





Abstract
Chronic cardiometabolic assaults during type 2 diabetes (T2D) and obesity induce a progenitor cell imbalance in the circulation characterized by overproduction and release of pro-inflammatory monocytes and granulocytes from the bone marrow alongside aberrant differentiation and mobilization of pro-vascular progenitor cells that generate downstream progeny for the coordination of blood vessel repair. This imbalance can be detected in the peripheral blood of individuals with established T2D and severe obesity using multiparametric flow cytometry analyses to discern pro-inflammatory vs. pro-angiogenic progenitor cell subsets identified by high aldehyde dehydrogenase activity, a conserved progenitor cell protective function, combined with lineage-restricted cell surface marker analyses. Recent evidence suggests that progenitor cell imbalance can be reversed by treatment with pharmacological agents or surgical interventions that reduce hyperglycaemia or excess adiposity. In this state-of-the-art review, we present current strategies to assess the progression of pro-vascular regenerative cell depletion in peripheral blood samples of individuals with T2D and obesity and we summarize novel clinical data that intervention using sodium-glucose co-transporter 2 inhibition or gastric bypass surgery can efficiently restore cell-mediated vascular repair mechanisms associated with profound cardiovascular benefits in recent outcome trials. Collectively, this thesis generates a compelling argument for early intervention using current pharmacological agents to prevent or restore imbalanced circulating progenitor content and maintain vascular regenerative cell trafficking to sites of ischaemic damage. This conceptual advancement may lead to the design of novel therapeutic approaches to prevent or reverse the devastating cardiovascular comorbidities currently associated with T2D and obesity.
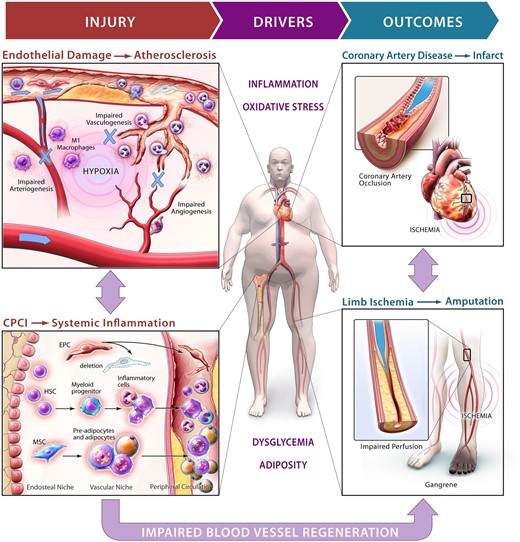
During cardiometabolic based chronic disease, dysglycemia and excess adipocity induce a differentiate imbalance in the bone marrow that contribute to overproduction of inflammatory cells and impaired mobilization of progenitor cells that mediate endogenous mechanisms for vessel repair and regeneration.
Introduction to chronic cardiometabolic disease
Link between chronic cardiometabolic disease and cardiovascular comorbidities
The clinical outcomes of ischaemic cardiovascular diseases (CVD), including coronary and peripheral artery disease, myocardial infarction, heart failure and stroke, are often considered unavoidable complications that compromises quality of life in patients with type 2 diabetes (T2D) and obesity. CVD was responsible for nearly 18 million global deaths in 2015,1 , 2 and collectively CVD remains the leading cause of death worldwide.3 , 4 It is estimated that 420 million individuals are living with diabetes and 2 billion individuals are considered overweight, 650 million of which are affected by obesity [body mass index (BMI) >30 kg/m2]. Due to the increased prevalence of T2D and obesity in an ageing population worldwide, and despite improved pharmacologic and surgical intervention, the incidence of CVD is expected to continue to rise over the next decade.5 The global financial burden of treating diabetes was estimated at $1.31 trillion in 2015,6 and CVD comorbidities were responsible for nearly one-third ($437 billion) of the cost of therapy.7 Thus, there exists a compelling need to better understand the pathophysiological interrelationship between T2D, obesity and the subsequent development of CVD.
In a recent review, Mechanick et al.8 conceptualized that ischaemic CVD represents the endpoint of an accumulation of clinical risk factors including hyperglycaemia and insulin resistance during T2D and the elevation of systemic inflammation during obesity. Because T2D and obesity are often highly entangled, comorbid states, chronic dysglycaemia, and increased adiposity can be considered primary drivers of vascular damage that overwhelm endogenous vessel repair mechanisms.9–13 This unmanaged cycle of excessive vascular damage combined with gradually compromised vessel regenerative response can intensify atherosclerotic plaque instability and elevate the risk of experiencing ischaemic macrovascular events,14 including myocardial infarction and stroke. Therefore, strategies to stimulate the recovery of endogenous blood vessel repair represent a novel target to prevent and treat CVD progression15 (See Graphical Abstract).
Progenitor cells that mediate blood vessel repair
Endogenous processes for blood vessel repair and regeneration
In response to tissue hypoxia or ischaemic injury, bone marrow (BM)-derived haematopoietic progenitor cells (HPC) and rare endothelial progenitor cells (EPC) are stimulated by neurohormonal signals and chemokines in the BM to mobilize into the peripheral circulation. As such, endogenous vascular regeneration is bolstered by haemangiogenic precursor cells and their downstream progeny that recruit to sites of vessel damage. Blood vessel repair and regeneration is typically classified into THREE overlapping multicellular processes: (i) vasculogenesis, the de novo synthesis of new blood vessels by circulating EPC first identified by Asahara and Isner;16 , 17 (ii) angiogenesis, the sprouting of new blood vessels from pre-existing vessels stimulated by angiocrine secretion from endothelial cells and circulating haematopoietic cells; and (iii) arteriogenesis, the remodelling of pre-existing collateral vessels by monocytes that polarize into regenerative macrophages in peripheral tissues.18 , 19 These complementary processes occur simultaneously to synergize blood vessel regeneration, repair and anastomoses.20 Thus, healthy human blood contains circulating BM-derived progenitor cells that contribute to the maintenance of blood vessel homeostasis throughout life. However, in individuals with T2D and obesity, hyperglycaemia and adiposity elevate oxidative stress and inflammation to generate a ‘perfect storm’ that alters progenitor cell differentiation upsetting the execution of blood vessel repair mechanisms.
