





Abstract
Excess dietary sodium intake and retention lead to hypertension. Impaired dermal lymphangiogenesis and lymphatic dysfunction–mediated sodium and fluid imbalance are pathological mechanisms. The adenosine A2A receptor (A2AR) is expressed in lymphatic endothelial cells (LECs), while the roles and mechanisms of LEC–A2AR in skin lymphangiogenesis during salt-induced hypertension are not clear.
The expression of LEC–A2AR correlated with lymphatic vessel density in both high-salt diet (HSD)–induced hypertensive mice and hypertensive patients. Lymphatic endothelial cell–specific A2AR knockout mice fed HSD exhibited 17 ± 2% increase in blood pressure and 17 ± 3% increase in Na+ content associated with decreased lymphatic density (−19 ± 2%) compared with HSD-WT mice. A2AR activation by agonist CGS21680 increased lymphatic capillary density and decreased blood pressure in HSD-WT mice. Furthermore, this A2AR agonist activated MSK1 directly to promote VEGFR2 activation and endocytosis independently of VEGF as assessed by phosphoprotein profiling and immunoprecipitation assays in LECs. VEGFR2 kinase activity inhibitor fruquintinib or VEGFR2 knockout in LECs but not VEGF-neutralizing antibody bevacizumab suppressed A2AR activation–mediated decrease in blood pressure. Immunostaining revealed phosphorylated VEGFR2 and MSK1 expression in the LECs were positively correlated with skin lymphatic vessel density and A2AR level in hypertensive patients.
The study highlights a novel A2AR-mediated VEGF-independent activation of VEGFR2 signaling in dermal lymphangiogenesis and sodium balance, which might be a potential therapeutic target in salt-sensitive hypertension.
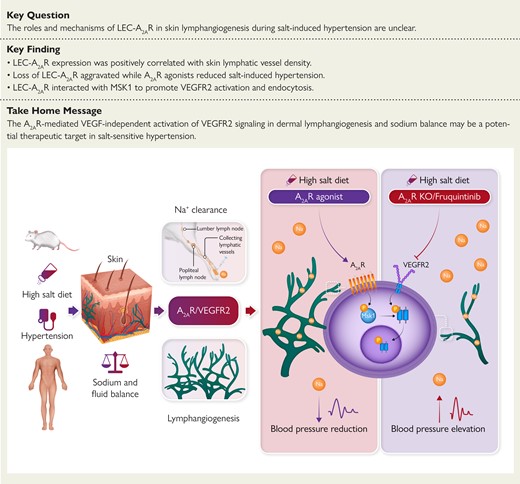
Impaired dermal lymphangiogenesis and lymphatic dysfunction contribute to the pathogenesis of salt-sensitive hypertension. Modulation of lymphatic endothelial A2AR-mediated VEGF-independent activation of VEGFR2 signaling, to regulate dermal lymphangiogenesis and sodium balance, might be a potential therapeutic target in salt-sensitive hypertension. A2AR, adenosine A2A receptor; KO, knock out; LEC, lymphatic endothelial cell; MSK1, mitogen- and stress-activated kinase 1; Na+, sodium; VEGF, vascular endothelial growth factor; VEGFR2, vascular endothelial growth factor receptor 2.
See the editorial comment for this article ‘Skin sodium, lymphatics, and blood pressure: a non-canonical mechanism of salt-sensitive hypertension', by R. Nosalski and T.J. Guzik, https://doi.org/10.1093/eurheartj/ehad290.
Excess dietary sodium intake and retention lead to hypertension. Impaired lymphangiogenesis and lymphatic dysfunction cause sodium and fluid imbalance which contribute to the development of salt-sensitive hypertension. A2AR-mediated activation of VEGFR2 signaling in dermal lymphangiogenesis and sodium balance might be a potential therapeutic target in salt-sensitive hypertension. Moreover, the hypertensive effect of the antineoplastic agent fruquintinib is partly due to its antagonism of the lymphangiogenic response to elevated salt intake.
Introduction
Excess dietary sodium intake and retention play a central role in the pathogenesis of hypertension.1 However, plasma sodium levels are maintained with a narrow range in both normotensive and hypertensive mammals,2,3 and the mechanisms for salt-sensitive hypertension remain unclear. Recent studies implied that impaired lymphangiogenesis and lymphatic dysfunction–mediated sodium and fluid imbalance contribute to the development of salt-sensitive hypertension.4,5 There is growing evidence that skin interstitium and the skin lymphatics are important regulators of both sodium (Na+) balance and blood pressure (BP).6 Sodium accumulates in the skin during a high-salt diet (HSD), which stimulates dermal macrophages to secrete vascular endothelial growth factor C (VEGFC), activating lymphangiogenesis locally.5 Inhibition of lymphangiogenesis exacerbates the hypertensive effect of HSD.7,8 However, the mechanisms by which the lymphatic endothelial cells (LECs) participate in mitigating salt-sensitive hypertension are largely unstudied.
The G protein–coupled adenosine receptors comprise A1, A2A, A2B, and A3 subtypes, which are activated upon binding with adenosine.9 The A2A receptor (A2AR) is abundantly expressed in various cell types, including endothelial cells, macrophages, and adipocytes, and regulates cardiovascular response.10–14 Activation of A2AR inhibits the release from immune cells of inflammatory factors, promotes vasodilation, regulates the secretion of adipokines, and thereby promotes cardiovascular homeostasis.15–17 Endothelial A2AR activation induces angiogenesis in wound healing and retinopathy.14 In normal rats, A2AR antagonists reduce renal sodium and water excretion.18 Furthermore, deletion of A2AR augments salt-induced BP elevation in Dahl salt-sensitive hypertensive animals.19
Herein, we report our discovery that A2AR expression in LECs is positively correlated with lymphatic vessel density in the skin in HSD-induced hypertensive mice as well as in hypertensive patients. We observe that a reduction in A2AR expression in LECs contributes to hypertension, because LEC-specific A2AR knockout mice (A2ARLECKO) have increased BP associated with decreased lymphangiogenesis and reduced skin Na+ clearance. We further observe a VEGF-independent activation of VEGFR2 during A2AR-mediated lymphangiogenesis. Consistent with this mechanism, we find that the VEGFR2 inhibitor fruquintinib or genetic VEGFR2 deletion in LEC, but not the VEGF antibody bevacizumab, reverses dermal lymphangiogenesis induced by the A2AR agonist and increases BP in HSD mice. These observations are consistent with our hypothesis that A2AR activation in LECs protects against high salt–induced hypertension by activating dermal lymphangiogenesis and restoring sodium balance in the skin.
Methods
Detailed materials and methods are presented in the supplementary material online.
