





Abstract
To appraise the clinical and genetic evidence that low-density lipoproteins (LDLs) cause atherosclerotic cardiovascular disease (ASCVD).
We assessed whether the association between LDL and ASCVD fulfils the criteria for causality by evaluating the totality of evidence from genetic studies, prospective epidemiologic cohort studies, Mendelian randomization studies, and randomized trials of LDL-lowering therapies. In clinical studies, plasma LDL burden is usually estimated by determination of plasma LDL cholesterol level (LDL-C). Rare genetic mutations that cause reduced LDL receptor function lead to markedly higher LDL-C and a dose-dependent increase in the risk of ASCVD, whereas rare variants leading to lower LDL-C are associated with a correspondingly lower risk of ASCVD. Separate meta-analyses of over 200 prospective cohort studies, Mendelian randomization studies, and randomized trials including more than 2 million participants with over 20 million person-years of follow-up and over 150 000 cardiovascular events demonstrate a remarkably consistent dose-dependent log-linear association between the absolute magnitude of exposure of the vasculature to LDL-C and the risk of ASCVD; and this effect appears to increase with increasing duration of exposure to LDL-C. Both the naturally randomized genetic studies and the randomized intervention trials consistently demonstrate that any mechanism of lowering plasma LDL particle concentration should reduce the risk of ASCVD events proportional to the absolute reduction in LDL-C and the cumulative duration of exposure to lower LDL-C, provided that the achieved reduction in LDL-C is concordant with the reduction in LDL particle number and that there are no competing deleterious off-target effects.
Consistent evidence from numerous and multiple different types of clinical and genetic studies unequivocally establishes that LDL causes ASCVD.
Introduction
Atherosclerotic cardiovascular disease (ASCVD) and its clinical manifestations, such as myocardial infarction (MI) and ischaemic stroke, are the leading cause of morbidity and mortality throughout the world. Multiple exposures have been reported to be associated with an increased risk of cardiovascular events.1 The most extensively studied of these exposures by far is low-density lipoprotein (LDL). Multiple lines of evidence have established that cholesterol-rich LDL and other apolipoprotein B (apoB)-containing lipoproteins, including very low-density lipoproteins (VLDL) and their remnants, intermediate density lipoproteins (IDL), and lipoprotein(a) [Lp(a)], are directly implicated in the development of ASCVD.2
Despite this extensive body of evidence, however, some still express scepticism of the causal nature of the relationship between LDL and the development of ASCVD.3 With the availability of new, highly efficacious LDL-lowering agents and the development of additional novel lipid-lowering agents with prolonged duration of action, there is a need for a consensus as to whether LDL causes ASCVD in order to inform treatment guidelines and help shape regulatory agency guidance for the approval of new medicines.
This Consensus Statement, the first of two that evaluates the case for LDL causality, appraises evidence from genetic, epidemiologic, and clinical intervention studies. The second paper discusses the evidence for LDL causality based on the current understanding of the pathophysiology of ASCVD. While our focus is on LDL, this does not diminish the importance of the role of other apoB-containing lipoproteins on the development of ASCVD nor does it exclude potential atherogenic actions of the individual components of the lipidome and proteome of LDL beyond cholesterol and apoB.
Most publications that question the causal effect of LDL on the development of ASCVD tend to cite evidence from individual studies or a small group of highly selected studies, often without a quantitative synthesis of the presented evidence.4 Therefore, to avoid this type of selection bias, we have based our conclusions on the totality of evidence from separate meta-analyses of genetic studies, prospective epidemiologic studies, Mendelian randomization studies, and randomized clinical trials. This evidence base includes over 200 studies involving over 2 million participants with over 20 million person-years of follow-up and more than 150 000 cardiovascular events. Together these studies provide remarkably consistent and unequivocal evidence that LDL causes ASCVD as summarized in Table 1.
Criteria for causality: low-density lipoprotein (LDL) and atherosclerotic cardiovascular disease (ASCVD)
| Criterion (modified from reference5) | Evidence grade | Summary of the evidence (references) |
|---|---|---|
| 1. Plausibility | 1 | LDL and other apolipoprotein (apo) B-containing lipoproteins (very low-density lipoprotein their remnants, intermediate-density lipoprotein and lipoprotein(a)) are directly implicated in the initiation and progression of ASCVD; experimentally induced elevations in plasma LDL and other apoB-containing lipoproteins lead to atherosclerosis in all mammalian species studied.2 , 5–12 |
| 2. Strength | 1 | Monogenic and polygenic-mediated lifelong elevations in LDL lead to markedly higher lifetime risk.13–20 , 27–31 , 40 , 43 |
| 3. Biological gradient | 1 | Monogenic lipid disorders, prospective cohort studies, Mendelian randomization studies, and randomized intervention trials uniformly demonstrate a dose-dependent, log-linear association between the absolute magnitude of exposure to LDL and risk of ASCVD13–22 , 27–36 , 38–40 , 42–47 |
| 4. Temporal sequence | 1 | Monogenic lipid disorders and Mendelian randomization studies demonstrate that exposure to elevated LDL precedes the onset of ASCVD13–20 , 27–31 , 40 , 43 |
| 5. Specificity | 1 | Mendelian randomization studies and randomized intervention trials both provide unconfounded randomized evidence that LDL is associated with ASCVD independent of other risk factors28 , 31–33 , 40 , 43 |
| 6. Consistency | 1 | Over 200 studies involving more than 2 million participants with over 20 million person-years of follow-up and more than 150 000 cardiovascular events consistently demonstrate a dose-dependent, log-linear association between the absolute magnitude of exposure to LDL and risk of ASCVD13–22 , 27–36 , 38–40 , 42–47 |
| 7. Coherence | 1 | Monogenic lipid disorders, prospective cohort studies, Mendelian randomization studies, and randomized intervention trials all show a dose-dependent, log-linear association between the absolute magnitude of exposure to LDL and risk of ASCVD15–18 , 21 , 22 , 28 , 30–32 , 35 , 36 , 43 , 44 , 47 |
| 8. Reduction in risk with intervention | 1 | More than 30 randomized trials involving over 200 000 participants and 30 000 ASCVD events evaluating therapies specifically designed to lower LDL (including statins, ezetimibe, and PCSK9 inhibitors) consistently demonstrate that reducing LDL cholesterol (LDL-C) reduces the risk of ASCVD events proportional to the absolute reduction in LDL-C32–34 , 38 , 39 , 42 , 45–47 |
| Criterion (modified from reference5) | Evidence grade | Summary of the evidence (references) |
|---|---|---|
| 1. Plausibility | 1 | LDL and other apolipoprotein (apo) B-containing lipoproteins (very low-density lipoprotein their remnants, intermediate-density lipoprotein and lipoprotein(a)) are directly implicated in the initiation and progression of ASCVD; experimentally induced elevations in plasma LDL and other apoB-containing lipoproteins lead to atherosclerosis in all mammalian species studied.2 , 5–12 |
| 2. Strength | 1 | Monogenic and polygenic-mediated lifelong elevations in LDL lead to markedly higher lifetime risk.13–20 , 27–31 , 40 , 43 |
| 3. Biological gradient | 1 | Monogenic lipid disorders, prospective cohort studies, Mendelian randomization studies, and randomized intervention trials uniformly demonstrate a dose-dependent, log-linear association between the absolute magnitude of exposure to LDL and risk of ASCVD13–22 , 27–36 , 38–40 , 42–47 |
| 4. Temporal sequence | 1 | Monogenic lipid disorders and Mendelian randomization studies demonstrate that exposure to elevated LDL precedes the onset of ASCVD13–20 , 27–31 , 40 , 43 |
| 5. Specificity | 1 | Mendelian randomization studies and randomized intervention trials both provide unconfounded randomized evidence that LDL is associated with ASCVD independent of other risk factors28 , 31–33 , 40 , 43 |
| 6. Consistency | 1 | Over 200 studies involving more than 2 million participants with over 20 million person-years of follow-up and more than 150 000 cardiovascular events consistently demonstrate a dose-dependent, log-linear association between the absolute magnitude of exposure to LDL and risk of ASCVD13–22 , 27–36 , 38–40 , 42–47 |
| 7. Coherence | 1 | Monogenic lipid disorders, prospective cohort studies, Mendelian randomization studies, and randomized intervention trials all show a dose-dependent, log-linear association between the absolute magnitude of exposure to LDL and risk of ASCVD15–18 , 21 , 22 , 28 , 30–32 , 35 , 36 , 43 , 44 , 47 |
| 8. Reduction in risk with intervention | 1 | More than 30 randomized trials involving over 200 000 participants and 30 000 ASCVD events evaluating therapies specifically designed to lower LDL (including statins, ezetimibe, and PCSK9 inhibitors) consistently demonstrate that reducing LDL cholesterol (LDL-C) reduces the risk of ASCVD events proportional to the absolute reduction in LDL-C32–34 , 38 , 39 , 42 , 45–47 |
Criteria are graded by a modification of the quality criteria adopted by the European Society of Cardiology system.