Circulating CD34+VEGFR-2+ cells are decreased during type 2 diabetes and obesity
The seminal discovery by Asahara et al. 16 and Isner et al. 17 first defined EPC as highly proliferative CD34+ cells that could be mobilized from the BM and integrate into vascular structures in vivo. Because mature endothelial cells within blood vessels are remarkably quiescent with an estimated half-life of ∼3 years, Asahara et al. 21 first suggested that post-natal vasculogenesis, a process initially thought to be restricted to the developing embryo, could be mediated in adult organisms after transplantation or mobilization of EPC from the BM. Later, cell surface markers such as CD133 and vascular endothelial growth factor receptor-2 (VEGFR-2) were added to better define the putative EPC population. In 2001, Vasa et al. 22 were the first to report that the number and migratory function of EPC (identified as CD34+/VEGFR-2+ cells) was severely reduced in patients with coronary artery disease (CAD). Individuals with CAD had ∼50% fewer circulating CD34+/VEGFR-2+ cells compared to healthy controls. Furthermore, common risk factors associated with CAD correlated with decreased circulating CD34+/VEGFR-2+ content. Smoking was the most significant predictor for EPC depletion, alongside hypertension, serum low-density lipoprotein (LDL) cholesterol, age and previous family history of CAD.22 Reciprocally, Werner et al.23 observed that in subjects with CAD, individuals in the top tertile for circulating CD34+/VEGFR-2+ cells demonstrated a reduced risk of major cardiovascular events. Zafar et al. recently enrolled a human cohort with variable CVD risk and demonstrated an association between different progenitor cell subsets within indices of T2D. Subjects with a lower frequency of CD34+/CD133+ cells had a significantly higher incidence of T2D, higher glycated haemoglobin (HbA1c), higher CVD risk scores, and impaired vascular function.24 Circulating CD34+/CD133+ cells from patients with T2D demonstrated reduced CXCR4 and VEGFR-2 expression, suggesting a homing deficit to areas of ischaemia. Surprisingly, existing CD34+/CD133+ cells in the high-risk group compensated with increased expression of angiopoietins, VEGF-A, PlGF, HGF, EPO, and MMP-9 compared to the lower risk cohort. Thus, reduced circulating progenitor cell content and recruitment to sites of vascular injury were implicated with T2D pathology without an observed deficit in angiogenic cytokine expression.
In a series of studies, Fadini et al. 25 , 26 sought to determine if hyperglycaemia resulted in EPC functional impairment. In hyperglycaemic mice with hind-limb ischaemic injury, CD34+/VEGFR2+ cells were ineffectively mobilized into the periphery compared to normoglycaemic mice. Insulin administration to promote glycaemic control induced partial recovery of circulating CD34+/VEGFR2+ cells. In individuals with T2D, peripheral artery disease was independently associated with reduced circulating CD34+/VEGFR2+ content,25 and CD34+/VEGFR2+ isolated from individuals with peripheral artery disease demonstrated reduced ability to form endothelial cell colonies in vitro. In contrast, a mild elevation in circulating CD34+ cells in healthy individuals was associated with increasing adiposity and insulin resistance.27 Collectively, these studies established CD34, CD133, and VEGFR-2 as biomarkers to document changes in circulating progenitor cell content during T2D and obesity. However, the identity and function of ‘true’ EPC remained elusive and soon met controversy surrounding the physical contribution of BM-derived progenitor cells into the growing vasculature in healthy and diseased conditions. For a comprehensive review on circulating stem cells and cardiovascular outcomes refer to Fadini et al.28
The endothelial progenitor cell controversy: CD34+/VEGFR-2+ cells primarily represent progenitor cells of the myeloid lineage
Haematopoiesis is highly integrated with blood vessel repair and maintenance. HPC differentiate into myeloid progeny that give rise to reticulated erythrocytes, monocytes and granulocytes, and megakaryocytes, essential for the transport of oxygen, innate immune surveillance, and platelet production, respectively.29 In addition, primitive HPC and monocytes also possess potent pro-angiogenic secretory function and stimulate microvascular capillary formation to recover perfusion in mice with hindlimb ischaemia.30 Indeed, mouse embryos deficient in acute myeloid leukaemia 1 protein (AML1), a transcription factor necessary for myeloid cell differentiation, were incapable of stimulating angiogenesis resulting in embryonic lethality.30 Notably, embryo lethality could be rescued by incubation with BM-derived HPC, suggesting the profound role of myeloid progenitor cell secretions to stimulate blood vessel regeneration throughout adult life.