Results
The lymphatic endothelial cell–A2AR mediates skin lymphangiogenesis and Na+ clearance
We observed that A2AR was the dominant adenosine receptor in mouse skin LECs (see Supplementary data online, Figure S1A). In cultured human LECs (HLECs), the adenosine receptor with the greatest increase in expression responsive to NaCl stimulation (40 mM) is A2AR. In contrast, the expression of A1R, A2BR, and A3R were unchanged in response to high NaCl (see Supplementary data online, Figure S1B). The expression of A2AR was dose-dependently increased in HLECs by NaCl (see Supplementary data online, Figure S1C). Moreover, we found no apparent LEC death when NaCl concentration is below 40 mM. However, at high-concentration NaCl (80 mM), we observed an increase in surface markers of apoptosis and necrosis (see Supplementary data online, Figure S1D and E). Therefore, all in vitro experiments were performed under 40 mM. We next observed the time-dependent A2AR expression in NaCl-treated HLECs and found that A2AR expression increased during the first 6 h and then decreased (see Supplementary data online, Figure S1F).
Similarly, we observed an increase in A2AR expression (66 ± 6%) in the cutaneous lymphatic endothelium, as well as serum adenosine in mice administered HSD, in parallel with increased dermal lymphatic capillary density (52 ± 11%) (Figure 1A–C). Intriguingly, in hypertensive patients, A2AR expression (−33 ± 9%) was decreased in skin lymphatic vessels (−28 ± 9%, podoplanin+ cell), associated with decreased lymphatic capillary density as observed in 3D images (Figure 1D and Supplementary data online, Videos S1 and S2) and coimmunostaining (Figure 1E–G). These findings are consistent with the hypothesis that lymphatic endothelial A2AR participates in HSD-driven lymphatic capillary density and sodium clearance in skin. We therefore hypothesized that the increased A2AR expression in mice may enhance sodium clearance by stimulating lymphangiogenesis.
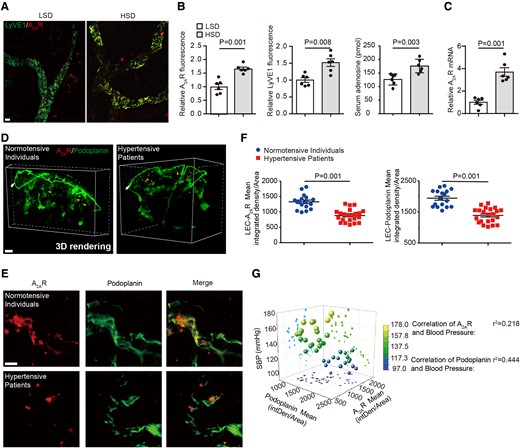
Lymphatic endothelial A2AR expression is altered in high-salt diet-induced hypertensive mice and patients. (A) Representative whole-mount staining of A2AR and LyVE1 in ears of mice fed with low-salt diet and high-salt diet. Scale bar: 100 μm. (B) Quantification of the fluorescent intensity of A2AR and LyVE1 and serum adenosine concentration in mice fed with low-salt diet and high-salt diet. n = 6. (C) A2AR expression by quantitative polymerase chain reaction in lymphatic endothelial cells of skin tissues of mice fed with low-salt diet and high-salt diet. n = 6. (D) Representative images of A2AR and podoplanin coimmunolabeling-enabled 3D imaging of solvent-cleared normotensive and hypertensive human skin tissue. Arrowheads indicate lymphatic endothelial cells. Scale bar: 300 μm. (E, F) Representative coimmunostaining of A2AR and podoplanin in normotensive and hypertensive human skin tissue (E) with quantification of the fluorescent intensity of A2AR and podoplanin immunostaining in lymphatic endothelial cells (F). Scale bar: 50 μm. (G) Relationship between A2AR or podoplanin expression and blood systolic blood pressure in human. Data in (B), (C), and (F) are presented as mean ± standard error mean and an unpaired two-tailed t test was performed to calculate significance between two groups. Data in (G) was analyzed by simple linear regression and visualized using the software Origin Pro 9.0, and the Pearson correlation was calculated as the square root of r2. P < 0.05 was considered as statistically significant. LSD, low-salt diet; HSD, high-salt diet.
The lymphatic endothelial cell–A2AR protects against salt-induced hypertension
We performed flow cytometric analyses of disaggregated skin from HSD mice and found an increased F4/80-LyVE1+ LECs, but not F4/80+LyVE1+ macrophages (see Supplementary data online, Figure S2A). Although it has been reported that macrophage deletion of A2AR had no effect on hypertension,20 we performed bone marrow (BM) transplantation assay from A2AR global knockout mice (A2ARKO-BM)16 or control WT mice (A2ARWT-BM) to wild-type C57 mice to exclude the potential roles of macrophage A2AR in high salt–induced hypertension. No statistical difference of BP, lymphatic vessel density, skin VEGF mRNA expression, and Na+ content was observed between A2ARWT-BM and A2ARKO-BM mice (see Supplementary data online, Figure S3A–E). These data indicate that Lyve1-Cre is appropriate for studying skin lymphatic system.
To explore the role of the lymphatic endothelial A2AR in hypertension, we generated the LEC-specific Adora2a receptor gene deficient (Lyve1-Cre+ A2ARfl/fl; abbreviated as A2ARLECKO) and control littermate (Lyve1-Cre− A2ARfl/fl; abbreviated as WT) mice. The A2AR expression was decreased (−77 ± 5%) in LECs, but not significantly altered in skin macrophages and vascular ECs from A2ARLECKO mice (see Supplementary data online, Figure S2B). A2AR deletion in LECs had no effect on dermal lymphatic capillary density, BP, skin Na+, and Cl− content and concentration with low-salt diet (LSD) (Figures 2A and B and S2C).