For reference, see http://www.escardio.org/Guidelines-&-Education/Clinical-Practice-Guidelines/Guidelinesdevelopment/Writing-ESC-Guidelines (31 January 2017).
These are defined as follows:
Class 1: Evidence and/or general agreement that the criterion for causality is fulfilled.
Class 2: Conflicting evidence and/or a divergence of opinion about whether the criterion indicated causality.
Class 3: Evidence or general agreement that the criterion for causality is not fulfilled.
Criteria for causality: low-density lipoprotein (LDL) and atherosclerotic cardiovascular disease (ASCVD)
| Criterion (modified from reference5) | Evidence grade | Summary of the evidence (references) |
|---|---|---|
| 1. Plausibility | 1 | LDL and other apolipoprotein (apo) B-containing lipoproteins (very low-density lipoprotein their remnants, intermediate-density lipoprotein and lipoprotein(a)) are directly implicated in the initiation and progression of ASCVD; experimentally induced elevations in plasma LDL and other apoB-containing lipoproteins lead to atherosclerosis in all mammalian species studied.2 , 5–12 |
| 2. Strength | 1 | Monogenic and polygenic-mediated lifelong elevations in LDL lead to markedly higher lifetime risk.13–20 , 27–31 , 40 , 43 |
| 3. Biological gradient | 1 | Monogenic lipid disorders, prospective cohort studies, Mendelian randomization studies, and randomized intervention trials uniformly demonstrate a dose-dependent, log-linear association between the absolute magnitude of exposure to LDL and risk of ASCVD13–22 , 27–36 , 38–40 , 42–47 |
| 4. Temporal sequence | 1 | Monogenic lipid disorders and Mendelian randomization studies demonstrate that exposure to elevated LDL precedes the onset of ASCVD13–20 , 27–31 , 40 , 43 |
| 5. Specificity | 1 | Mendelian randomization studies and randomized intervention trials both provide unconfounded randomized evidence that LDL is associated with ASCVD independent of other risk factors28 , 31–33 , 40 , 43 |
| 6. Consistency | 1 | Over 200 studies involving more than 2 million participants with over 20 million person-years of follow-up and more than 150 000 cardiovascular events consistently demonstrate a dose-dependent, log-linear association between the absolute magnitude of exposure to LDL and risk of ASCVD13–22 , 27–36 , 38–40 , 42–47 |
| 7. Coherence | 1 | Monogenic lipid disorders, prospective cohort studies, Mendelian randomization studies, and randomized intervention trials all show a dose-dependent, log-linear association between the absolute magnitude of exposure to LDL and risk of ASCVD15–18 , 21 , 22 , 28 , 30–32 , 35 , 36 , 43 , 44 , 47 |
| 8. Reduction in risk with intervention | 1 | More than 30 randomized trials involving over 200 000 participants and 30 000 ASCVD events evaluating therapies specifically designed to lower LDL (including statins, ezetimibe, and PCSK9 inhibitors) consistently demonstrate that reducing LDL cholesterol (LDL-C) reduces the risk of ASCVD events proportional to the absolute reduction in LDL-C32–34 , 38 , 39 , 42 , 45–47 |
| Criterion (modified from reference5) | Evidence grade | Summary of the evidence (references) |
|---|---|---|
| 1. Plausibility | 1 | LDL and other apolipoprotein (apo) B-containing lipoproteins (very low-density lipoprotein their remnants, intermediate-density lipoprotein and lipoprotein(a)) are directly implicated in the initiation and progression of ASCVD; experimentally induced elevations in plasma LDL and other apoB-containing lipoproteins lead to atherosclerosis in all mammalian species studied.2 , 5–12 |
| 2. Strength | 1 | Monogenic and polygenic-mediated lifelong elevations in LDL lead to markedly higher lifetime risk.13–20 , 27–31 , 40 , 43 |
| 3. Biological gradient | 1 | Monogenic lipid disorders, prospective cohort studies, Mendelian randomization studies, and randomized intervention trials uniformly demonstrate a dose-dependent, log-linear association between the absolute magnitude of exposure to LDL and risk of ASCVD13–22 , 27–36 , 38–40 , 42–47 |
| 4. Temporal sequence | 1 | Monogenic lipid disorders and Mendelian randomization studies demonstrate that exposure to elevated LDL precedes the onset of ASCVD13–20 , 27–31 , 40 , 43 |
| 5. Specificity | 1 | Mendelian randomization studies and randomized intervention trials both provide unconfounded randomized evidence that LDL is associated with ASCVD independent of other risk factors28 , 31–33 , 40 , 43 |
| 6. Consistency | 1 | Over 200 studies involving more than 2 million participants with over 20 million person-years of follow-up and more than 150 000 cardiovascular events consistently demonstrate a dose-dependent, log-linear association between the absolute magnitude of exposure to LDL and risk of ASCVD13–22 , 27–36 , 38–40 , 42–47 |
| 7. Coherence | 1 | Monogenic lipid disorders, prospective cohort studies, Mendelian randomization studies, and randomized intervention trials all show a dose-dependent, log-linear association between the absolute magnitude of exposure to LDL and risk of ASCVD15–18 , 21 , 22 , 28 , 30–32 , 35 , 36 , 43 , 44 , 47 |
| 8. Reduction in risk with intervention | 1 | More than 30 randomized trials involving over 200 000 participants and 30 000 ASCVD events evaluating therapies specifically designed to lower LDL (including statins, ezetimibe, and PCSK9 inhibitors) consistently demonstrate that reducing LDL cholesterol (LDL-C) reduces the risk of ASCVD events proportional to the absolute reduction in LDL-C32–34 , 38 , 39 , 42 , 45–47 |
Criteria are graded by a modification of the quality criteria adopted by the European Society of Cardiology system.
For reference, see http://www.escardio.org/Guidelines-&-Education/Clinical-Practice-Guidelines/Guidelinesdevelopment/Writing-ESC-Guidelines (31 January 2017).
These are defined as follows:
Class 1: Evidence and/or general agreement that the criterion for causality is fulfilled.
Class 2: Conflicting evidence and/or a divergence of opinion about whether the criterion indicated causality.
Class 3: Evidence or general agreement that the criterion for causality is not fulfilled.
Pathophysiology of atherosclerosis
The key events in the initiation of ASCVD are the retention and accumulation of cholesterol-rich apoB-containing lipoproteins within the arterial intima at sites of predilection for plaque formation.5–9 Notably, LDL and other apoB-containing lipoproteins <70 nm in diameter (including VLDL, their remnants, IDL, and Lp(a)) efficiently enter and exit the arterial intima.9 , 10 At what is now considered physiologically relevant levels of LDL cholesterol [LDL-C; ∼0.5–1.0 mmol/L (20–40 mg/dL), typical of newborns11 and a wide range of mammalian species12], the probability of LDL particle retention and the risk of developing atherosclerosis is low.6 However, as the concentration of LDL-C increases above this level, the probability of intimal retention of LDL leading to the initiation and progressive development of atherosclerotic plaque increases in a dose-dependent manner.2 This topic will be discussed in detail in the second Consensus Statement on this topic.
Cholesterol, LDL, and LDL-C
The terms ‘cholesterol’, ‘LDL’, and ‘LDL cholesterol (LDL-C)’ are frequently conflated or used interchangeably, potentially leading to confusion. Cholesterol is an essential component of cell membranes and a precursor of bile acids and steroid hormones. Importantly, cholesterol of both exogenous and endogenous origin is transported to peripheral cells largely by the apoB-containing lipoproteins in plasma. In most people, LDL particles constitute ∼90% of circulating apoB-containing lipoproteins in fasting blood (Figure 1). However, in clinical practice, the plasma LDL level is generally not measured directly but instead is estimated from its cholesterol concentration—LDL-C—a measure of the total amount of cholesterol contained in LDL particles. As a result, calculated plasma LDL-C has become the focus for assessing cardiovascular risk and for evaluating therapeutic benefit in randomized clinical trials.
![Relative concentration of apolipoprotein B (ApoB) contained in circulating lipoproteins in normolipidaemic individuals. ApoB content was calculated in nanomoles per litre using 500 000 as the defined molecular mass [i.e. low-density lipoprotein (LDL) 100 mg/dL or 2000 nmol/L, very low-density lipoprotein (VLDL) 5 mg/dL or 100 nmol/L, intermediate density lipoprotein (IDL) remnants 5 mg/dL or 100 nmol/L and lipoprotein(a) 10 nmol/l*]. *Based on population median.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/eurheartj/38/32/10.1093_eurheartj_ehx144/4/m_eurheartj_38_32_2459_f1.jpeg?Expires=1749552643&Signature=Wt9nWgfcbVilHkdxdWzCSrawCblgE5as24XLe~oCNXcQAOZcdQEEb9wDcIkR5J22CQUmzl41GcQcm0d3llKcsXjvZmuIDoigEB4nUA5ms5fin51vlWcWOjlg7rlfvoswciSTgHvYBanN3el~8ri2LfaOmvhPeEH7XlMn2q~tFO3yJ-N5w3Ni4mf9ZgVeWzq28NU8Ev3xWyCrF1jI~xS7Z11ncH61xRpNL2ZYFCpdiEBfHyvbYErYCzCpdNPhwKdE4tx0~2a-2fRjXJcDbDQonpuBSusFL42kCmLNLD5yOvd9SVUDNlTfePY8W5U2LvKsmP2vIoJt8zQexsHPSPazGA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Relative concentration of apolipoprotein B (ApoB) contained in circulating lipoproteins in normolipidaemic individuals. ApoB content was calculated in nanomoles per litre using 500 000 as the defined molecular mass [i.e. low-density lipoprotein (LDL) 100 mg/dL or 2000 nmol/L, very low-density lipoprotein (VLDL) 5 mg/dL or 100 nmol/L, intermediate density lipoprotein (IDL) remnants 5 mg/dL or 100 nmol/L and lipoprotein(a) 10 nmol/l*]. *Based on population median.