Canonically, classification of EPC based on CD34 or VEGFR-2 expression has generated confusion in the field since myeloid HPC also express these markers. In seminal findings by the Dimmeler group, Urbich et al.31 demonstrated that CD34+/VEGFR-2+ cells, referred to as EPC in previous work, consisted primarily of cells from the haematopoietic lineage. Termed circulating angiogenic cells (CAC), endothelial-cell-like cultures could be established from CD14+ and CD14- mononuclear cells, both cell types demonstrated lectin uptake in vitro, and stimulated neovascularization after transplantation into mouse models of hind-limb ischaemia. The CD14+ cells were comprised of a mixture of monocytes, macrophages, and T lymphocytes, that did not produce endothelial cell progeny, but contributed to capillary formation with potent angiocrine function. Yoder et al. first delineated the functional differences between ‘true EPC’ termed endothelial colony-forming cells (ECFC) and CAC based on fundamental stem and progenitor cell principles.32 CAC formed early outgrowth (7–10 days) colonies in culture, co-expressed endothelial (CD34, VEGFR-2) and haematopoietic cell markers (CD45+/CD14+), and secreted cytokines to promote tip cell formation during angiogenic vessel sprouting but did not integrate into vessels in vivo. In contrast, ECFC were exclusively CD45−/CD14−, formed late outgrowth (>14 days) endothelial cell colonies in vitro, and could integrate into blood vessels in vivo. Nonetheless, these findings suggested novel roles for CD34+ HPC and CD14+ monocytes to stimulate vessel repair alongside circulating or vessel-resident EPC as a potential source of endothelial cells.
Arteriogenic remodelling is impaired during type 2 diabetes and obesity
Angiogenic and arteriogenic vessel repair mechanisms are distinguished based on distinct physiological drivers. Angiogenesis occurs primarily as a response to hypoxia, driving hypoxia-inducible factor 1α (HIF-1α)-mediated transcription of VEGF and chemokines that stimulate surveillant CAC to recruit to areas of ischaemia and secrete a variety of pro-angiogenic cytokines. In contrast, the predominant process to bypass vessels occluded during arteriogenesis occurs in response to fluid shear stress. Although arteriogenesis is not associated with increases in HIF-1α or VEGF,33 reduced luminal diameter in atherosclerotic vessels increases shear stress on the vascular endothelium, stimulating the secretion of powerful chemokines (such as monocyte chemoattractant protein-1, MCP-1)34 and expression of adhesion molecules (such as integrins) to recruit and adhere to CAC. Risk factors such as smoking35 and T2D36–38 have been shown to impair both angiogenic capillary formation and arteriogenic collateral vessel remodelling. Thus, better understanding of the relationship between circulating monocytes and development of tissue remodelling macrophages that synergize arteriogenic collateral vessel formation represents an ongoing focus to modulate large vessel regeneration.
Monocytes are largely considered precursors to matrix-remodelling macrophages. Within tissues, monocyte populations can adopt pro-inflammatory M1-like or pro-regenerative M2-like fates (further subdivided into M2a, M2b, and M2c groups).39 Notably, this oversimplified description of macrophage subtypes is better described as a continuum of highly plastic cells that play a vital role in arteriogenic tissue remodelling. In concept, M1-like macrophages primarily represent phagocytic cells that secrete pro-inflammatory cytokines such as interleukin (IL)-1β, IL-6, IL-12, and tumor necrosis factor-α and recruit neutrophils that propagate inflammation. Conversely, M2-like polarized macrophages are considered anti-inflammatory and mediate extracellular remodelling via the secretion of growth factors such as transforming growth factor-β (TGF-β) and fibroblast growth factor (FGF). Together, collateral artery activation relies on the recruitment of monocytes that polarize along the M1/M2 continuum to dictate the delicate balance between tissue inflammation vs. regenerative arteriogenesis.
Troidl et al.40 sought to determine the contribution of M1- and M2-polarized macrophages to arteriogenesis using femoral artery ligation in rodent models to induce shear stress. M2-polarized macrophages were scarcely distributed throughout the perivascular space prior to femoral artery ligation. After occlusion, both M1 and M2 polarized macrophage content was increased. M2-like macrophages augmented matrix degradation at the leading edge of remodelling vessels, highlighting a critical role for M2 macrophages to facilitate collateral activation. Takeda et al.41 observed that M2-polarized macrophages also secreted SDF-1 and PDGFB to recruit pericytes involved in blood vessel stabilization. Thus, collateral activation via arteriogenesis represents a novel target for blood vessel repair by altering the balance between M1-like and M2-like macrophage content associated with altered blood flow parameters.
