





The National Heart Institute
In early 1958, shortly after my appointment as head of the cardiac catheterization laboratory of the National Heart Institute (now the NHLBI) in Bethesda, MD, I faced a challenging problem. We had studied a 27-year-old man with progressively severe precordial pain and exertional dyspnoea; a heart murmur had been detected some years earlier. He exhibited a prominent left ventricular (LV) lift and a Grade 4/6 systolic murmur at the apex and along the left sternal border. His ECG showed left ventricular hypertrophy (LVH) and the chest X-ray suggested LV enlargement. Catheterization showed a subaortic gradient of 74 mmHg.
We considered congenital membranous subaortic stenosis as the most likely diagnosis. Andrew Glenn Morrow, the Chief of Cardiac Surgery, felt that he could readily resect the obstructing membrane with the patient on cardiopulmonary bypass and the heart arrested. However, we were shocked that at operation no intraventricular obstruction was found. Indeed, Morrow’s bimanual examination of the LV revealed only hypertrophy. The patient recovered uneventfully, but we remained quite puzzled by the nature of his condition. Several months later, another young man presented with very similar findings, i.e. a high intraventricular pressure gradient, with LVH on palpation in the operating room but no subaortic obstruction. Three months postoperatively, a left ventriculogram revealed that ‘The LV cavity appeared to be encroached upon and almost divided by an intrinsic mass, presumably hypertrophied cardiac muscle’.
In our paper describing these two patients,1 we concluded that ‘the obstruction to ventricular outflow is of such a nature that it is only operative in the contracting heart and not apparent during the paralysis induced by potassium citrate. “These features can only be explained by muscular hypertrophy of the LV outflow tract of sufficient severity that flow is actually impeded during contraction” (my emphasis). We misnamed the condition ‘Functional Aortic Stenosis’ but soon adopted a better name—‘Idiopathic Hypertrophic Subaortic Stenosis (IHSS)’.2 After it became apparent that some patients with this condition had no subaortic obstruction,3 we accepted the name ‘hypertrophic cardiomyopathy (HCM) with or without obstruction’.
After looking into the matter, I learned that individual cases of idiopathic LVH had been described as early as the 18th century. More recently, unexplained LVH was found at autopsy in young victims of sudden cardiac death. Also, LVH was reported to be familial in some instances. Importantly, in 1958, Donald Teare, a London pathologist, described nine patients with LVH showing marked asymmetric hypertrophy involving the interventricular septum, with myocyte hypertrophy and interstitial fibrosis. Eight of these patients had died suddenly; two were siblings.4 Sir (later Lord) Russell Brock, a distinguished British cardiac surgeon, described patients with subaortic muscular stenosis that he initially attributed to prolonged hypertension or severe aortic stenosis. We sent Brock a copy of our paper,1 which he cited in his report of similar patients with primary muscular subaortic obstruction.5
One of our key early findings was that the obstruction was dynamic2 and varied on a day-to-day basis. It could be intensified or provoked by beta-adrenergic stimulation (Figure 1), exercise, and by the Valsalva manoeuver. In 1962, Sir James Black, a Nobel Prize-winning British pharmacologist, described the first β-adrenergic blockers. We administered pronethalol to patients with HCM and were excited to observe that it prevented the provocation and intensification of obstruction by isoproterenol and exercise and often reduced symptoms.6 After scattered surgical attempts elsewhere, Morrow developed myotomy–myectomy for symptomatic patients with severe obstruction despite beta-blockade.7 These two therapies, introduced almost 60 years ago, continue to hold Class IB indications in the current practice guidelines for HCM.8
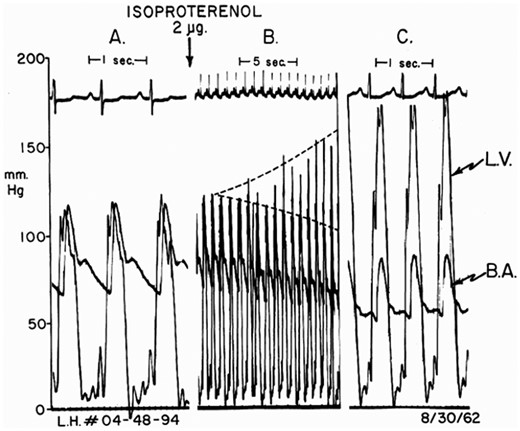
