the Berlin Institute of Health.
References
Author notes
Conflict of interest: Dr. Hazen reports being named as co-inventor on pending and issued patents held by the Cleveland Clinic relating to cardiovascular diagnostics and therapeutics, being a paid consultant formerly for Procter & Gamble in the past, and currently with Zehna Therapeutics. He also reports having received research funds from Procter & Gamble, Zehna Therapeutics and Roche Diagnostics, and being eligible to receive royalty payments for inventions or discoveries related to cardiovascular diagnostics or therapeutics from Procter & Gamble, Zehna Therapeutics, and Cleveland HeartLab, a wholly owned subsidiary of Quest Diagnostics. Jennifer Buffa reports having received royalty payments from Proctor & Gamble. Dr. Fischbach is a co-founder and director of Federation Bio and Viralogic, a co-founder of Revolution Medicines, a member of the scientific advisory. M. Fischbach also reports Consultancy: NGM Bio; Ownership Interest: Kelonia, NGM Bio; Patents or Royalties: Federation Bio; and Advisory or Leadership Role: Federation Bio, Kelonia, Board of NGM Biopharmaceuticals, and an innovation partner at The Column Group. Dr. Tang reports being a consultant for Sequana Medical A.G., Owkin Inc, Relypsa Inc, and PreCardiac Inc, having received honorarium from Springer Nature for authorship/editorship and American Board of Internal Medicine for exam writing committee participation—all unrelated to the subject and contents of this paper. The other authors report they have no relationships relevant to the contents of this paper to disclose.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}