High ALDH activity identifies progenitor cells with potent angiocrine function
To identify blood-derived progenitor cells with vascular regenerative potential, our group and others purify and expand human progenitor cell subsets based on selection for high aldehyde dehydrogenase (ALDH) activity.42–45 ALDH is a intracellular detoxification enzyme highly expressed by progenitor cells from multiple mesodermal lineages that protects differentiating progenitor cells from damage by alkylating agents or oxidative stress. As progenitor cells with high ALDH activity differentiate towards more expendable effector cells, ALDH activity is decreased up to 100-fold. By assessing the distribution of peripheral blood cells with high vs. low ALDH activity (Aldefluor™ assay; StemCell Technologies) by flow cytometry, we have identified a unique functional biomarker for progenitor cells (ALDHhi cells) vs. more differentiated cell progeny (ALDHlow cells). Storms et al.45 , 46 first identified human BM cells with high ALDH activity as myeloid HPC demonstrating multipotent colony formation in vitro. Because ALDHhiSSClow cells highly co-expressed CD34 (>75%) and CD133 (>55%), BM-derived ALDHhiSSClow cells demonstrated robust haematopoietic repopulation capacity in immunodeficient mice.47 , 48 In Capoccia et al., we revealed that human BM-derived ALDHhiSSClow cells represented a heterogeneous mixture of progenitor cells with haematopoietic (1 HCFC in 4 cells), endothelial (∼1 in ECFC 1500 cells), and fibroblast (∼1 FCFC in 1500 cells) colony-forming cell activity in vitro. Compared to mature mononuclear cells, purified BM ALDHhiSSClow cells possessed a pro-angiogenic secretory profile and accelerated the recovery from ischaemia when transplanted into immunodeficient mice with unilateral hind-limb ischaemia induced after femoral artery ligation and excision.49 Thus, ALDH activity represented a valuable tool to purify progenitor cells with vessel regenerative capacity based on a tightly conserved progenitor cell function. These findings were immediately translated into a clinical trial by Perin et al. 50 for severe critical limb ischaemia demonstrating that intramuscular implantation of autologous BM ALDHhi cells improved Rutherford category scores and ankle-brachial index at 12 weeks post-transplantation. However, in a study of 82 patients, the PACE trial recently reported that transplantation of autologous ALDHhi cells in patients with intermittent claudication showed no improvement in capillary perfusion compared to placebo, but ALDHhi cell administration was associated with an increase in the number of collateral arteries.51
The assessment of ALDH activity alongside cell granularity (increasing side scatter property) and the co-expression of lineage-specific cell surface markers within peripheral blood of individuals with T2D, has also provided a high-throughput method to compare circulating progenitor cell subsets previously associated with pro-inflammatory vs. pro-angiogenic properties.15 Our rationale was to assess1 circulating ALDHhiSSClow cells, representing uncommitted HPC with a pro-angiogenic secretome,52 alongside primitive cell surface marker (CD34, CD133, and VEGFR-2) co-expression; circulating ALDHhiSSCmid cells, representing monocyte precursors with M1 (CD80) vs. M2 (CD163, CD206) polarization, and ALDHhiSSChi cells, representing granulocytic precursors with pro-inflammatory (CD15, CD16) or pro-angiogenic marker (CD49d) co-expression. Importantly, ALDHlow cells from each SSC classification represented more differentiated cells from each lineage.
Reversal of progenitor cell imbalance during metabolic diseases
Circulating progenitor cell imbalance during type 2 diabetes
Homeostatic vascular remodelling occurs continually throughout growth and ageing. Indeed, the revascularization of damaged or ischaemic tissues represents a central component of all regenerative processes. Derived in the BM and mobilized to areas of hypoxia via the bloodstream, ALDHhiSSClow progenitor cells generate and direct the myriad of effector cells that participate in angiogenic, vasculogenic, and arteriogenic processes. Although human organs possess an intrinsic capacity for vessel regeneration, hyperglycaemia and increasing adiposity during T2D and obesity result in relentless damage that depletes the pro-angiogenic cell reservoir and forces the production of pro-inflammatory granulocyte progeny53 (Figure 1). This circulating progenitor cell imbalance encompasses the depletion or dysfunction of cells associated with vessel repair,54–57 and the overproduction of granulocytes and monocytes that heighten inflammation during T2D and obesity. Thus, progressive vessel damage driven by hyperglycaemia or atherosclerosis is compounded by significant deficits in vessel repair, ultimately elevating the risk of developing ischaemic CVD.58
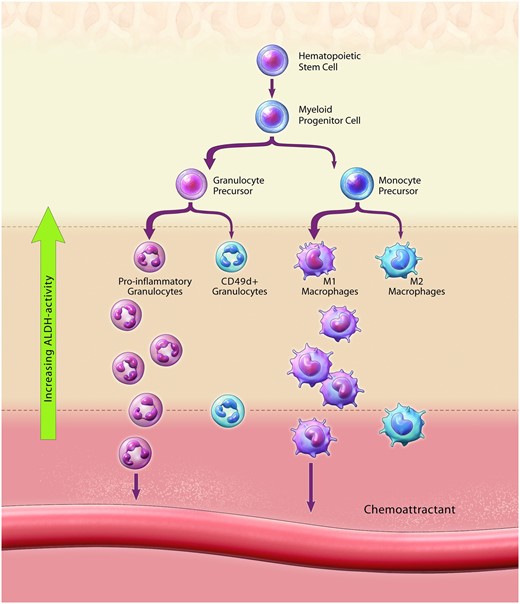
Circulating Progenitor Cell Imbalance elevates systemic inflammation and reduces circulating cell types that mediate blood vessel repair. Circulating progenitor cell imbalance results in reduced maintenance of ALDHhiSSClow progenitor cells with CD34 and CD133 co-expression and altered haematopoietic differentiation towards the overproduction of pro-inflammatory granulocytes at the expense of vessel regenerative cell types. Altered monocyte differentiation promoted the production of ALDHhiSSCmid monocytes with M1-polarization vs. M2-polarization. Collectively, altered circulating progenitor cell content impairs endogenous cell-mediated blood vessel repair and elevates systemic inflammation during type 2 diabetes and severe obesity.
Because a non-invasive assay was needed to accurately document the circulating progenitor cell imbalance in patients with T2D and obesity, we combined ALDH activity with cell type-specific surface marker analyses to develop a high-throughput flow cytometry platform to quantify lineage-restricted progenitor cell content within peripheral blood samples from patients with T2D compared to sex- and age-matched control subjects.59 Compared to non-diabetic controls, individuals with established T2D for a mean duration of >15 years exhibited: (i) an increased frequency of circulating ALDHhiSSChi granulocyte precursors, (ii) an increased frequency of ALDHhiSSCmid monocytes with M1 polarization, and (iii) depletion of ALDHhiSSClow precursor cells that co-expressed CD34 or CD133 with pro-angiogenic function.47 Notably, the duration of T2D directly correlated with the severity of circulating progenitor cell imbalance. Collectively, these observations further refined the progenitor cell imbalance as a reduction in circulating HPC combined with myelopoietic overproduction of inflammatory monocytes and granulocytes consistent with published mechanistic studies in mouse models of hyperglycaemia60 , 61 and obesity.62 This novel assessment of circulating ALDHhi progenitor cell subsets represented a promising approach to identify T2D patients with compromised blood vessel repair capacity.