Under most conditions, LDL-C concentration and LDL particle number are highly correlated, and therefore plasma LDL-C is a good surrogate for LDL particle concentration. However, in certain conditions (e.g. the metabolic syndrome, diabetes, and hypertriglyceridaemia), plasma LDL-C and LDL particle concentration can become discordant as a result of the predominance of small, dense cholesterol-poor LDL, and therefore plasma LDL-C may not accurately reflect LDL particle concentration or its effect on cardiovascular risk. Under these conditions, direct measurement of LDL particle number or apoB concentration (recognizing that each LDL particle contains a single apoB molecule) may more accurately reflect the causal effect of LDL on ASCVD. In this Consensus Statement, we assess the evidence that LDL causes ASCVD by critically appraising the clinical evidence, with the understanding that the majority of clinical studies have used LDL-C as an estimate of LDL concentration.
Evidence from inherited disorders of lipid metabolism
Familial hypercholesterolaemia (FH) is an autosomal co-dominant disorder that usually results from a loss-of-function (LOF) mutation in the LDL receptor (LDLR) gene, or less commonly, from LOF mutations in the APOB gene that reduce the binding of apoB-containing lipoproteins to the LDL receptor or a gain-of-function (GOF) mutation in the PCSK9 gene. Regardless of the underlying genetic defect, FH is characterized by markedly elevated levels of LDL-C and premature atherosclerosis, particularly coronary artery disease.13 , 14
In the most common form of FH, a mutation in the LDLR gene causes decreased LDL receptor function leading to a markedly increased concentration of circulating LDL particles and the cholesterol carried by those particles (i.e. LDL-C). Heterozygous FH (HeFH) affects between 1:200 and 1:300 people worldwide15 , 16 and when untreated is characterized by LDL-C levels in the range of 4.5–12 mmol/L and a marked increase in risk of ASCVD.13 Homozygous FH (HoFH) is a much rarer condition, with an extreme phenotype characterized by untreated plasma LDL-C levels often in excess of 13 mmol/L from birth and almost universal development of ASCVD in childhood or early adolescence.14 Although the phenotypic expression of FH is variable, the extent of atherosclerosis and the risk of cardiovascular events in both HeFH and HoFH is proportional to both the absolute magnitude and the duration of exposure to elevated LDL-C levels.15 , 17 , 18
Within any affected family, each child has an equal and random 50% probability of inheriting a mutation that causes FH. The fact that siblings who inherit an FH mutation have markedly elevated plasma LDL-C levels and a corresponding dose-dependent markedly elevated lifetime risk of ASCVD as compared to their unaffected siblings provides powerful evidence that LDL causes ASCVD.19 This conclusion is substantially strengthened by the complementary observations that GOF mutations in PCSK9 result in a markedly elevated LDL-C concentration and a corresponding markedly elevated risk of ASCVD, while LOF mutations in PCSK9 result in a lower LDL-C concentration and a corresponding markedly lower lifetime risk of ASCVD.20
Evidence from prospective epidemiologic studies
Several large meta-analyses of prospective observational epidemiologic studies using individual participant data have consistently reported a continuous log-linear association between the absolute magnitude of exposure to plasma LDL-C levels and the risk of ASCVD.
The Emerging Risk Factors Collaboration (ERFC) reported the results of a meta-analysis of individual participant data from 302 430 persons without prevalent vascular disease at the time of enrolment in 68 prospective studies during which 8857 non-fatal MI and 928 coronary heart disease (CHD) deaths accrued over 2.79 million person-years of follow-up.21 In these studies, plasma LDL-C concentration was log-linearly associated with increased risk of non-fatal MI or CHD death. Although the authors reported the association between non-HDL-C concentration and risk of CHD in the primary analysis, all studies included in this meta-analysis measured total cholesterol, high-density lipoprotein cholesterol (HDL-C), and triglycerides and reported the calculated LDL-C concentration as estimated by the Friedewald equation. The ERFC authors point out that any regression model that includes terms for non-HDL-C, HDL-C, and triglycerides is a simple mathematical rearrangement of a model that includes terms for calculated LDL-C, HDL-C, and triglycerides. Therefore, in the ERFC analysis, the effect of LDL-C is exactly equal to the effect of non-HDL-C on the risk of CHD by definition in the analysis. The authors confirmed this fact by demonstrating that in a subsample of 8 studies involving 44 234 individuals, the effect of directly measured LDL-C on the risk of CHD was nearly identical to the effect of non-HDL-C (and calculated LDL-C) per millimole per litre.
Similarly, the Prospective Studies Collaboration reported a meta-analysis of individual participant data on 892 337 persons without cardiovascular disease at baseline who had been enrolled in 61 prospective cohort studies during which 33 744 ischaemic heart disease deaths accrued over nearly 12 million person-years of follow-up. This meta-analysis reported a strong, graded log-linear association between total plasma cholesterol and the risk of ischaemic heart disease mortality.22 Importantly, in a subsample of 153 798 participants for whom HDL-C measurements were available, the effect of non-HDL-C on the risk of ischaemic heart disease mortality was nearly identical to the effect of total cholesterol per millimole per litre.
Together, these meta-analyses of prospective epidemiologic cohort studies provide coherent and consistent evidence that plasma LDL-C concentration is strongly and log-linearly associated with a dose-dependent increase in the risk of incident ASCVD events (Figure 2).

Log-linear association per unit change in low-density lipoprotein cholesterol (LDL-C) and the risk of cardiovascular disease as reported in meta-analyses of Mendelian randomization studies, prospective epidemiologic cohort studies, and randomized trials. The increasingly steeper slope of the log-linear association with increasing length of follow-up time implies that LDL-C has both a causal and a cumulative effect on the risk of cardiovascular disease. The proportional risk reduction (y axis) is calculated as 1−relative risk (as estimated by the odds ratio in Mendelian randomization studies, or the hazard ration in the prospective epidemiologic studies and randomized trials) on the log scale, then exponentiated and converted to a percentage. The included meta-analyses were identified from (i) MEDLINE and EMBASE using the search terms meta-analysis, LDL, and ‘cardiovascular or coronary’; (ii) the reference lists of the identified meta-analyses; (iii) public data from GWAS consortia; and (iv) by discussion with members of the EAS Consensus Panel. We included the most updated meta-analyses available, giving preference to meta-analyses that used individual participant data. Trial acronyms: AF/TexCAPS, Air Force/Texas Coronary Atherosclerosis Prevention Study; ALERT, Assessment of LEscol in Renal Transplantation; ALLHAT-LLT, Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial Lipid Lowering Trial; ALLIANCE, Aggressive Lipid-Lowering Initiation Abates New Cardiac Events; ASPEN, Atorvastatin Study for Prevention of Coronary Heart Disease Endpoints in non-insulin-dependent diabetes mellitus; ASCOT LLA, Anglo Scandinavian Cardiac Outcomes Trial Lipid Lowering Arm; AURORA, A Study to Evaluate the Use of Rosuvastatin in Subjects on Regular Hemodialysis: An Assessment of Survival and Cardiovascular Events; CARE, Cholesterol and Recurrent Events; CARDS, Collaborative Atorvastatin Diabetes Study; CHGN, Community Health Global Network; 4D Deutsche Diabetes Dialyse Studies; ERFC, Emerging Risk Factors Collaboration; GISSI, Gruppo Italiano per lo Studio della Sopravvivenza nell'Infarto Miocardico; HOPE, Heart Outcomes Prevention Evaluation Study; HPS, Heart Protection Study; IDEAL, Incremental Decrease in End Points Through Aggressive Lipid Lowering; IMPROVE-IT, Examining Outcomes in Subjects With Acute Coronary Syndrome: Vytorin (Ezetimibe/Simvastatin) vs Simvastatin; JUPITER, Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin trial; LIPID,, Long-Term Intervention with Pravastatin in Ischemic Disease; LIPS, Lescol Intervention Prevention Study; MEGA, Management of Elevated Cholesterol in the Primary Prevention Group of Adult Japanese; POST-CABG, Post Coronary Artery Bypass Graft; PROSPER, Pravastatin in elderly individuals at risk of vascular disease; PROVE-IT, Pravastatin or Atorvastatin Evaluation and Infection Therapy; SHARP, Study of Heart and Renal Protection; TNT, Treating to New Targets; WOSCOPS, West of Scotland Coronary Prevention Study.