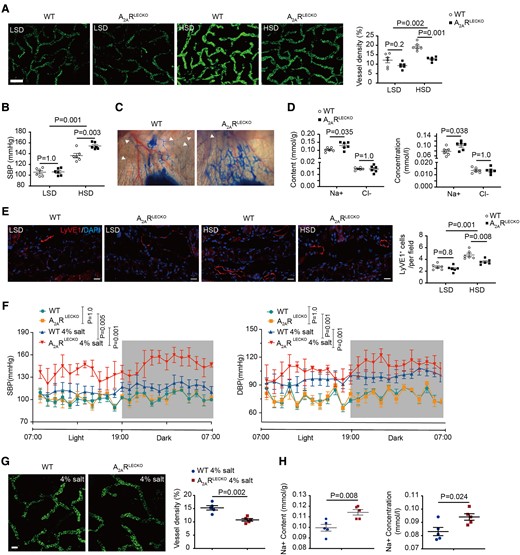
Lymphatic endothelial-specific A2AR deletion increases blood pressure, decreases lymphangiogenesis, and skin Na+ clearance in response to high-salt diet. (A) Representative whole-mount staining of LyVE1 in ears of A2ARLECKO and wild-type mice fed with low-salt diet and high-salt diet, with quantification of percentage of LyVE1-positive area at the right. n = 6. Scale bar: 200 μm. (B) Systolic blood pressure in A2ARLECKO and wild-type mice after low-salt diet and high-salt diet. n = 6. (C) Representative images of Evans blue lymphangiography in ears of A2ARLECKO and wild-type mice fed with high-salt diet. In the A2ARLECKO mice, the lymphatic vessels are dilated, and extravasation of Evans blue is observed. Arrows indicate lymphatic vessels. (D) Na+ and Cl− content and concentrations in skin of the same mice fed with high-salt diet. n = 6. (E) Representative images of LyVE1 immunostaining in ear section of A2ARLECKO and wild-type mice fed with low-salt diet and high-salt diet. Scale bar: 50 μm. (F) Radiotelemetry BP monitoring (DSI) assay to detect blood pressure in A2ARLECKO and wild-type mice with 4% salt chow diet plus 0.9% saline ad libitum. n = 3. (G) Representative whole-mount staining of LyVE1 in ears of A2ARLECKO and wild-type mice fed with 4% salt chow diet plus 0.9% saline ad libitum, with quantification of percentage of LyVE1-positive area at the right. n = 5. Scale bar: 50 μm. (H) Na+ content and concentrations in skin of A2ARLECKO and wild-type mice fed with 4% salt chow diet plus 0.9% saline to drink ad libitum. All data are presented as mean ± standard error mean. Data in (A), (B), (E), and (F) were analyzed by two-way ANOVA. Data in (D), (G), and (H) were analyzed by unpaired two-tailed t test. P < 0.05 was considered as statistically significant. LSD, low-salt diet; HSD, high-salt diet; WT, wild-type.
Intriguingly, with HSD, we observed an increase in dermal lymphatic capillary density (59 ± 7%) and an increase in BP (29 ± 4%) in WT mice compared with LSD-fed WT mice. In comparison, in the A2ARLECKO mice, there was a blunted increase in lymphatic capillary density (37 ± 5%) and an exaggerated increase in BP (45 ± 2%) with HSD (Figure 2A and B). Thus, A2AR deficiency in LECs inhibits cutaneous lymphangiogenesis and increases BP in response to HSD. We also visualized lymphatic vessels by Evans blue dye lymphangiography and found fine distinct lymphatic vessels in HSD-treated WT mice. In contrast, HSD-treated A2ARLECKO mice showed dilated lymphatic vessels and Evans blue dye extravasated from these dilated vessels (Figure 2C and Supplementary data online, Videos S3 and S4). Na+ content and concentration were both increased, but Cl− content and concentration were unchanged in the skin of HSD-A2ARLECKO mice compared with HSD-WT mice (Figure 2D). No statistical differences in plasma Na+ and Cl− concentration were observed between HSD-A2ARLECKO and HSD-WT littermate mice (see Supplementary data online, Figure S2D). The decreased cutaneous lymphatic capillary density in A2AR-deficient mice was further confirmed by quantitative immunofluorescence microscopy analysis using the lymphatic marker LyVE1 (Figure 2E).
We also utilized a tamoxifen-inducible Prox1CreERT2 mouse line to specifically delete LEC–A2AR in adult and generated Prox1CreERT2+ A2ARfl/fl (abbreviated as A2ARPLECKO) and control littermate A2ARfl/fl mice. Tamoxifen administration extinguished A2AR expression in LECs of A2ARPLECKO mice (see Supplementary data online, Figure S4A). In response to HSD, A2ARPLECKO mice exhibited an attenuated dermal lymphatic density and exaggerated increase in systolic BP (SBP) and cutaneous Na+ accumulation compared in comparison to control A2ARfl/fl mice (see Supplementary data online, Figure S4B–D).
We next used a mild mouse model with 4% salt diet plus 0.9% saline water to drink and detected change in BP of A2ARLECKO and littermate WT mice by radiotelemetry BP monitoring. Both SBP and diastolic BP were elevated in WT mice (Figure 2F). Blood pressure was elevated to a greater degree in the A2ARLECKO mice, accompanied with reduced dermal lymphatic capillary density (−30 ± 3%), and increased skin Na+ content (from 0.10 ± 0.003 to 0.11 ± 0.003 mmol/g) and Na+ concentration (from 0.083 ± 0.003 to 0.094 ± 0.002 mmol/L, Figure 2F–H).
To determine if other causes of hypertension were exacerbated in the A2ARLECKO mouse, we induced hypertension using angiotensin II infusion. We observed no differences in dermal lymphatic capillary density or BP between A2ARLECKO and WT littermates treated with angiotensin II (see Supplementary data online, Figure S5A–C).
To determine if there is a direct link between lymphangiogenesis and BP, we treated mice with recombinant VEGFC. As expected, VEGFC infusion significantly increased skin lymphatic capillary density in HSD-WT mice from 56 ± 1 to 74 ± 2 AU and normalized the decreased skin lymphangiogenesis in HSD-A2ARLECKO mice from 44 ± 2 to 60 ± 1 AU (see Supplementary data online, Figure S6A). Similarly, VEGFC administration normalized skin Na+ clearance and BP in HSD-A2ARLECKO mice (see Supplementary data online, Figure S6B and C). The effect of LEC–A2AR on lymphangiogenesis was confirmed in A2AR homozygous knockout HLECs generated by CRISPR/Cas9 methodology [documented by quantitative polymerase chain reaction (qPCR) and western blot; Supplementary data online, Figure S7A]. Consistent with the in vivo data, NaCl inhibited HLEC proliferation, migration, and tube formation with reduction of branch points and total tube length (see Supplementary data online, Figure S7B–D). This effect of NaCl was exacerbated by A2AR knockout (see Supplementary data online, Figure S7B–D).
Lymphatic endothelial cell–A2AR deletion decreases Na+ transport caused by lymphatic dysfunction
To provide direct evidence that A2AR regulates lymphatic function, especially Na+ removal, we measured the transport of Evans blue from the peripheral tissue to the popliteal lymph node (LN) and then lumbar LN. Transport of Evans blue, quantified by VISQUE fluorescence imaging system, from foot to popliteal and lumbar LN was unchanged in LSD-A2ARLECKO compared with LSD-WT mice (Figure 3A and C). We observed decreased transport of Evans blue (−30 ± 3%) in HSD-A2ARLECKO mice compared with HSD-WT mice (Figure 3A and C–E). Furthermore, we evaluated Na+ transport by detecting Na+ concentration in the LN at specified times after subcutaneous injection of NaCl. Na+ concentrations in both popliteal (0.016 ± 0.0003 mmol/L vs. 0.014 ± 0.0005 mmol/L) and lumbar (0.014 ± 0.0004 vs. 0.013 ± 0.0004 mmol/L) LNs of HSD-A2ARLECKO mice were significantly reduced in comparison to HSD-WT mice, although Na+ concentrations between LSD-A2ARLECKO and LSD-WT mice were not changed (Figure 3A and B).