SGLT2 inhibition reversed the progenitor cell imbalance in individuals with T2D
Chronic hyperglycaemia (HbA1c >6.5%) represents an important driver for the development of microvascular risk in patients with T2D. Sodium-glucose co-transporter 2 inhibitors (SGLT2i), including dapagliflozin, canagliflozin, and empagliflozin, represent a novel class of glucose-lowering medications that block the reabsorption of glucose and sodium from the kidney to the bloodstream, increasing urinary glucose excretion and thereby lowering HbA1c levels.63 However, the benefits of SGLT2i may extend beyond glycaemic control. The EMPA-REG OUTCOME trial established that empagliflozin treatment in patients with T2D correlated with significant reductions in major adverse cardiovascular events (MACE) and all-cause mortality.64 However, the mechanisms driving these improved outcomes remain undetermined.
In human BM, hyperglycaemia has been shown to induce premature inflammatory cell differentiation and inhibition of vascular regenerative progenitor cell egression into the periphery65 (Figure 1). Performed as a sub-study of the EMPA-Heart CardioLink-6 trial, we investigated whether administration of empagliflozin could impact the frequencies of circulating ALDHhi progenitor cell subsets in patients with established T2D over a 6-month period.66 Remarkably, individual with T2D administered empagliflozin demonstrated reduced circulating ALDHhiSSChi granulocyte precursor burden, an improved ratio of ALDHhiSSCmid M1- vs. M2-polarized monocytes, and increased ALDHhiSSClow CD34+/CD133+ progenitor cell content compared to subjects receiving standard medical care. In the plasma of subjects administered empagliflozin, we also documented stabilization of NADPH oxidase 1 (NOX1), an enzyme dedicated to the production of reactive oxygen species (ROS), alongside increased catalase, an anti-oxidant that accelerates ROS decomposition. Collectively, empagliflozin treatment contributed to reversal of the circulating progenitor cell imbalance in patients with T2D (Figure 2), correlated with reduced CVD risk as observed in the EMPA-REG OUTCOME trial. Thus, restoration of progenitor cell content in the circulation represented a treatable target to improve vascular homeostasis during T2D.
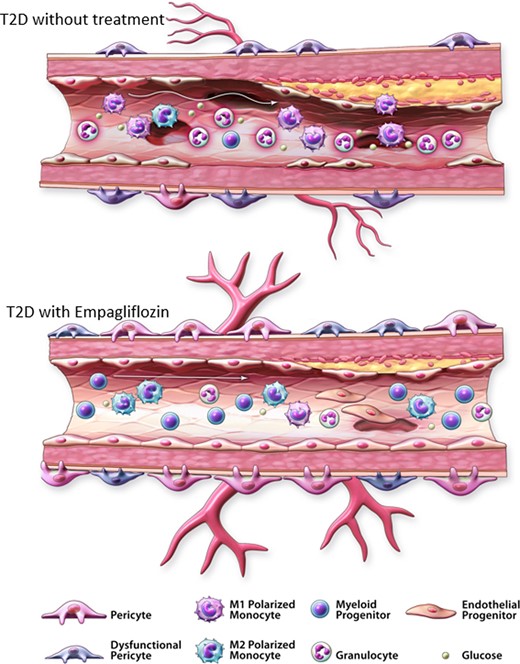
Reversal of circulating progenitor cell imbalance associated with type 2 diabetes. Circulating progenitor cell imbalance is characterized by increased pro-inflammatory progenitor cells and the depletion of pro-angiogenic haematopoietic and monocytes in the peripheral blood. (A) In individuals with type 2 diabetes, increased circulating ALDHhiSSChi granulocyte precursors and increased ALDHhiSSCmid monocytes with M1 polarization contribute to elevated systemic inflammation associated with endothelial dysfunction. Reduced circulating ALDHhiSSClow cells with pro-angiogenic secretory capacity will interfere with the repair of damaged vessels. After administration of the SGLT2i empagliflozin, decreased circulating ALDHhiSSChi granulocyte content alongside improved ALDHhiSSCmid monocyte balance will reduce systemic inflammation. Increased circulating ALDHhiSSClow cells with CD34/CD133 co-expression will promote vessel repair.
In contrast, Bonora et al. reported that in patients with T2D, circulating CD34+ and CD34+/VEGFR2+ cells was not reduced in subjects that received dapagliflozin compared to placebo,67 suggesting the significant reversal of the progenitor cell imbalance observed in our studies66 may not be conserved between different formulations within the SGLT2i class, and/or additional assessment identifying progenitor cell subsets with high ALDH activity expression was required to document circulating progenitor cell recovery.