Evidence from Mendelian randomization studies
Although the association between LDL-C and the risk of ASCVD is strong, graded, and reproducible in meta-analyses of prospective cohort studies, these studies are not randomized and are therefore unavoidably vulnerable to confounding, reverse causation, and other forms of bias. Mendelian randomization studies introduce a randomization scheme into an observational study specifically to assess whether an observed association between an exposure and an outcome is likely to be causal.23
Numerous variants in multiple genes have been reported to be associated with lower LDL-C levels.24 , 25 Each of these variants is inherited approximately randomly at the time of conception in a process sometimes referred to as Mendelian randomization. Therefore, inheriting an LDL-C lowering allele in one of these genes is analogous to being randomly allocated to treatment with an LDL-C-lowering therapy, while inheriting the other allele is analogous to being randomly allocated to ‘usual care’. If the variant under study is associated solely with LDL-C and not with other lipid or non-lipid pleiotropic effects, and if allocation is indeed random, then comparing the risk of ASCVD among persons with and without such a variant should provide an unconfounded estimate of the causal effect of lower LDL-C levels on the risk of ASCVD in a manner analogous to a long-term randomized trial.26
Mendelian randomization studies have consistently demonstrated that variants in over 50 genes that are associated with lower LDL-C levels (but not with other potential predictors or intermediates for ASCVD) are also associated with a correspondingly lower risk of CHD,20 , 27–30 thus providing powerful evidence that LDL is causally associated with the risk of CHD. Indeed, when the effect of each LDL-C variant is plotted against its effect on CHD, there is a continuous, dose-dependent, and log-linear causal association between the magnitude of the absolute change in LDL-C level and the lifetime risk of CHD (Figure 2).28 , 30
Furthermore, when adjusted for a standard decrement in LDL-C, each of the genetic variants associated with LDL-C has a remarkably similar effect on the risk of CHD per unit lower LDL-C, including variants in the genes that encode the targets of pharmacological agents commonly used to lower LDL-C [i.e. 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGCR), the target of statins; Niemann-Pick C1-like 1 (NPC1L1), the target of ezetimibe; and proprotein convertase subtilisin/kexin type 9 (PCSK9), the target of the monoclonal antibodies alirocumab and evolocumab; see Figure 3], with no evidence of any heterogeneity of effect (I 2 = 0%).28 , 30 , 31 This observation strongly implies that the causal effect of these variants on the risk of CHD is mediated essentially entirely through LDL, because it would be implausible that variants in numerous different genes involving multiple distinct biological pathways by which LDL is lowered would each have directionally concordant and quantitatively similar pleiotropic effects on the risk of ASCVD.

Effect of exposure to lower low-density lipoprotein cholesterol (LDL-C) by mechanism of LDL-C lowering. Panel A shows the effect of genetic variants or genetic scores combining multiple variants in the genes that encode for the targets of currently available LDL-C-lowering therapies, adjusted for a standard decrement of 0.35 mmol/L lower LDL-C, in comparison with the effect of lower LDL-C mediated by variants in the LDL receptor gene. Panel B shows the effect of currently available therapies that act to primarily lower LDL-C through the LDL receptor pathway, adjusted per millimole per litre lower LDL-C. Both the naturally randomized genetic data in Panel A and the data from randomized trials in Panel B suggest that the effect of LDL-C on the risk of cardiovascular events is approximately the same per unit change in LDL-C for any mechanism that lowers LDL-C via up-regulation of the LDL receptor where the change in LDL-C (which is used in clinical medicine to estimate the change in LDL particle concentration) is likely to be concordant with changes in LDL particle concentration.
Taken together, meta-analyses of Mendelian randomization studies involving more than 300 000 participants and 80 000 CHD cases provide compelling evidence that LDL is causally associated with the risk of ASCVD and that the causal effect of LDL on ASCVD is largely independent of the mechanism by which LDL is ‘lowered’.
Evidence from randomized controlled trials
Perhaps the most compelling clinical evidence for causality is provided by randomized clinical trials evaluating the effect of therapies that reduce LDL-C on the risk of cardiovascular events. (For reference, Figure 4 shows the principal sites of action of therapeutic interventions that act mainly to lower LDL.) It is important to note, however, that the interpretation of any given individual trial can be substantially affected by its design. In general, studies that are underpowered due to small sample sizes or few accrued events, that do not produce a substantial difference in LDL-C concentration between treatment arms, and those with short-term follow-up (2 years or less) are more likely to produce results that do not show statistically significant differences. Furthermore, some therapies that lower LDL-C (e.g. oestrogen) also have adverse effects that increase the risk of ASCVD which can attenuate or erase the clinical benefit of lowering LDL-C. Over-interpretation of these individual trials may lead to biased conclusions. For that reason, we focus on the totality of the evidence derived from meta-analyses of randomized trials.
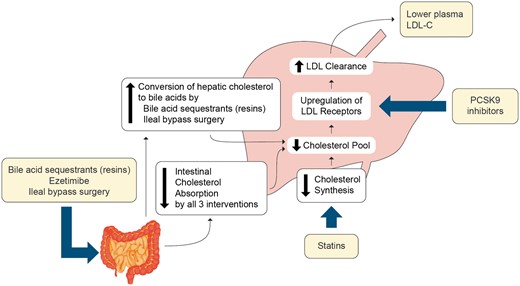
Schematic figure showing that all therapies that act predominantly to lower low-density lipoprotein (LDL) act via the LDL receptor pathway to up-regulate LDL receptors and thus increase LDL clearance.
In a meta-analysis of individual-participant data from 26 statin trials including almost 170 000 individuals, treatment with a statin was associated with a log-linear 22% proportional reduction in the risk of major cardiovascular events per millimole per litre reduction in LDL-C over a median of 5 years of treatment.32 The effect was somewhat less during the first year of treatment, followed by a consistent 22-24% proportional reduction in cardiovascular events per millimole per litre reduction in LDL-C during each subsequent year of treatment.33 The magnitude of this effect was independent of baseline LDL-C level, similar among persons with and without pre-existing cardiovascular disease at baseline, and remarkably consistent in all subgroups studied.32 , 33 This meta-analysis therefore provides powerful evidence that reducing plasma LDL-C levels by inhibiting HMG-CoA reductase with a statin leads to dose-dependent reduction in the risk of major cardiovascular events that is proportional to the absolute magnitude of the reduction in LDL-C (Figure 2).
Notably, in the statin trials, the absolute yearly event rate observed in each randomized treatment arm was strongly and linearly associated with the absolute achieved LDL-C level (Figure 5A).34 In these studies, LDL-C and apoB concentrations had very similar effects on the risk of cardiovascular events per millimole per litre, thus confirming that LDL-C is a satisfactory surrogate for LDL particle number under most circumstances. Furthermore, intravascular ultrasound studies of coronary atherosclerosis involving statin-treated patients have consistently demonstrated that progression of coronary atherosclerotic plaque volume can be substantially arrested at achieved LDL-C levels of ∼1.8 mmol/L (70 mg/dL) (Figure 5B).35 , 36
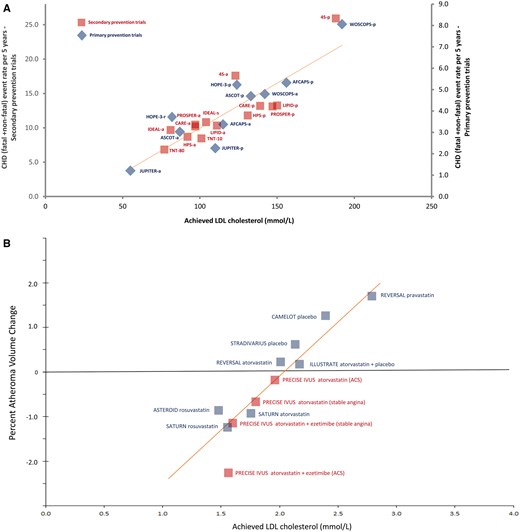
Linear association between achieved low-density lipoprotein cholesterol (LDL-C) level and absolute coronary heart disease (CHD) event rate or progression of atherosclerosis. Panel A shows absolute cardiovascular event rates in randomized statin trials and Panel B shows progression of atherosclerosis as measured by intravascular ultrasound. In Panel A, achieved LDL-C in primary prevention trials and secondary prevention trials in stable CHD patients was related to the end point of CHD events (fatal plus non-fatal myocardial infarction, sudden CHD death) proportioned to 5 years assuming linear rates with time. Trendlines for primary and secondary prevention associations are virtually superimposable. Key: p, placebo; a, active treatment arm, except for IDEAL, where s, simvastatin and a, atorvastatin; and HOPE-3, where r, rosuvastatin; and TNT where reference is made to atorvastatin 10 and 80 mg dose. Trial acronyms: AFCAPS, Air Force Coronary Atherosclerosis Prevention Study; ASCOT, Anglo Scandinavian Cardiac Outcomes Trial; ASTEROID, A Study To Evaluate the Effect of Rosuvastatin on Intravascular Ultrasound-Derived Coronary Atheroma Burden; CARE, Cholesterol and Recurrent Events; CAMELOT, Comparison of Amlodipine vs. Enalapril to Limit Occurrence of Thrombosis; HOPE, Heart Outcomes Prevention Evaluation Study; HPS, Heart Protection Study; IDEAL, Incremental Decrease in End Points Through Aggressive Lipid Lowering; ILLUSTRATE, Investigation of Lipid Level Management Using Coronary Ultrasound To Assess Reduction of Atherosclerosis by CETP Inhibition and HDL Elevation; JUPITER, Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin trial; LIPID, Long-Term Intervention with Pravastatin in Ischemic Disease; PRECISE IVUS, Plaque REgression with Cholesterol absorption Inhibitor or Synthesis inhibitor Evaluated by IntraVascular UltraSound; PROSPER, Pravastatin in elderly individuals at risk of vascular disease; REVERSAL, Reversal of Atherosclerosis With Aggressive Lipid Lowering; 4S Scandinavian Simvastatin Survival Study; SATURN, Study of Coronary Atheroma by Intravascular Ultrasound: Effect of Rosuvastatin vs. Atorvastatin; STRADIVARIUS, Strategy To Reduce Atherosclerosis Development InVolving Administration of Rimonabant—the Intravascular Ultrasound Study; TNT, Treating to New Targets; WOSCOPS, West Of Scotland Coronary Prevention Study.