A2AR deletion in lymphatic endothelial cells decreases the transport of Na+ and Evans blue under high-salt diet treatment. (A) Schematic diagram of in vivo experiment to show the transport of Na+ and Evans blue. (B) Na+ content in popliteal lymph node (left) and lumbar lymph node (right) of high-salt diet–treated A2ARLECKO and wild-type mice. n = 5. (C) Quantification of relative fluorescent intensity of Evans blue in popliteal lymph node. n = 5. (D) Representative images of Evans blue in popliteal lymph node (arrow) and lumbar lymph node (arrow). Scale bar: 5 mm. (E) Representative images of Evans blue in popliteal lymph node (arrow) and lumbar lymph node (arrow) captured by VISQUE fluorescence imaging system. (F) Plasma renin, angiotensin II, and aldosterone concentration in A2ARLECKO and wild-type mice. n = 5. (G) Urine Na+ and Cl− concentration in in A2ARLECKO and wild-type mice. n = 5. (H) Feed (left) and water (right) consumption in A2ARLECKO and wild-type mice. n = 5. (I) Na+ consumption of feed (left) and drink (right) in A2ARLECKO and wild-type mice. n = 5. All data are presented as mean ± standard error mean. Data in (B), (C), (H), and (I) were analyzed by two-way ANOVA. Data in (F) and (G) were analyzed by unpaired two-tailed t test. P < 0.05 was considered as statistically significant. LN, lymph node; LV, lymphatic vessel; LSD, low-salt diet; HSD, high-salt diet; WT, wild-type; pLN, popliteal lymph node.
Because A2AR did not affect angiotensin II–induced hypertension, we also assessed renin–angiotensin–aldosterone system and renal function. The plasma renin activity, angiotensin II, and aldosterone were not changed in HSD-treated A2ARLECKO and WT mice (Figure 3F). Functional renal analyses by metabolic cage experiments revealed no significant difference of feed and water consumption, Na+ consumption of feed and drink, and urine Na+ and Cl− between A2ARLECKO and WT mice with HSD (Figure 3G–I). These data indicate that the LEC–A2AR deficiency exacerbates salt-induced hypertension via aggravating lymphatic dysfunction to decrease dermal lymphangiogenesis and Na+ transport.
Pharmacological activation of A2AR induces lymphangiogenesis and skin Na+ clearance and decreases blood pressure
To determine whether A2AR activation could protect against HSD-induced hypertension, we treated HSD-fed A2ARLECKO and WT mice with the A2AR-specific agonist CGS21680. This A2AR agonist increased lymphatic capillary density (40 ± 9%) and decreased BP (−23 ± 12%) in HSD-WT mice (Figure 4A and B). Decreased Na+ (−19 ± 11%) and unchanged Cl− content and concentration were observed in the skin of HSD-WT mice treated with CGS21680 (Figure 4C). More importantly, these beneficial effects of A2AR activation including lymphangiogenesis, skin Na+ clearance, and BP reduction were absent in HSD-treated A2ARLECKO mice (Figure 4A–C). Consistently, A2AR agonist CGS21680 could increase HLEC proliferation, migration, and tube formation in vitro, which were also attenuated after A2AR knockout (Figure 4D–F). These observations suggest that pharmacological activation of A2AR protects against HSD-induced hypertension via inducing dermal lymphangiogenesis and promoting skin Na+ clearance.
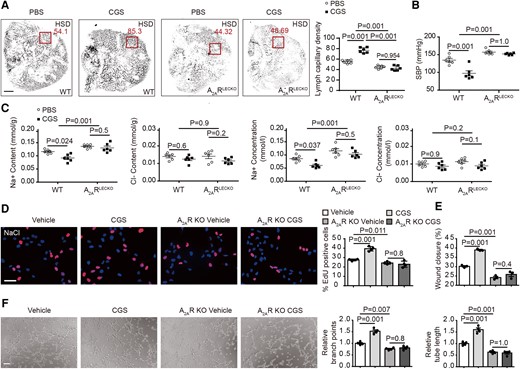
Pharmacological stimulation of A2AR decreases blood pressure and increases lymphangiogenesis and skin Na+ clearance. (A) Representative whole-mount staining of LyVE1 in ears of high-salt diet–fed A2ARLECKO and wild-type mice with and without A2AR agonist CGS21680, with quantification of lymphatic capillary density at the right. n = 6. The squares indicate the typical area used for quantification of lymph capillary density in ears. Scale bar: 2 mm. (B) Systolic blood pressure in A2ARLECKO and wild-type mice fed with high-salt diet, with and without CGS21680. n = 6. (C) Skin Na+ and Cl− content and concentration in the same mice fed with high-salt diet. n = 6. (D) Cell proliferation assessed by EdU staining with quantification at the right in NaCl-treated A2AR knockout and control wild-type human lymphatic endothelial cells, with and without CGS21680. n = 4. Scale bar: 50 μm. (E) Cell migration assessed by wound healing in NaCl-treated A2AR knockout and control wild-type human lymphatic endothelial cells, with and without CGS21680. n = 3. (F) Representative images of tube formation with quantification of branch points and tube length at the right in NaCl-treated A2AR knockout and control wild-type human lymphatic endothelial cells, with and without CGS21680. n = 4. Scale bar: 100 μm. All data are presented as mean ± standard error mean. Data in (A)–(C) were analyzed by two-way ANOVA. Data in (D)–(F) were analyzed by one-way ANOVA. P < 0.05 was considered as statistically significant. CGS, A2AR agonist CGS21680; HSD, high-salt diet; WT, wild-type; KO, knockout.
A2AR regulates VEGFR2 activity via promoting MSK1 phosphorylation
To gain insight into of the mechanisms of LEC–A2AR-mediated lymphangiogenesis, we performed phosphoprotein profiling assay in HLECs following pharmacological A2AR activation. The protein microarray identified phosphorylated VEGFR2 (Tyr1175) expression was increased 1.8-fold in HLECs, whereas VEGFR2 protein expression was not changed, after activation of A2AR (see Supplementary data online, Table S4 and Figure 5A and B), which were confirmed by western blot in both skin LECs and NaCl-treated HLECs with A2AR agonist stimulation (Figure 5C). In contrast, the expression of phosphorylated VEGFR2 (Tyr1175) and mitogen- and stress-activated kinase 1 (MSK1) (Ser376) were not changed after treatment of A2ARLECKO mice or A2AR knockout HLECs with angiotensin II (see Supplementary data online, Figure S5D–F). It is known that VEGFR2 undergoes endocytosis and that phosphorylated VEGFR2 could translocate to the nucleus.21,22 We found that A2AR activation induces VEGFR2 endocytosis and increases phosphorylated VEGFR2 in the nucleus (see Supplementary data online, Figure S8C and D). The expression of VEGF family, including VEGFA, VEGFB, VEGFC, and VEGFD, in NaCl-treated HLECs were not changed following A2AR knockout (Figure 5D) or A2AR activation (Figure 5E). These studies indicate that VEGFR2 phosphorylation and internalization induced by A2AR might be independent of VEGF.