Gastric bypass surgery improved the circulating progenitor cell imbalance associated with severe obesity
Abnormal adiposity increases the risk of developing T2D and adverse cardiovascular events.68 Roux-en-Y gastric bypass surgery is an effective intervention to induce sustained weight loss compared to non-surgical diet restriction with best medical therapy.69 Remarkably, gastric bypass surgery has also been associated with spontaneous remission of T2D and profound CVD risk reductions.69 , 70 Because the metabolic and cellular mechanisms driving these benefits were undetermined, we analysed circulating ALDHhi progenitor cell frequencies in normal-weight controls (BMI <25 kg/m2) compared to patients living with severe obesity (BMI >40 kg/m2) prior to and 3 months after gastric bypass surgery.71 Blood from individuals with severe obesity consistently demonstrated elevated circulating ALDHhiSSChi granulocyte precursor content compared to normal-weight controls. Remarkably, normal granulocyte distribution was restored 3 months post-surgery. ALDHhiSSCmid monocytes with pro-inflammatory M1 polarization (CD14+/CD80+ cells) were also elevated in bariatric surgery patients compared to normal-weight controls, and were significantly reduced 3 months after surgery. In contrast, ALDHhiSSCmid monocytes with M2 polarization (CD14+/CD163+) were increased post-surgery. Finally, ALDHhiSSClow cells co-expressing CD34 and CD133 were depleted in individuals with obesity and significantly increased after surgery (Figure 2). Thus, increased inflammatory cell burden and vessel regenerative progenitor subset depletion during obesity was remarkably improved within 3 months of bariatric surgery.
Proposed mechanisms implicated in blood vessel regeneration
While detailed functional evaluation of purified ALDHhi progenitor cell subsets in patients with T2D and obesity remain ongoing in our laboratory, seminal contributions by several groups have investigated further into analogous cellular subtypes integral to endogenous blood vessel repair during T2D. In pre-clinical mouse studies, the Fadini group has clearly established that during the metabolic consequences of T2D, the BM undergoes extensive remodelling, including microvessel rarefaction and fat accumulation, generating a hostile environment for resident progenitor cell differentiation.72 Fadini further suggested that HPC mobilopathy and enhanced myelopoietic differentiation into progeny secreting pro-inflammatory and anti-angiogenic mediators occurred as a result of T2D.73 , 74 The observed mobilopathy effect was attributed to an accumulation of CD169+ macrophages in the BM that prevented the mobilization of haematopoietic cells.75
Cappellari et al.76 focused on the role of a novel granulocyte subset expressing the integrin receptor CD49d as a potent mediator of vessel repair in response to endothelial damage. CD49d+ granulocytes augmented tubule formation via physical interactions with endothelial cells in vitro, and granulocytes from subjects with T2D had profoundly impaired capacity to stimulate endothelial cell tube formation compared to non-diabetic control populations. CD49d+ granulocytes were 30–40% reduced in the peripheral blood of human subjects with T2D and were two-fold increased after dapagliflozin treatment. This study first established reduced vessel-regenerative granulocyte content during T2D could be modulated by dapagliflozin treatment.
Currently, there is little evidence in the literature to support that SGLT2i or gastric bypass surgery demonstrate direct effects on BM-derived progenitor cells. However, in recent mechanistic studies performed in streptozotocin (STZ)-treated hyperglycaemic mice, Albiero et al.77 demonstrated that despite low levels of SGLT2 receptor expression on BM-derived cells, SGLT2 inhibition with dapagliflozin improved endothelial repair through enhanced mobilization and recruitment of haematopoietic cells from the BM directly to the site of carotid artery injury. In hyperglycaemic mice, dapagliflozin administration reduced the frequency of CD169+ macrophages in mouse BM by 70% and partially rescued the mobilization defect in response to granulocyte colony-stimulating factor. Next, the authors examined the effect of improved haematopoietic cell mobilization after the induction of carotid artery wound healing. Remarkably, re-endothelialization after denudation injury was 10-fold decreased in hyperglycaemic mice, and endothelial healing was significantly improved after dapagliflozin treatment. In elegant experiments using chimeric mice reconstituted with GFP+ BM cells to effectively trace GFP+ cells localized to the site of injury, reduced recruitment of GFP+ cells to the site of carotid injury observed in STZ-treated diabetic chimeras was reversed by dapagliflozin treatment. Mature GFP+ monocytes and neutrophils were abundant in the damaged carotid artery, with both F4/80+ macrophage and CD49d+ granulocyte recruitment significantly increased by dapagliflozin treatment. Thus, monocyte and CD49d+ granulocyte mobilization, recruitment, and adhesion at the site of injury promoted re-endothelialization after dapagliflozin administration, identifying CD49d+ granulocytes as a novel therapeutic target to promote endothelial repair during diabetes. These findings suggested that modulation of granulocyte subset generation and mobilization using SGLT2i treatment may represent a therapeutic strategy to repair damaged vessels during T2D.
Clinical perspectives and future directions
Novel pharmacological interventions to reverse the circulating progenitor cell imbalance during T2D and obesity
Rational selection of therapeutic agents with the potential to improve endogenous vessel repair has been repeatedly demonstrated by agents that promote glucose control. However, in our relatively small substudy of 30 patients, ongoing metformin or insulin therapy did not improve the circulating progenitor cell imbalance phenotype observed during T2D,59 despite previous reports that these agents improve HSPC content in the peripheral blood.28 In addition, pioglitazone and dipeptidyl peptidase-4 inhibitors have been previously reported to directly improve CD34+ cell trafficking.78 , 79 The widespread metabolic stabilization following gastric bypass surgery has also provided insight into potential mechanisms driving improved circulating progenitor cell content. Even before significant weight loss has occurred after surgery, ∼40% patients experienced metabolic stabilization including improved glycaemic control and increased insulin sensitivity that can result in discontinuation of T2D medication.69 Due to direct gastric emptying into a distal segment of the gastrointestinal tract, gastric bypass surgery consistently results in a sharp postprandial rise in gut peptides, including the incretin hormone and satiety protein glucagon-like peptide-1 (GLP-1). A recent meta-analysis evaluating 368 individuals across 24 studies after gastric bypass surgery indicated that postprandial GLP-1 levels were significantly increased following surgery, providing evidence that GLP-1 receptor (GLP-1R) signalling axis may play an important role in the metabolic benefits observed following bariatric surgery.80 GLP-1 is a hormone produced primarily in the ileum and colon and secreted into the circulation in response to increased glucose load. GLP-1 enhances insulin secretion by activating GLP-1R on the surface of β cells in a glucose-dependent manner. GLP-1R is expressed throughout the gastrointestinal tract and in the satiety control centre of the hypothalamus. Therefore, in addition to insulinotropic effects, GLP-1R activity has been shown to delay gastric emptying, increase appetite suppression, and reduce glucagon activity in the clinical setting. Furthermore, GLP-1R is surprisingly expressed on many mesodermal cell types, including arterial smooth muscle cells, endothelial cells, and various haematopoietic cell lineages.81 In addition, animal models have shown infusion of GLP-1 increased HPC proliferation and differentiation,82 , 83 protected of endothelial cells from oxidative stress,84 and improved endothelial cell function.85 We postulate that increased GLP-1 release following gastric bypass surgery may contribute towards improved vessel repair via modulation of the progenitor cell imbalance paradigm86.