Importantly, other therapies that reduce LDL-C without inhibiting HMG-CoA reductase have also been shown to reduce the risk of cardiovascular events in large randomized cardiovascular outcome trials. Ezetimibe inhibits intestinal absorption of cholesterol by binding to the NPC1L1 transporter protein.37 Among the 18 144 persons with acute coronary syndrome enrolled in the IMPROVE-IT trial, treatment with ezetimibe plus a statin vs. statin monotherapy reduced LDL-C levels by 0.40 mmol/L over the trial duration, and was associated with a 6.5% proportional reduction in major cardiovascular events.38 Supportive evidence is available from the SHARP trial in 9270 persons with chronic kidney disease, which compared treatment with the combination of simvastatin and ezetimibe with placebo. Combination treatment reduced LDL-C levels by 0.85 mmol/L (33 mg/dL) and reduced the incidence of major cardiovascular events by 17% relative to placebo.39 In both trials, the magnitude of the observed proportional risk reduction was consistent with the clinical benefit that would be expected from similar absolute reductions in LDL-C during treatment with statin monotherapy. These data suggest that statins and ezetimibe have therapeutically equivalent effects on the risk of cardiovascular events per unit lower LDL-C (Figure 3B). These findings also agree closely with the naturally randomized genetic data that have demonstrated that variants in the HMGCR and NPC1L1 genes have biologically equivalent effects on the risk of cardiovascular disease per unit lower LDL-C31 , 40 (Figure 3A).
In addition, among 27,564 participants with cardiovascular disease enrolled in the recently reported FOURIER trial, treatment with the PCSK9 monoclonal antibody evolocumab added to a statin reduced LDL-C by 1.4 mmol/L (53.4 mg/dl) to a mean LDL-C level of 0.78 mmol/L (30 mg/dl) and reduced the incidence of cardiovascular death, MI or stroke by 20% during a median of 2.2 years of follow-up.41 , 42 When analysed by each year of treatment, evolocumab had nearly identical effects on the risk of multiple different cardiovascular endpoints per millimole per litre reduction in LDL-C compared to treatment with a statin as reported by the Cholesterol Treatment Trialists Collaboration.31 The results of this trial are remarkably consistent with the results of a recent Mendelian randomization study that showed that variants in the PCSK9 and HMGCR genes have nearly identical effects on the risk of cardiovascular events per unit lower LDL-C, and therefore accurately predicted that PCSK9 inhibitors would reduce the risk of cardiovascular events by approximately the same amount as statins per unit reduction in LDL-C.43 The results of the FOURIER trial are also consistent with the results of the recent GLAGOV trial in which treatment with evolocumab added to a statin reduced LDL-C levels by 1.45 mmol/L and induced plaque regression that appeared to be directly proportional to the achieved absolute LDL-C level, even at LDL-C levels as low as 0.95 mmol/L (36.6 mg/dl).44 Taken together, the results of these studies strongly suggest that PCSK9 inhibitors and statins have biologically equivalent effects on the risk of cardiovascular events per unit change in LDL-C.
Furthermore, in the Lipid Research Clinics Coronary Primary Prevention Trial, treatment with the bile acid sequestrant cholestyramine reduced LDL-C by 0.7 mmol/L and reduced the relative risk of cardiovascular death or MI by 19%.45 Similarly, in the Program on Surgical Control of Hyperlipidemia (POSCH) study, ileal bypass surgery reduced LDL-C by 1.85 mmol/L and reduced the relative risk of cardiovascular death or MI by 35%.46 In both trials, the observed risk reduction was consistent with the proportional risk reduction per unit lower LDL-C observed during treatment with a statin or with combined statin and ezetimibe therapy (Figure 2). Indeed, in a recent meta-analysis of 49 studies including 312 175 participants and 39 645 major vascular events that compared the effects of 9 different types of lipid-lowering therapies, nearly all of these therapeutic approaches to lowering LDL-C were associated with a consistent 20–25% proportional reduction in vascular events per millimole per litre reduction in LDL-C.47 These findings are in line with those from the Mendelian randomization studies and demonstrate that the clinical benefit of lowering LDL-C appears to be independent of the mechanism by which LDL-C is lowered (Figure 3).
The one notable exception to this finding is the effect of the cholesteryl ester transfer protein (CETP) inhibitors. In the recently reported ACCELERATE Trial, the CETP inhibitor evacetrapib plus a statin reduced LDL-C by 0.75 mmol/L compared with statin monotherapy but did not reduce the risk of cardiovascular events.48 To date, we lack a mechanistic explanation for this finding, although deleterious effects on CHD due to a 1.5 mmHg mean increase in systolic blood pressure and/or possible dysfunctional HDL phenotypes on evacetrapib may constitute contributory factors. In addition, the LDL particle-lowering effect of CETP inhibitors appears to become attenuated when a CETP inhibitor is added to a statin, thus leading to discordance between the measured LDL-C and apoB reductions.48 The ongoing 30 000-person REVEAL trial with the CETP inhibitor anacetrapib will provide more definitive evidence of the effect of adding a CETP inhibitor to a statin on the risk of cardiovascular events.
Criteria for causality
A critical appraisal of the evidence discussed in this review demonstrates that the association between plasma LDL-C concentration and the risk of ASCVD satisfies all the criteria for causality (Table 1).49 Indeed, the prospective epidemiologic studies, Mendelian randomization studies, and randomized intervention trials all demonstrate a remarkably consistent dose-dependent log-linear association between the absolute magnitude of exposure to LDL-C and the risk of ASCVD, and together demonstrate that the effect of LDL-C on the risk of ASCVD increases with increasing duration of exposure (Figure 2). This concordance between multiple lines of evidence, most notably the remarkable concordance between the unbiased naturally randomized genetic data and the results of numerous randomized intervention trials using multiple different agents to reduce LDL-C, provides overwhelming clinical evidence that LDL causes ASCVD and that lowering LDL reduces the risk of cardiovascular events.
Evidence for the cumulative effect of exposure to LDL on ASCVD
Mendelian randomization studies suggest that long-term exposure to lower LDL-C is associated with up to a three-fold greater proportional reduction in the risk of cardiovascular disease per unit reduction in LDL-C, when compared with shorter term treatment with a statin started later in life after atherosclerosis has developed.28 This finding suggests that the causal effect of LDL on the risk of ASCVD is determined by both the absolute magnitude and the cumulative duration of exposure to LDL-C. This finding is consistent with increasing benefit observed over time in the long-term follow-up of the WOSCOPS trial.50
Because the effect of LDL-C on the risk of ASCVD appears to be both causal and cumulative over time, lowering the plasma LDL-C level earlier than is currently recommended, may result in proportionally greater reductions in the lifetime risk of ASCVD than which is estimated in short-term randomized trials. Integrating the available evidence from Mendelian randomization studies and randomized trials suggests that each millimole per litre reduction in LDL-C reduces the relative risk of ASCVD events by ∼10% during the first year of treatment, by ∼16% after 2 years of treatment and by ∼20% after 3 years of treatment, presumably related to the stabilization of any existing underlying plaque burden.33 Each subsequent year of treatment after the third might then be expected to result in a further 1.5% proportional reduction in ASCVD events per millimole per litre per year (Tables 2–4). Therefore, 5 years of treatment with a lipid-lowering agent should reduce the relative risk of ASCVD by ∼20–25% per millimole per litre reduction in LDL-C, while 40 years of treatment (or approximately equivalently 40 years of exposure to lower LDL-C) would be expected to reduce ASCVD events by ∼50–55% per millimole per litre lower LDL-C (Figure 2).