Provocation of obstruction in a patient with left ventricular hypertrophy without obstruction catheterized by the author in 1962. (A) Baseline pressure; (B) 16 cardiac cycles following isoproterenol bolus; dashed lines show increases in left ventricle and reductions in brachial artery systolic pressures. (C) Maximal effect = 90 mmHg gradient. B.A., brachial artery; L.V., left ventricle. Reproduced from Ref.2
In 1964, we reviewed our observations on 64 patients and pulled them together into a specific entity that had several components. Hypertrophic cardiomyopathy patients had characteristic clinical and ECG findings.2 They usually exhibited hypercontractile LV function, dynamic obstruction to LV outflow, and impaired LV relaxation. Sudden cardiac death was the most common cause of death in adolescents and young adults, while in older patients deaths were usually from heart failure. In about one-third of our patients, the HCM was familial with autosomal dominant inheritance. A small number of patients had no provokable obstruction.3
My post National Heart Institute years
After I left the NIH, the development of echocardiography provided an accurate, non-invasive, routine approach to diagnosis that led to the discovery of large numbers of patients with HCM worldwide. Also, the implantation of cardioverter-defibrillators in HCM patients at high risk of developing ventricular fibrillation greatly reduced this frequent cause of death. In 1995, alcohol septal ablation using percutaneous coronary intervention was introduced. This technique has been effective in relieving severe obstruction and has been widely employed.
At Harvard and the Brigham, I had the good fortune to get to know a brilliant wife-husband team of cardiac geneticists, Drs Christine and Jonathan Seidman. This gave me a bird’s eye view of further progress in understanding HCM. In 1990, the Seidmans and their colleagues provided the first molecular basis for familial HCM,9 a missense mutation of the myosin-heavy chain gene, the first of at least eight sarcomeric genes whose variants are responsible for familial HCM. These discoveries have made possible both genetic testing and counselling in this condition.
MyoKardia
In 2016, Green, a scientist at the biotech company MyoKardia (since 2018, Dr Braunwald has served as a Science Advisor to MyoKardia, which was acquired by Bristol Myers Squibb in 2020), together with the Seidmans and others screened small molecular inhibitors of cardiac myosin ATPase. These inhibitors reduce actin-myosin cross-bridge formation, which inhibits sarcomere hypercontractility characteristic of HCM.10 The most promising of these inhibitors, named mavacamten, was studied in patients with obstructive HCM. A phase 3 placebo-controlled, double-blind trial showed that mavacamten reduced subaortic obstruction, myocardial mass, and symptoms. It was well tolerated11 and is now under consideration by regulatory authorities.
Conclusions
It has been a privilege to participate in and follow the enormous advances that have occurred in this condition for the 63 years since I studied the two young men with the intraventricular pressure gradients without obstruction.1 Hypertrophic cardiomyopathy is a well-known, important condition,8,12 the most common monogenic cardiac disorder for which there are specialized care centres in many parts of the world. However, there are still many important questions to be answered. For example, what molecular mechanisms are responsible for the ∼60% of HCM patients who are non-familial and without detectable genetic variants? Conversely, what causes many carriers of typical HCM variants to never express the phenotype? Will mavacamten treatment begun at an early age before full phenotypic expression change the natural history of HCM? Will it reduce the need for septal reduction therapy, and of implanted cardioverter-defibrillators, as well as the late development of heart failure? Many of the answers to these and other important questions are likely to appear in the pages of this journal.
Conflict of interest: Research grant support through Brigham and Women’s Hospital from: AstraZeneca, Daiichi-Sankyo, Merck, and Novartis; consulting for: Amgen, Boehringer-Ingelheim/Lilly, Cardurion, MyoKardia, NovoNordisk, and Verve.


{kind=link}