![A2AR regulates VEGFR2 activity via binding to MSK1. (A) Representative image (left) and quantification (right) of protein array showing spots for total and phosphorylated VEGFR2 (boxes) and MSK1 (boxes) in NaCl-treated human lymphatic endothelial cells following vehicle or A2AR agonist CGS21680 stimulation. (B) Signal pathway analysis showing phosphorylated signals regulated by A2AR activation. (C) Representative images of total and phosphorylated VEGFR2 and MSK1 expression in the skin of mice (left) and cultured human lymphatic endothelial cells (right) with and without A2AR agonist CGS21680 treatment evaluated by western blot, with quantification of phosphorylated/total VEGFR2 and MSK1 at the right. (D) VEGF mRNA expression in A2AR knockout and control wild-type human lymphatic endothelial cells with NaCl treatment. n = 3. (E) VEGF mRNA expression in NaCl-treated high-salt diet, with or without A2AR agonist CGS21680. n = 3. (F) Representative images of immunoprecipitation in the skin of high-salt diet–treated mice (left) and 293T cells transfected with V5-tagged A2AR and Flag-tagged MSK1 vector (right), with images of Ponceau staining showing heavy chain below. (G) Representative images of phosphorylated VEGFR2 and MSK1 immunoblots in the nucleus of NaCl-treated human lymphatic endothelial cells with MSK1 and Ser376 mutated MSK1 [MSK1(376A)] overexpression. (H) Representative images of phosphorylated VEGFR2 and MSK1 immunocytochemistry in NaCl-treated human lymphatic endothelial cells with MSK1 and MSK1(376A) overexpression. Scale bar: 10 μm. (I, J) Representative images (I) of EdU staining (up) and wound healing (below) in Empty-, MSK1-, and MSK1(376A)-transfected human lymphatic endothelial cells under NaCl stimulation, with quantification of positive EdU cells and wound closure below (J). n = 4. Scale bar: 100 μM. All data are presented as mean ± standard error mean. Data in (C), (I), and (J) were analyzed by one-way ANOVA. Data in (D) and (E) were analyzed by unpaired two-tailed t test. P < 0.05 was considered as statistically significant. CGS, A2AR agonist CGS21680; LSD, low-salt diet; HSD, high-salt diet; WT, wild-type; KO, knockout; IP, immunoprecipitation; IB, immunoblot.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/eurheartj/44/29/10.1093_eurheartj_ehad377/1/m_ehad377f5.jpeg?Expires=1750432232&Signature=XzedN5lW2OcaL6xwnM2-Ct6cSYJf20fi4dFnBQlBuVdt4kVJhb45ze5kbDMeDWYo9nEKOOA211FKoq~x0-nNGd4z13FoCFYXyjpVP0z8MOorBw-1HCHoUrpIPnboBgZ4lW-MTE-5dZ5q-ImuxZLYXZk1n-osEQhOPamfAfbDPMpYXJwohlddEV3qMgx40IPE-IywQ8HmfWXw2pU4i-~4YqA-ohtzxDiMuMxNgzYDr6VLlkS6~dxDtZzY3C0CAlIcGYmzF3rK0Ic7U50Htv4zpvTOXbTim1xGAPFqz9rgsLLQgbQNru7KIDPi4OWQKxRx2R-Zsjfk9~Nw~lK62nf3SQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
A2AR regulates VEGFR2 activity via binding to MSK1. (A) Representative image (left) and quantification (right) of protein array showing spots for total and phosphorylated VEGFR2 (boxes) and MSK1 (boxes) in NaCl-treated human lymphatic endothelial cells following vehicle or A2AR agonist CGS21680 stimulation. (B) Signal pathway analysis showing phosphorylated signals regulated by A2AR activation. (C) Representative images of total and phosphorylated VEGFR2 and MSK1 expression in the skin of mice (left) and cultured human lymphatic endothelial cells (right) with and without A2AR agonist CGS21680 treatment evaluated by western blot, with quantification of phosphorylated/total VEGFR2 and MSK1 at the right. (D) VEGF mRNA expression in A2AR knockout and control wild-type human lymphatic endothelial cells with NaCl treatment. n = 3. (E) VEGF mRNA expression in NaCl-treated high-salt diet, with or without A2AR agonist CGS21680. n = 3. (F) Representative images of immunoprecipitation in the skin of high-salt diet–treated mice (left) and 293T cells transfected with V5-tagged A2AR and Flag-tagged MSK1 vector (right), with images of Ponceau staining showing heavy chain below. (G) Representative images of phosphorylated VEGFR2 and MSK1 immunoblots in the nucleus of NaCl-treated human lymphatic endothelial cells with MSK1 and Ser376 mutated MSK1 [MSK1(376A)] overexpression. (H) Representative images of phosphorylated VEGFR2 and MSK1 immunocytochemistry in NaCl-treated human lymphatic endothelial cells with MSK1 and MSK1(376A) overexpression. Scale bar: 10 μm. (I, J) Representative images (I) of EdU staining (up) and wound healing (below) in Empty-, MSK1-, and MSK1(376A)-transfected human lymphatic endothelial cells under NaCl stimulation, with quantification of positive EdU cells and wound closure below (J). n = 4. Scale bar: 100 μM. All data are presented as mean ± standard error mean. Data in (C), (I), and (J) were analyzed by one-way ANOVA. Data in (D) and (E) were analyzed by unpaired two-tailed t test. P < 0.05 was considered as statistically significant. CGS, A2AR agonist CGS21680; LSD, low-salt diet; HSD, high-salt diet; WT, wild-type; KO, knockout; IP, immunoprecipitation; IB, immunoblot.