Expected proportional risk reduction based on pre-treatment low-density lipoprotein cholesterol (LDL-C), absolute magnitude of LDL-C reduction, and total duration of therapy
| Baseline | Absolute reduction | Duration of treatment exposure | Guideline | ||||
|---|---|---|---|---|---|---|---|
| LDL-C (mmol/L) | LDL-C (mmol/L) | [expected proportional risk reduction (%)] | recommended treatment51 , 52 | ||||
| 5 years | 10 years | 20 years | 30 years | 40 years | |||
| 7 | 3.5 | 58 | 68 | 81 | 89 | 93 | Yes (due to markedly elevated LDL-C) |
| 7 | 3.0 | 53 | 62 | 76 | 85 | 90 | |
| 7 | 2.5 | 46 | 56 | 70 | 79 | 86 | |
| 7 | 2.0 | 39 | 48 | 61 | 71 | 79 | |
| 7 | 1.5 | 31 | 39 | 51 | 61 | 69 | |
| 5 | 2.5 | 46 | 56 | 70 | 79 | 86 | Yes (due to markedly elevated LDL-C) |
| 5 | 2.0 | 39 | 48 | 61 | 71 | 79 | |
| 5 | 1.5 | 31 | 39 | 51 | 61 | 69 | |
| 5 | 1.0 | 22 | 28 | 38 | 46 | 54 | |
| 3 | 1.5 | 31 | 39 | 51 | 61 | 69 | Depends on risk of ASCVD |
| 3 | 1.0 | 22 | 28 | 38 | 46 | 54 | |
| 3 | 0.5 | 12 | 15 | 21 | 27 | 32 | |
| 2 | 1.0 | 22 | 28 | 38 | 46 | 54 | Depends on risk of ASCVD |
| 2 | 0.5 | 12 | 15 | 21 | 27 | 32 | |
| Baseline | Absolute reduction | Duration of treatment exposure | Guideline | ||||
|---|---|---|---|---|---|---|---|
| LDL-C (mmol/L) | LDL-C (mmol/L) | [expected proportional risk reduction (%)] | recommended treatment51 , 52 | ||||
| 5 years | 10 years | 20 years | 30 years | 40 years | |||
| 7 | 3.5 | 58 | 68 | 81 | 89 | 93 | Yes (due to markedly elevated LDL-C) |
| 7 | 3.0 | 53 | 62 | 76 | 85 | 90 | |
| 7 | 2.5 | 46 | 56 | 70 | 79 | 86 | |
| 7 | 2.0 | 39 | 48 | 61 | 71 | 79 | |
| 7 | 1.5 | 31 | 39 | 51 | 61 | 69 | |
| 5 | 2.5 | 46 | 56 | 70 | 79 | 86 | Yes (due to markedly elevated LDL-C) |
| 5 | 2.0 | 39 | 48 | 61 | 71 | 79 | |
| 5 | 1.5 | 31 | 39 | 51 | 61 | 69 | |
| 5 | 1.0 | 22 | 28 | 38 | 46 | 54 | |
| 3 | 1.5 | 31 | 39 | 51 | 61 | 69 | Depends on risk of ASCVD |
| 3 | 1.0 | 22 | 28 | 38 | 46 | 54 | |
| 3 | 0.5 | 12 | 15 | 21 | 27 | 32 | |
| 2 | 1.0 | 22 | 28 | 38 | 46 | 54 | Depends on risk of ASCVD |
| 2 | 0.5 | 12 | 15 | 21 | 27 | 32 | |
Recommendations for treatment: expected clinical benefit per unit reduction in LDL-C expressed as the expected proportional risk reduction (%). The proportional reduction in short-term risk is based on an expected 22% reduction in risk per millimole per litre reduction in LDL-C over 5 years as estimated from randomized trials and is calculated as [(1−0.78 (absolute reduction in LDL-C in mmol/L)) × 100]. The proportional reduction in long-term risk is based on an expected 54% reduction in risk per millimole per litre reduction in LDL-C over 40 years of exposure as estimated from Mendelian randomization studies and is calculated as [(1−0.46 (absolute reduction in LDL-C in mmol/L)) × 100]. The expected proportional risk reduction per mmol/L reduction in LDL-C for any specific treatment duration is calculated as: [(1−e(−0.249 + (number of years of treatment − 5) × (−0.0152))) × 100].
ASCVD, atherosclerotic cardiovascular disease.
Expected proportional risk reduction based on pre-treatment low-density lipoprotein cholesterol (LDL-C), absolute magnitude of LDL-C reduction, and total duration of therapy
| Baseline | Absolute reduction | Duration of treatment exposure | Guideline | ||||
|---|---|---|---|---|---|---|---|
| LDL-C (mmol/L) | LDL-C (mmol/L) | [expected proportional risk reduction (%)] | recommended treatment51 , 52 | ||||
| 5 years | 10 years | 20 years | 30 years | 40 years | |||
| 7 | 3.5 | 58 | 68 | 81 | 89 | 93 | Yes (due to markedly elevated LDL-C) |
| 7 | 3.0 | 53 | 62 | 76 | 85 | 90 | |
| 7 | 2.5 | 46 | 56 | 70 | 79 | 86 | |
| 7 | 2.0 | 39 | 48 | 61 | 71 | 79 | |
| 7 | 1.5 | 31 | 39 | 51 | 61 | 69 | |
| 5 | 2.5 | 46 | 56 | 70 | 79 | 86 | Yes (due to markedly elevated LDL-C) |
| 5 | 2.0 | 39 | 48 | 61 | 71 | 79 | |
| 5 | 1.5 | 31 | 39 | 51 | 61 | 69 | |
| 5 | 1.0 | 22 | 28 | 38 | 46 | 54 | |
| 3 | 1.5 | 31 | 39 | 51 | 61 | 69 | Depends on risk of ASCVD |
| 3 | 1.0 | 22 | 28 | 38 | 46 | 54 | |
| 3 | 0.5 | 12 | 15 | 21 | 27 | 32 | |
| 2 | 1.0 | 22 | 28 | 38 | 46 | 54 | Depends on risk of ASCVD |
| 2 | 0.5 | 12 | 15 | 21 | 27 | 32 | |
| Baseline | Absolute reduction | Duration of treatment exposure | Guideline | ||||
|---|---|---|---|---|---|---|---|
| LDL-C (mmol/L) | LDL-C (mmol/L) | [expected proportional risk reduction (%)] | recommended treatment51 , 52 | ||||
| 5 years | 10 years | 20 years | 30 years | 40 years | |||
| 7 | 3.5 | 58 | 68 | 81 | 89 | 93 | Yes (due to markedly elevated LDL-C) |
| 7 | 3.0 | 53 | 62 | 76 | 85 | 90 | |
| 7 | 2.5 | 46 | 56 | 70 | 79 | 86 | |
| 7 | 2.0 | 39 | 48 | 61 | 71 | 79 | |
| 7 | 1.5 | 31 | 39 | 51 | 61 | 69 | |
| 5 | 2.5 | 46 | 56 | 70 | 79 | 86 | Yes (due to markedly elevated LDL-C) |
| 5 | 2.0 | 39 | 48 | 61 | 71 | 79 | |
| 5 | 1.5 | 31 | 39 | 51 | 61 | 69 | |
| 5 | 1.0 | 22 | 28 | 38 | 46 | 54 | |
| 3 | 1.5 | 31 | 39 | 51 | 61 | 69 | Depends on risk of ASCVD |
| 3 | 1.0 | 22 | 28 | 38 | 46 | 54 | |
| 3 | 0.5 | 12 | 15 | 21 | 27 | 32 | |
| 2 | 1.0 | 22 | 28 | 38 | 46 | 54 | Depends on risk of ASCVD |
| 2 | 0.5 | 12 | 15 | 21 | 27 | 32 | |
Recommendations for treatment: expected clinical benefit per unit reduction in LDL-C expressed as the expected proportional risk reduction (%). The proportional reduction in short-term risk is based on an expected 22% reduction in risk per millimole per litre reduction in LDL-C over 5 years as estimated from randomized trials and is calculated as [(1−0.78 (absolute reduction in LDL-C in mmol/L)) × 100]. The proportional reduction in long-term risk is based on an expected 54% reduction in risk per millimole per litre reduction in LDL-C over 40 years of exposure as estimated from Mendelian randomization studies and is calculated as [(1−0.46 (absolute reduction in LDL-C in mmol/L)) × 100]. The expected proportional risk reduction per mmol/L reduction in LDL-C for any specific treatment duration is calculated as: [(1−e(−0.249 + (number of years of treatment − 5) × (−0.0152))) × 100].
ASCVD, atherosclerotic cardiovascular disease.