Over the past 12 years, recombinant human GLP-1R agonists including liraglutide (2009), dulaglutide (2014), and semaglutide (2017), among others, have been sequentially released. Delivery of GLP-1R agonists has been by weekly subcutaneous injections. However, novel advancements in the formulation of oral semaglutide (2019) have recently been developed.87 Notably, the LEADER trial revealed that the GLP-1R agonist (liraglutide) significantly reduced MACE and all-cause mortality in individuals with T2D.88 Because gastric bypass surgery is highly invasive and recommended only to patients meeting the selection criteria for bariatric surgery (typically BMI ≥40 kg/m2), and because liraglutide is indicated for weight loss during obesity (BMI >30 kg/m2), we have initiated a clinical trial to determine whether treatment with liraglutide can improve circulating progenitor cell content in patients with more moderate obesity (BMI 30–35 kg/m2).
Extracellular vesicles: a novel therapeutic opportunity to induce vessel repair
Human HPC, EPC, and MSC are highly secretory and abundantly produce extracellular vesicles (EVs), including exosomes and microvesicles, that contain potent regenerative cargo in the form of proteins, lipids, and microRNAs. EVs are released by membrane blebbing or exocytotic mechanisms and used for intracellular communication and delivery of signals over long distances via the blood stream.89 A primary target for microparticle uptake is the vascular endothelial lining. As such, investigating the role of extracellular vesicles in vessel repair is warranted as an exciting modality to improve blood vessel regeneration during chronic T2D and obesity.
EVs secreted by CD34+ HPC have been evaluated for their ability to induce blood vessel growth.89–91 Furthermore, the pro-vascular functions of a plethora of miRNAs have been associated with endothelial cell homeostasis, including repression of senescence, increasing adhesion molecule expression, and activation of the developmental Wnt-signalling pathway central to progenitor cell differentiation. Specifically, van Balkom et al.91 demonstrated that miR-214 plays a central role in the induction of angiogenesis in mice. Because CD34+ HPC stimulate capillary formation through pro-angiogenic cytokine release, Mathiyalagan et al.90 recently examined the exosomal contents of CD34+ cells in mice after femoral artery ligation-induced unilateral hind-limb ischaemia. Excitingly, mice injected with purified CD34+ cell exosomes demonstrated improved hind-limb perfusion and preserved muscle mass. MicroRNA screening uncovered CD34+ cell exosomes contained high levels of miR-10a and miR-130 with previously demonstrated anti-fibrotic activities in the ischaemic myocardium.90 , 92 , 93 Fibrotic remodelling after myocardial infarct can limit stroke volume and cardiac output due to myocardial thickening and progression towards heart failure.94 , 95 Furthermore, fibrosis in the vasculature can lead to hypertension and increased mechanical stress on the heart.94 , 95 Although the use of designer EVs to impact blood vessel regeneration in the clinical setting requires further mechanistic experimentation and extensive regulatory oversight, the potential to improve vessel health and augment cardiac output after myocardial infarction by the delivery of progenitor cell-engineered EVs represents an exciting opportunity to combat CVD in the future.
Supplementation of vascular grafts with regenerative progenitor cells
Management of cardiovascular remodelling after sub-lethal myocardial infarction remains a critical challenge worldwide. Native vessel grafts from autologous mammary arteries or saphenous veins during coronary artery bypass graft surgery are often unsuitable for grafting, fuelling the search for a more mechanically robust alternative.96 , 97 Dahl et al.98 , 99 elegantly reviewed the advancements made in the previous three decades in the generation of bioengineered vascular grafts. Most processes involve the creation of new vascular conduits employ in vitro growth from autologous progenitor cells grown in 3D bioscaffolds. Hoerstrup et al.100 demonstrated 100% primary patency at 20–100 weeks following pulmonary artery replacement with bioengineered vascular grafts in bovine models. While the generation of autologous grafts using progenitor cells in vitro has the potential to confer benefit in the maintenance and integration of vascular grafts, there have been challenges with the time needed to generate the vessels. For example, the use of bioengineered vascular grafts could increase patient wait time by weeks or months to grow a sufficient conduit, calling for the examination of more efficient vessel growth strategies in vitro. As such, the applicability of engineered vessels as an alternative to native vessels during revascularization procedures remains arguably premature.
A more immediate strategy to improve the integrity of vessels appropriate for grafting may involve supplementation of compromised grafts with autologous ALDHhi subpopulations, CD49d+ granulocytes, M2-polarized monocytes, to promote angiogenic or arteriogenic remodelling within the graft itself. Consequently, improving upon transplanted vessel survival and the risk of ischaemia re-establishment, specifically in micro-vascular capillaries, may be instrumental in improving vessel patency in the future. Finally, supplementation of native vessels with bioengineered materials such as autologous pro-angiogenic progenitor cell subsets or designer EVs could serve to improve graft integrity while stimulating the repair of native vessels. This union could bolster graft stability to become more effective for functional revascularization after surgery.