We next determined whether A2AR directly interacted with VEGFR2. Immunoprecipitation in 293T cells transfected with V5-A2AR and HA-VEGFR2 revealed that A2AR did not directly interact with VEGFR2 (see Supplementary data online, Figure S8A). Therefore, we employed protein microarray to identify potential A2AR downstream signaling that may activate VEGFR2 phosphorylation. We found that phosphorylated MSK1 (Ser376) was increased following A2AR activation (Figure 5A and B), which was confirmed by western blot in both skin LECs and HLECs treated with A2AR agonist and NaCl (Figure 5C and D). Immunostaining data showed that in CGS-treated mice, there was increased phosphorylation of MSK1 (Ser376) and VEGFR2 (Tyr1175) in LECs (see Supplementary data online, Figure S8B and C). Immunoprecipitation assay in 293T cells transfected with V5-tagged A2AR and Flag-tagged MSK1 vector showed that A2AR directly interacted with MSK1 (Figure 5F). To further confirm the result in vivo, we performed immunoprecipitation in HSD-treated mice and found that A2AR directly bound to MSK1 (Figure 5F).
To investigate the effects of phosphorylated site Ser376 of MSK1 on VEGFR2 activity and endocytosis, we mutated Ser376 of MSK1 to Ala376 [MSK1(376A)] and performed western blots in MSK1 or mutated MSK1(376A)-transfected HLECs under NaCl stimulation. An increase in phosphorylated MSK1 (Ser376) and phosphorylated VEGFR2 (Tyr1175) was observed in the nucleus of MSK1-transfected HLECs compared with Empty-transfected HLECs (Figure 5G). In comparison, phosphorylated MSK1 (Ser376) and phosphorylated VEGFR2 (Tyr1175) in the nucleus of MSK1(376A)-transfected HLECs were significantly decreased compared with MSK1-transfected HLECs. Similar results were observed in the nucleus of HLECs by utilizing confocal immunostaining (Figure 5H). We also performed in vitro EdU and wound healing assays in MSK1 or mutated MSK1(376A)-transfected HLECs and found that MSK1 overexpression promoted cell proliferation and migration, while mutated MSK1(376A) had no effect on HLEC proliferation and migration (Figure 5I and J). These studies suggest that MSK1(376A) plays an important role in VEGFR2 activation and internalization and HLEC function.
We also utilized the MSK1 antagonist (CKI7) and found that MSK1 antagonist inhibited phosphorylation of MSK1 and VEGFR2 (see Supplementary data online, Figure S9A) and decreased HLEC proliferation, migration, and tube formation (see Supplementary data online, Figure S9B–D). These studies suggest that A2AR activation regulates LEC functions via MSK1-dependent, VEGF-independent VEGFR2 activation and internalization.
Pharmacological inhibition or lymphatic endothelial-specific genetic deletion of VEGFR2 blocks the beneficial effects of A2AR activation in high-salt diet mice
To determine whether VEGF-independent VEGFR2 signaling is involved in A2AR activation–mediated phenotype in HSD mice, we utilized the antineoplastic agent VEGFR2 tyrosine kinase inhibitor fruquintinib or VEGF-neutralizing antibody bevacizumab. Bevacizumab scavenges VEGF to prevent its activation of VEGFR2, whereas fruquintinib inhibits processes downstream of VEGFR2 activation. Mice fed HSD were pretreated with the antagonists and then received the A2AR agonist CGS21680. CGS21680 treatment increased lymphatic capillary density (from 19.0 ± 0.3% to 27.0 ± 0.4%) and skin Na+ clearance (Na+ content from 0.1 ± 0.001 to 0.0794 ± 0.004 mmol/g) and decreased BP (from 120 ± 3 to 97 ± 5) in HSD-WT mice. Fruquintinib pretreatment decreased lymphatic capillary density (by −36 ± 1%) and skin Na+ clearance (by −19 ± 3%) and increased BP (by 42 ± 8%). In contrast, bevacizumab did not influence these parameters in CGS21680-treated HSD mice (Figures 6A–C and S10A).

VEGFR2 inhibition or genetic deletion in lymphatic endothelial cell blocks the beneficial effects of A2AR activation in high-salt diet mice. (A) Representative whole-mount staining of LyVE1 in ears of vehicle or A2AR agonist CGS21680–treated mice fed with high-salt diet, with and without fruquintinib or bevacizumab, with quantification of percentage of LyVE1-positive area at the right. Scale bar: 100 μm. n = 4. (B) Systolic blood pressure in vehicle or A2AR agonist CGS21680–treated mice fed with high-salt diet, with and without fruquintinib or bevacizumab. n = 4. (C) Na+ content and concentrations in the skin of the same mice. n = 4. (D) Blood pressure in bland and high-salt diet patients before and after fruquintinib (left) or bevacizumab (right) treatment. (E) VEGFR2 expression in lymphatic endothelial cells of VEGFR2PLECKO and wild-type mice evaluated by quantitative polymerase chain reaction. n = 5. (F) Representative images of phosphorylated VEGFR2 immunostaining in mice. Scale bar: 100 μm. (G) Representative whole-mount staining of LyVE1 in ears of vehicle or A2AR agonist CGS21680–treated wild-type and A2AR agonist CGS21680–treated VEGFR2PLECKO mice fed with high-salt diet, with quantification of percentage of LyVE1-positive area at the right. Scale bar: 100 μm. n = 5. (H) Systolic blood pressure in vehicle or A2AR agonist CGS21680–treated wild-type and A2AR agonist CGS21680–treated VEGFR2PLECKO mice fed with high-salt diet. n = 5. (I) Na+ content and concentrations in the skin of the same mice fed with high-salt diet. n = 5. (J) Representative images and quantification of Evans blue in popliteal lymph node (arrow) of low-salt diet wild-type mice, vehicle or A2AR agonist CGS21680–treated wild-type and A2AR agonist CGS21680–treated VEGFR2 PLECKO mice after high-salt diet. n = 5. All data are presented as mean ± standard error mean. Data in (A)–(C) and (G)–(J) were analyzed by one-way ANOVA. Data in (D) and (E) were analyzed by unpaired two-tailed t test. P < 0.05 was considered as statistically significant. CGS, A2AR agonist CGS21680; Bev, bevacizumab; Fru, fruquintinib; pLN, popliteal lymph node; LSD, low-salt diet; HSD, high-salt diet; BD, bland.
We next observed the effect of fruquintinib or bevacizumab on BP of colorectal cancer patients without history of hypertension before hospitalization. The BP of patients was increased after fruquintinib or bevacizumab treatment (see Supplementary data online, Table S2). We then grouped the patients into bland diet group and HSD group according to their dietary preferences. We found a significant increase in BP in high-salt group compared with bland diet patients with fruquintinib, while no difference of BP was observed between bland diet and high-salt patients with bevacizumab (Figure 6D). These data demonstrate that fruquintinib increases BP mainly in patients with HSD preference.