Expected short-term absolute risk reduction and number needed to treat based on baseline absolute risk of cardiovascular disease and pre-treatment low-density lipoprotein cholesterol (LDL-C) with 5 years of treatment to lower LDL-C
| 10-year absolute risk of CVD (%) | Baseline LDL-C mmol/L (mg/dL) | LDL-C after 50% reduction mmol/L (mg/dL) | Proportional risk reduction (%) | 10-year Absolute risk (%) after 50% LDL-C reduction | ARR (%) | NNT | Guideline recommended treatment51 , 52 |
|---|---|---|---|---|---|---|---|
| 20 | 5 (200) | 2.5 (100) | 42.8 | 11.4 | 8.6 | 11.7 | Yes (based on 10-year risk of ASCVD) |
| 20 | 4 (160) | 2.0 (80) | 36 | 12.8 | 7.2 | 13.9 | |
| 20 | 3 (120) | 1.5 (60) | 28.4 | 14.3 | 5.7 | 17.6 | |
| 20 | 2 (80) | 1.0 (40) | 20 | 16 | 4 | 25 | |
| 15 | 5 (200) | 2.5 (100) | 42.8 | 8.6 | 6.4 | 15.6 | Yes (based on 10-year risk of ASCVD) |
| 15 | 4 (160) | 2.0 (80) | 36 | 9.6 | 5.4 | 18.5 | |
| 15 | 3 (120) | 1.5 (60) | 28.4 | 10.7 | 4.3 | 23.4 | |
| 15 | 2 (80) | 1.0 (40) | 20 | 12 | 3 | 33.3 | |
| 10 | 5 (200) | 2.5 (100) | 42.8 | 5.7 | 4.3 | 23.4 | Yes (based on 10-year risk of ASCVD) |
| 10 | 4 (160) | 2.0 (80) | 36 | 6.4 | 3.6 | 27.8 | |
| 10 | 3 (120) | 1.5 (60) | 28.4 | 7.2 | 2.8 | 35.2 | |
| 10 | 2 (80) | 1.0 (40) | 20 | 8 | 2 | 50 | |
| 5 | 5 (200) | 2.5 (100) | 42.8 | 2.9 | 2.1 | 46.8 | No (based on 10-year risk of ASCVD) |
| 5 | 4 (160) | 2.0 (80) | 36 | 3.2 | 1.8 | 55.6 | |
| 5 | 3 (120) | 1.5 (60) | 28.4 | 3.6 | 1.4 | 70.3 | |
| 5 | 2 (80) | 1.0 (40) | 20 | 4 | 1 | 100 |
| 10-year absolute risk of CVD (%) | Baseline LDL-C mmol/L (mg/dL) | LDL-C after 50% reduction mmol/L (mg/dL) | Proportional risk reduction (%) | 10-year Absolute risk (%) after 50% LDL-C reduction | ARR (%) | NNT | Guideline recommended treatment51 , 52 |
|---|---|---|---|---|---|---|---|
| 20 | 5 (200) | 2.5 (100) | 42.8 | 11.4 | 8.6 | 11.7 | Yes (based on 10-year risk of ASCVD) |
| 20 | 4 (160) | 2.0 (80) | 36 | 12.8 | 7.2 | 13.9 | |
| 20 | 3 (120) | 1.5 (60) | 28.4 | 14.3 | 5.7 | 17.6 | |
| 20 | 2 (80) | 1.0 (40) | 20 | 16 | 4 | 25 | |
| 15 | 5 (200) | 2.5 (100) | 42.8 | 8.6 | 6.4 | 15.6 | Yes (based on 10-year risk of ASCVD) |
| 15 | 4 (160) | 2.0 (80) | 36 | 9.6 | 5.4 | 18.5 | |
| 15 | 3 (120) | 1.5 (60) | 28.4 | 10.7 | 4.3 | 23.4 | |
| 15 | 2 (80) | 1.0 (40) | 20 | 12 | 3 | 33.3 | |
| 10 | 5 (200) | 2.5 (100) | 42.8 | 5.7 | 4.3 | 23.4 | Yes (based on 10-year risk of ASCVD) |
| 10 | 4 (160) | 2.0 (80) | 36 | 6.4 | 3.6 | 27.8 | |
| 10 | 3 (120) | 1.5 (60) | 28.4 | 7.2 | 2.8 | 35.2 | |
| 10 | 2 (80) | 1.0 (40) | 20 | 8 | 2 | 50 | |
| 5 | 5 (200) | 2.5 (100) | 42.8 | 2.9 | 2.1 | 46.8 | No (based on 10-year risk of ASCVD) |
| 5 | 4 (160) | 2.0 (80) | 36 | 3.2 | 1.8 | 55.6 | |
| 5 | 3 (120) | 1.5 (60) | 28.4 | 3.6 | 1.4 | 70.3 | |
| 5 | 2 (80) | 1.0 (40) | 20 | 4 | 1 | 100 |
Recommendations for treatment: expected clinical benefit per unit reduction in LDL-C. The proportional reduction in short-term risk is based on an expected 22% reduction in risk per millimole per litre reduction in LDL-C over 5 years as estimated from randomized trials and is calculated as [(1−0.78 (absolute reduction in LDL-C in mmol/L))×100]. The absolute risk reduction is calculated as [ARR = Column 1 - Column 5], and the number needed to treat is calculated as [(1 ÷ ARR) × 100].
CVD, cardiovascular disease; ASCVD, atherosclerotic cardiovascular disease; ARR, absolute risk reduction; NNT, number needed to treat.
Expected short-term absolute risk reduction and number needed to treat based on baseline absolute risk of cardiovascular disease and pre-treatment low-density lipoprotein cholesterol (LDL-C) with 5 years of treatment to lower LDL-C
| 10-year absolute risk of CVD (%) | Baseline LDL-C mmol/L (mg/dL) | LDL-C after 50% reduction mmol/L (mg/dL) | Proportional risk reduction (%) | 10-year Absolute risk (%) after 50% LDL-C reduction | ARR (%) | NNT | Guideline recommended treatment51 , 52 |
|---|---|---|---|---|---|---|---|
| 20 | 5 (200) | 2.5 (100) | 42.8 | 11.4 | 8.6 | 11.7 | Yes (based on 10-year risk of ASCVD) |
| 20 | 4 (160) | 2.0 (80) | 36 | 12.8 | 7.2 | 13.9 | |
| 20 | 3 (120) | 1.5 (60) | 28.4 | 14.3 | 5.7 | 17.6 | |
| 20 | 2 (80) | 1.0 (40) | 20 | 16 | 4 | 25 | |
| 15 | 5 (200) | 2.5 (100) | 42.8 | 8.6 | 6.4 | 15.6 | Yes (based on 10-year risk of ASCVD) |
| 15 | 4 (160) | 2.0 (80) | 36 | 9.6 | 5.4 | 18.5 | |
| 15 | 3 (120) | 1.5 (60) | 28.4 | 10.7 | 4.3 | 23.4 | |
| 15 | 2 (80) | 1.0 (40) | 20 | 12 | 3 | 33.3 | |
| 10 | 5 (200) | 2.5 (100) | 42.8 | 5.7 | 4.3 | 23.4 | Yes (based on 10-year risk of ASCVD) |
| 10 | 4 (160) | 2.0 (80) | 36 | 6.4 | 3.6 | 27.8 | |
| 10 | 3 (120) | 1.5 (60) | 28.4 | 7.2 | 2.8 | 35.2 | |
| 10 | 2 (80) | 1.0 (40) | 20 | 8 | 2 | 50 | |
| 5 | 5 (200) | 2.5 (100) | 42.8 | 2.9 | 2.1 | 46.8 | No (based on 10-year risk of ASCVD) |
| 5 | 4 (160) | 2.0 (80) | 36 | 3.2 | 1.8 | 55.6 | |
| 5 | 3 (120) | 1.5 (60) | 28.4 | 3.6 | 1.4 | 70.3 | |
| 5 | 2 (80) | 1.0 (40) | 20 | 4 | 1 | 100 |
| 10-year absolute risk of CVD (%) | Baseline LDL-C mmol/L (mg/dL) | LDL-C after 50% reduction mmol/L (mg/dL) | Proportional risk reduction (%) | 10-year Absolute risk (%) after 50% LDL-C reduction | ARR (%) | NNT | Guideline recommended treatment51 , 52 |
|---|---|---|---|---|---|---|---|
| 20 | 5 (200) | 2.5 (100) | 42.8 | 11.4 | 8.6 | 11.7 | Yes (based on 10-year risk of ASCVD) |
| 20 | 4 (160) | 2.0 (80) | 36 | 12.8 | 7.2 | 13.9 | |
| 20 | 3 (120) | 1.5 (60) | 28.4 | 14.3 | 5.7 | 17.6 | |
| 20 | 2 (80) | 1.0 (40) | 20 | 16 | 4 | 25 | |
| 15 | 5 (200) | 2.5 (100) | 42.8 | 8.6 | 6.4 | 15.6 | Yes (based on 10-year risk of ASCVD) |
| 15 | 4 (160) | 2.0 (80) | 36 | 9.6 | 5.4 | 18.5 | |
| 15 | 3 (120) | 1.5 (60) | 28.4 | 10.7 | 4.3 | 23.4 | |
| 15 | 2 (80) | 1.0 (40) | 20 | 12 | 3 | 33.3 | |
| 10 | 5 (200) | 2.5 (100) | 42.8 | 5.7 | 4.3 | 23.4 | Yes (based on 10-year risk of ASCVD) |
| 10 | 4 (160) | 2.0 (80) | 36 | 6.4 | 3.6 | 27.8 | |
| 10 | 3 (120) | 1.5 (60) | 28.4 | 7.2 | 2.8 | 35.2 | |
| 10 | 2 (80) | 1.0 (40) | 20 | 8 | 2 | 50 | |
| 5 | 5 (200) | 2.5 (100) | 42.8 | 2.9 | 2.1 | 46.8 | No (based on 10-year risk of ASCVD) |
| 5 | 4 (160) | 2.0 (80) | 36 | 3.2 | 1.8 | 55.6 | |
| 5 | 3 (120) | 1.5 (60) | 28.4 | 3.6 | 1.4 | 70.3 | |
| 5 | 2 (80) | 1.0 (40) | 20 | 4 | 1 | 100 |
Recommendations for treatment: expected clinical benefit per unit reduction in LDL-C. The proportional reduction in short-term risk is based on an expected 22% reduction in risk per millimole per litre reduction in LDL-C over 5 years as estimated from randomized trials and is calculated as [(1−0.78 (absolute reduction in LDL-C in mmol/L))×100]. The absolute risk reduction is calculated as [ARR = Column 1 - Column 5], and the number needed to treat is calculated as [(1 ÷ ARR) × 100].