Concluding remarks
Pro-vascular progenitor cells transplantation has been proposed as a novel modality to treat CVD through the induction vessel regeneration. Unfortunately, hyperglycaemia and excess adiposity associated with T2D and obesity generate a damaging microenvironment in the BM and impact the production and mobilization of progenitor cells required to combat vessel damage (See Graphical Abstract). Assessment of ALDHhi progenitor cell subsets in the circulation of patients with T2D and obesity establish that pro-oxidative and pro-inflammatory microenvironment negatively impact circulating pro-angiogenic progenitor cell content. Early intervention with strategies that prevent or reverse abnormalities in the circulating progenitor cell pool represents an under recognized mechanism to effectively augment vessel repair and regeneration. Therefore, early intervention in patients with developing T2D and obesity may provide immediately translatable therapeutic options to reduce blood vessel damage and preserve blood vessel health in patients with T2D or obesity.
Acknowledgements
The authors gratefully acknowledge Gail Rudkevich in the generation of the figures presented.
Conflict of interest: D.A.H. does not have any conflict of interest. S.V. holds a Tier 1 Canada Research Chair in Cardiovascular Surgery and reports receiving research grants and/or speaking honoraria from Amarin, Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly, EOCI Pharmacomm Ltd, HLS Therapeutics, Janssen, Merck, Novartis, Novo Nordisk, Pfizer, PhaseBio, Sanofi, Sun Pharmaceuticals, and the Toronto Knowledge Translation Working Group. D.B. discloses the following relationships—Advisory Board: Cardax, CellProthera, Cereno Scientific, Elsevier Practice Update Cardiology, Janssen, Level Ex, Medscape Cardiology, MyoKardia, Novo Nordisk, PhaseBio, PLx Pharma, and Regado Biosciences; Board of Directors: Boston VA Research Institute, Society of Cardiovascular Patient Care, TobeSoft; Chair: American Heart Association Quality Oversight Committee; Data Monitoring Committees: Baim Institute for Clinical Research (formerly Harvard Clinical Research Institute, for the PORTICO trial, funded by St. Jude Medical, now Abbott), Cleveland Clinic (including for the ExCEED trial, funded by Edwards), Contego Medical (Chair, PERFORMANCE 2), Duke Clinical Research Institute, Mayo Clinic, Mount Sinai School of Medicine (for the ENVISAGE trial, funded by Daiichi Sankyo), Population Health Research Institute; Honoraria: American College of Cardiology (Senior Associate Editor, Clinical Trials and News, ACC.org; Vice-Chair, ACC Accreditation Committee), Baim Institute for Clinical Research (formerly Harvard Clinical Research Institute; RE-DUAL PCI clinical trial steering committee funded by Boehringer Ingelheim; AEGIS-II executive committee funded by CSL Behring), Belvoir Publications (Editor in Chief, Harvard Heart Letter), Canadian Medical and Surgical Knowledge Translation Research Group (clinical trial steering committees), Duke Clinical Research Institute (clinical trial steering committees, including for the PRONOUNCE trial, funded by Ferring Pharmaceuticals), HMP Global (Editor-in-Chief, Journal of Invasive Cardiology), Journal of the American College of Cardiology (Guest Editor; Associate Editor), K2P (Co-Chair, interdisciplinary curriculum), Level Ex, Medtelligence/ReachMD (CME steering committees), MJH Life Sciences, Population Health Research Institute (for the COMPASS operations committee, publications committee, steering committee, and USA national co-leader, funded by Bayer), Slack Publications (Chief Medical Editor, Cardiology Today’s Intervention), Society of Cardiovascular Patient Care (Secretary/Treasurer), WebMD (CME steering committees); Other: Clinical Cardiology (Deputy Editor), NCDR-ACTION Registry Steering Committee (Chair), VA CART Research and Publications Committee (Chair); Research Funding: Abbott, Afimmune, Amarin, Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, Cardax, CellProthera, Cereno Scientific, Chiesi, CSL Behring, Eisai, Ethicon, Ferring Pharmaceuticals, Forest Laboratories, Fractyl, Garmin, HLS Therapeutics, Idorsia, Ironwood, Ischemix, Janssen, Lexicon, Lilly, Medtronic, MyoKardia, Novartis, Novo Nordisk, Owkin, Pfizer, PhaseBio, PLx Pharma, Regeneron, Roche, Sanofi, Synaptic, The Medicines Company, 89Bio; Royalties: Elsevier (Editor, Cardiovascular Intervention: A Companion to Braunwald’s Heart Disease); Site Co-Investigator: Abbott, Biotronik, Boston Scientific, CSI, St. Jude Medical (now Abbott), Svelte; Trustee: American College of Cardiology; and Unfunded Research: FlowCo, Merck, Takeda. E.B. and D.C.T. do not report potential conflicts of interest relevant to this article. F.C. reports receiving research grants from Swedish Research Council, Swedish Heart & Lung Foundation, and King Gustav V and Queen Victoria Foundation, as well as speaking/advisory board honoraria from Abbott, AstraZeneca, Bayer, Bristol-Myers Squibb, Merck Sharp & Dohme, Mundipharma, Novo Nordisk, Boehringer Ingelheim, and Pfizer.
Data availability
The data in this article will be shared upon reasonable request to the corresponding author.
References


{kind=link}
{kind=link}
{kind=link}