Moreover, we obtained conditional VEGFR2fl/fl mice and crossed with Prox1CreERT2 mice to generate Prox1CreERT2+VEGFR2fl/fl (VEGFR2PLECKO) mice. In these animals, tamoxifen treatment reduced VEGFR2 expression in LECs (by −70 ± 3%), which was confirmed by qPCR and immunostaining (Figure 6E and F). VEGFR2 deletion in LECs specifically reversed the effects of A2AR activation to increase lymphangiogenesis and skin Na+ clearance and to reduce BP in HSD-treated mice (Figure 6G and I). Furthermore, the increased transport of Evans blue from peripheral tissue to popliteal LN in HSD mice pretreated with A2AR agonist CGS21680 was blocked when VEGFR2 was deleted in LECs (Figure 6J). These data confirmed that the effects of A2AR activation on high salt–induced hypertension were through downstream VEGFR2 signaling.
In accordance with the in vivo studies, fruquintinib reversed A2AR agonist–mediated increase of LEC proliferation and migration, whereas bevacizumab has no effect on LEC functions (see Supplementary data online, Figure S10B and C). Knockdown of VEGFR2 in vitro cultured HLEC also attenuated CGS-induced proliferation, migration, and tube formation (see Supplementary data online, Figure S11A–D). These studies further indicate that A2AR activates VEGF-independent VEGFR2 signaling, which increases dermal lymphangiogenesis to protect against HSD-induced hypertension.
Phosphorylated VEGFR2 and MSK1 levels positively correlate with lymphatic markers in the skin of hypertensive patients
We finally evaluated whether phosphorylated VEGFR2 and MSK1 expression correlated with markers of lymphatic vessels (podoplanin) and Na+ content and BP in the skin tissue of hypertensive patients. We observed that phosphorylated MSK1 (Figure 7A) and VEGFR2 (Figure 7B) were decreased in hypertensive patients, which values also correlated with increased skin Na+ retention and decreased podoplanin expression (Figure 7C–F). These data provide evidence of morphological and functional changes in cutaneous lymphatics of hypertensive patients resulting in reduced sodium clearance. Thus, restoring the structure and function of cutaneous lymphatics may represent a novel therapeutic target for hypertension.
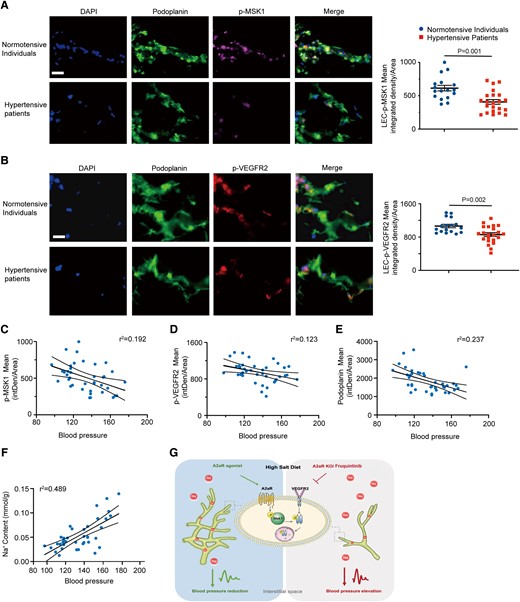
Expression levels of phosphorylated VEGFR2 and MSK1 positively correlate with lymphatic markers in human skin tissues with hypertension. (A, B) Representative immunostaining of p-MSK1 (A) and p-VEGFR2 (B) in normotensive and hypertensive patients with quantification of integrated density per area at right. Scale bar: 50 μm. (C–F) Relationship of skin phosphorylated MSK1 (C), p-VEGFR2 (D), and podoplanin (E) expression and Na+ content (F) with systolic blood pressure in normotensive and hypertensive patients. (G) Model of lymphatic endothelial A2AR-mediated VEGF-independent activation of VEGFR2 signaling in dermal lymphangiogenesis and sodium balance. Data in (A) and (B) are presented as mean ± standard error mean and were analyzed by unpaired two-tailed t test. Data in (C)–(F) were analyzed by simple linear regression, and the Pearson correlation was calculated as the square root of r2. P < 0.05 was considered as statistically significant. intDen, integrated density.
Discussion
We provide evidence for a role of the LEC–A2AR in mediating skin lymphangiogenesis, sodium clearance, and BP. Lymphatic endothelial cell–A2AR expression was increased in a positive relationship with skin lymphatics in HSD-induced hypertensive mice. Furthermore, A2ARLECKO have reduced lymphatic vessel density, increased skin sodium, and an exaggerated increase in BP with HSD. The A2AR-induced lymphangiogenesis in high-salt conditions is regulated by MSK1-mediated VEGF-independent VEGFR2 internalization and activation (Figure 7G). Finally, we show that in hypertensive patients, there is a decrease in lymphatic vessels and an increase in skin sodium concentration. Furthermore, the expression of A2AR, as well as phosphorylated VEGFR2 and MSK1, is reduced in skin tissue of hypertensive patients and is inversely correlated with Na+ content and concentration. Therefore, LEC–A2AR-mediated VEGF-independent activation of VEGFR2 signaling mediates dermal lymphangiogenesis, sodium balance, and BP.
There is a causal relationship between dietary salt intake and BP. Reduction in salt intake from the current world average of ∼10 g/day to the World Health Organization recommended level of <5 g/day lowers BP and reduces the risk of cardiovascular disease and all-cause mortality.23–25 Previous studies have confirmed high salt intake elevated BP in both rodents7,26 and humans.3,27 The molecular mechanisms of high salt intake and BP elevation are still unclear. There is growing evidence that skin is a large reservoir of Na+ ions which are stored in an osmotically inactive form on glycosaminoglycans.6 Local hypertonicity due to extensive accumulation of Na+ within the skin was reported to induce macrophage accumulation and subsequently to secrete VEGFC via upregulating transcription factor TonEBP, resulting in lymphangiogenesis within the skin.5 High levels of salt exposure also elevated BP via upregulating the NLRP3 inflammasome, pyroptotic and apoptotic caspases, and interleukin-1β transcription in monocytes.26
The increased lymphangiogenesis in the skin seems to be adaptive in maintaining BP in high salt-load states, though one recent study revealed that dermal lymphatic vessels are not involved in salt storage or BP regulation.28 However, more studies showed that administration of exogenous VEGFC, or VEGFC overexpression, attenuates HSD-induced BP elevation and related target organ damage.7,29,30 Furthermore, HSD-induced extracellular sodium accumulation causes lymph flow increase, effect which is blocked by depletion of macrophages/monocytes.7 These studies suggest that lymphangiogenesis has an important role in BP control in high-salt conditions. Herein, we utilized two models of HSD: an angiotensin II and 8% HSD model, and a less intense mouse model with 4% salt diet. Blood pressures in HSD-treated mice were assessed by the tail–cuff method, as well as radiotelemetry, as the latter method was more sensitive to the smaller BP changes in the 4% salt diet–treated mice. Both models confirmed the association of lymphangiogenesis with BP.
A related question is whether the function of the skin lymphatics is affected by high dietary salt, a question that had not been addressed previously. We herein provide direct evidence that high-salt treatment inhibits the transport of Na+ from the skin to the LNs, associated with an increase in skin sodium and BP. Lymphatic endothelial cell–A2AR activation mediates skin lymphangiogenesis and skin sodium clearance and downregulates BP. Our hypothesis was supported by observations of the effects of HSD or A2AR activation on lymphatic transport of Na+, as assessed by changes in Na+ concentration over time in regional LNs after subcutaneous injection of NaCl. Methods with greater temporal and spatial resolution are needed to confirm and refine our observations.
A2AR is abundantly expressed in immune cells and throughout the cardiovascular system. Activation of A2AR is known to induce a Th2 response and the production of antiinflammatory factors, attenuating cardiovascular inflammation and related organ damage.17 Furthermore, A2AR induces vasodilation by activating the release of endothelium-derived nitric oxide and by opening ATP-sensitive potassium channels to hyperpolarize vascular smooth muscle.31 We herein show that HLEC–A2AR is increased and then decreased with time-dependent high-salt conditions in vitro. Though the number of skin samples is small, and skin Na+ concentrations and lymphangiogenesis might be affected by antihypertensive drugs or concomitant diseases, we observed a reduced LEC–A2AR expression in the skin of hypertensive patients.
The molecular mechanism of high salt–mediated A2AR expression needs further investigation. The core promoter region of the A2AR gene has binding sites for the transcription factor cAMP response element–binding protein (CREB).32 Meanwhile, CREB is the target of salt-inducible kinases, which could be phosphorylated and activated responsive to high-salt conditions.33 The detailed mechanism of CREB in regulation of A2AR expression needs to be further explored in the future.
VEGF family members and their receptors are crucial for development and maintenance of both the blood and lymphatic vascular networks. Among these, VEGFC and VEGFD are the primary growth factors regulating the lymphatic vasculature via the VEGFR3 tyrosine kinase pathway.34,35 VEGFC also activates VEGFR2 in LECs by promoting heterodimerization between VEGFR2 and VEGFR3, and regulates LEC survival, proliferation, migration, and lymphangiogenesis.36–38 It is well established that VEGF-independent VEGFR upregulation in endothelial cells contributes to angiogenesis.39 We herein provide direct evidence that VEGF-independent VEGFR2 phosphorylation and activation in LECs promote lymphangiogenesis in high-salt conditions, which is mediated by the A2AR–MSK1 signaling pathway. MSK1 activates ERK1/2 and p38 MAPK pathways and endothelial cell function.40,41 MSK1 contains an N-terminal kinase domain (NTKD) in the AGC kinase family and a C-terminal kinase domain (CTKD) from the calmodulin kinase family. MSK1 activation causes CTKD autophosphorylation of NTKD which can phosphorylate substrates such as histone H3 and transcription factors CREB and ATF1 in the nucleus.40 Previous studies demonstrated that VEGFR2 internalization is required for downstream signaling to regulate endothelial cell migration, proliferation, and survival.42,43 Here, we show that A2AR mediates a VEGF-independent VEGFR2 endocytosis and phosphorylation and promotes LEC proliferation, migration, and tube formation in high-salt conditions.
Anti-VEGF–neutralizing antibody bevacizumab and VEGFR2 tyrosine kinase inhibitor fruquintinib are both used in the treatment of malignancies.44,45 They may each cause hypertension as a side effect, because they each antagonize VEGF-mediated vasodilation.44 However, they act differently on the VEGF pathway. Bevacizumab scavenges VEGF, whereas fruquintinib blocks phosphorylation of VEGFR.46 Consistent with a VEGF-independent action of A2AR as described above, we showed that during high-salt conditions, fruquintinib, but not bevacizumab, blocks A2AR activation–induced dermal lymphangiogenesis and increases BP. These results were supported by our observations in cancer patients with HSD, though the number of subjects was small. Greater understanding of the vascular effects of the VEGF/VEGFR pathway could lead to novel therapeutic approaches to reduce the hypertensive effects of antiangiogenic therapies in cancer patients. However, the animal sample size in our study was low, which limited adequate power to ensure clinically important meaning or difference.
Our results suggest that LEC–A2AR might be a potential therapeutic target in salt-sensitive hypertension because it mediates VEGF-independent activation of VEGFR2 signaling to increase dermal lymphangiogenesis and reduce skin sodium retention. However, A2AR agonists are reported to have detrimental effects, which may require the development of more selective A2AR ligands that preferentially act on skin lymphatics.47 Moreover, previous studies demonstrated that the effects of adenosine receptors differed according to sex.48,49 The animals used in the present study were male, and we will investigate the sex differences of LEC–A2AR receptor in dermal lymphangiogenesis and sodium balance in the future. Finally, it is important to note that, whereas we have documented a VEGF-independent effect of LEC–A2AR, our results do not exclude a role for VEGF in salt-sensitive hypertension as it enhances dermal lymphangiogenesis.
Our research reveals the importance of lymphatic endothelial A2AR in the progression of high salt–induced hypertension. Lymphatic endothelial cell–A2AR activation increases dermal lymphangiogenesis and skin sodium clearance by promoting phosphorylation of MSK1 and VEGFR2 endocytosis and activation. Notwithstanding the much more advanced complexity of human salt-sensitive hypertension in comparison with the mouse model, our novel findings suggest an attractive possibility that activating A2AR-mediated lymphangiogenesis may be a potential therapeutic target in the treatment of salt-sensitive hypertensive patients.
Supplementary data
Supplementary data are available at European Heart Journal online.
Declarations
Disclosure of Interest
All authors declare no conflict of interest for this contribution.
Data Availability
All data associated with are present in the paper or the supplementary material online. All resources generated in this study are available upon request from the corresponding author upon providing a completed material transfer agreement.
Funding
This study was supported by the National Key R&D Program of China (2022YFA1104200), National Natural Science Foundation of China (82070456, 81922004, 81870180, and 81900434), Shanghai Science and Technology Commission (21XD1420500 and 19JC1414600), Shanghai Rising-Star Program (21QA1401400), Young Health Talents of Shanghai Municipal Health Commission (2022YQ069), Innovative research team of high-level local universities in Shanghai and a key laboratory program of the Education Commission of Shanghai Municipality (ZDSYS14005), Huangpu Municipal Health Commission (HLQ202102), National Institutes of Health (NIH) R01s HL133254 and HL148338, Cancer Prevention and Research Institute of Texas CPRIT RP150611, and Medical Engineering Joint Fund of Fudan University (YG2022-26).
Ethical Approval
Ethical Approval was not required.
Pre-registered Clinical Trial Number
None supplied.
References
Author notes
Tao Zhuang, Yu Lei, Jin-Jia Chang and Yan-Ping Zhou contributed equally to the study.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}