CVD, cardiovascular disease; ASCVD, atherosclerotic cardiovascular disease; ARR, absolute risk reduction; NNT, number needed to treat.
Expected long-term absolute risk reduction and number needed to treat based on baseline absolute risk of cardiovascular disease and pre-treatment low-density lipoprotein cholesterol (LDL-C) with 30 years of treatment (or exposure) to lower LDL-C
| 30-year Absolute risk of CVD (%) | Baseline LDL-C mmol/L (mg/dL) | LDL-C after reduction mmol/L (mg/dL) | Proportional risk reduction (%) | 30-year absolute risk (%) after 50% LDL-C reduction | ARR(%) | NNT | Guideline recommended treatment51 , 52 |
|---|---|---|---|---|---|---|---|
| 60 | 5 (200) | 2.5 (100) | 82.3 | 10.6 | 49.4 | 2 | Individualized (based on lifetime risk of ASCVD) |
| 60 | 4 (160) | 2.0 (80) | 75 | 15 | 45 | 2.2 | |
| 60 | 3 (120) | 1.5 (60) | 64.6 | 21.2 | 38.9 | 2.6 | |
| 60 | 2 (80) | 1.0 (40) | 50 | 30 | 30 | 3.3 | |
| 45 | 5 (200) | 2.5 (100) | 82.3 | 8 | 37 | 2.7 | Individualized (based on lifetime risk of ASCVD) |
| 45 | 4 (160) | 2.0 (80) | 75 | 11.3 | 33.8 | 3 | |
| 45 | 3 (120) | 1.5 (60) | 64.6 | 15.9 | 29.1 | 3.4 | |
| 45 | 2 (80) | 1.0 (40) | 50 | 22.5 | 22.5 | 4.4 | |
| 30 | 5 (200) | 2.5 (100) | 82.3 | 5.3 | 24.7 | 4 | Individualized (based on lifetime risk of ASCVD) |
| 30 | 4 (160) | 2.0 (80) | 75 | 7.5 | 22.5 | 4.4 | |
| 30 | 3 (120) | 1.5 (60) | 64.6 | 10.6 | 19.4 | 5.2 | |
| 30 | 2 (80) | 1.0 (40) | 50 | 15 | 15 | 6.7 | |
| 15 | 5 (200) | 2.5 (100) | 82.3 | 2.7 | 12.3 | 8.1 | Individualized (based on lifetime risk of ASCVD) |
| 15 | 4 (160) | 2.0 (80) | 75 | 3.8 | 11.3 | 8.9 | |
| 15 | 3 (120) | 1.5 (60) | 64.6 | 5.3 | 9.7 | 10.3 | |
| 15 | 2 (80) | 1.0 (40) | 50 | 7.5 | 7.5 | 13.3 |
| 30-year Absolute risk of CVD (%) | Baseline LDL-C mmol/L (mg/dL) | LDL-C after reduction mmol/L (mg/dL) | Proportional risk reduction (%) | 30-year absolute risk (%) after 50% LDL-C reduction | ARR(%) | NNT | Guideline recommended treatment51 , 52 |
|---|---|---|---|---|---|---|---|
| 60 | 5 (200) | 2.5 (100) | 82.3 | 10.6 | 49.4 | 2 | Individualized (based on lifetime risk of ASCVD) |
| 60 | 4 (160) | 2.0 (80) | 75 | 15 | 45 | 2.2 | |
| 60 | 3 (120) | 1.5 (60) | 64.6 | 21.2 | 38.9 | 2.6 | |
| 60 | 2 (80) | 1.0 (40) | 50 | 30 | 30 | 3.3 | |
| 45 | 5 (200) | 2.5 (100) | 82.3 | 8 | 37 | 2.7 | Individualized (based on lifetime risk of ASCVD) |
| 45 | 4 (160) | 2.0 (80) | 75 | 11.3 | 33.8 | 3 | |
| 45 | 3 (120) | 1.5 (60) | 64.6 | 15.9 | 29.1 | 3.4 | |
| 45 | 2 (80) | 1.0 (40) | 50 | 22.5 | 22.5 | 4.4 | |
| 30 | 5 (200) | 2.5 (100) | 82.3 | 5.3 | 24.7 | 4 | Individualized (based on lifetime risk of ASCVD) |
| 30 | 4 (160) | 2.0 (80) | 75 | 7.5 | 22.5 | 4.4 | |
| 30 | 3 (120) | 1.5 (60) | 64.6 | 10.6 | 19.4 | 5.2 | |
| 30 | 2 (80) | 1.0 (40) | 50 | 15 | 15 | 6.7 | |
| 15 | 5 (200) | 2.5 (100) | 82.3 | 2.7 | 12.3 | 8.1 | Individualized (based on lifetime risk of ASCVD) |
| 15 | 4 (160) | 2.0 (80) | 75 | 3.8 | 11.3 | 8.9 | |
| 15 | 3 (120) | 1.5 (60) | 64.6 | 5.3 | 9.7 | 10.3 | |
| 15 | 2 (80) | 1.0 (40) | 50 | 7.5 | 7.5 | 13.3 |
Recommendations for treatment: expected clinical benefit per unit reduction in LDL-C. The proportional reduction in long-term risk is based on an expected 54% reduction in risk per millimole per litre reduction in LDL-C over 40 years of exposure as estimated from Mendelian randomization studies and is calculated as [(1−0.46 (absolute reduction in LDL-C in mmol/L))×100]. The absolute risk reduction is calculated as [ARR = Column 1 − Column 5], and the number needed to treat is calculated as [(1 ÷ ARR) × 100].
CVD, cardiovascular disease; ASCVD, atherosclerotic cardiovascular disease; ARR, absolute risk reduction; NNT, number needed to treat.
Expected long-term absolute risk reduction and number needed to treat based on baseline absolute risk of cardiovascular disease and pre-treatment low-density lipoprotein cholesterol (LDL-C) with 30 years of treatment (or exposure) to lower LDL-C
| 30-year Absolute risk of CVD (%) | Baseline LDL-C mmol/L (mg/dL) | LDL-C after reduction mmol/L (mg/dL) | Proportional risk reduction (%) | 30-year absolute risk (%) after 50% LDL-C reduction | ARR(%) | NNT | Guideline recommended treatment51 , 52 |
|---|---|---|---|---|---|---|---|
| 60 | 5 (200) | 2.5 (100) | 82.3 | 10.6 | 49.4 | 2 | Individualized (based on lifetime risk of ASCVD) |
| 60 | 4 (160) | 2.0 (80) | 75 | 15 | 45 | 2.2 | |
| 60 | 3 (120) | 1.5 (60) | 64.6 | 21.2 | 38.9 | 2.6 | |
| 60 | 2 (80) | 1.0 (40) | 50 | 30 | 30 | 3.3 | |
| 45 | 5 (200) | 2.5 (100) | 82.3 | 8 | 37 | 2.7 | Individualized (based on lifetime risk of ASCVD) |
| 45 | 4 (160) | 2.0 (80) | 75 | 11.3 | 33.8 | 3 | |
| 45 | 3 (120) | 1.5 (60) | 64.6 | 15.9 | 29.1 | 3.4 | |
| 45 | 2 (80) | 1.0 (40) | 50 | 22.5 | 22.5 | 4.4 | |
| 30 | 5 (200) | 2.5 (100) | 82.3 | 5.3 | 24.7 | 4 | Individualized (based on lifetime risk of ASCVD) |
| 30 | 4 (160) | 2.0 (80) | 75 | 7.5 | 22.5 | 4.4 | |
| 30 | 3 (120) | 1.5 (60) | 64.6 | 10.6 | 19.4 | 5.2 | |
| 30 | 2 (80) | 1.0 (40) | 50 | 15 | 15 | 6.7 | |
| 15 | 5 (200) | 2.5 (100) | 82.3 | 2.7 | 12.3 | 8.1 | Individualized (based on lifetime risk of ASCVD) |
| 15 | 4 (160) | 2.0 (80) | 75 | 3.8 | 11.3 | 8.9 | |
| 15 | 3 (120) | 1.5 (60) | 64.6 | 5.3 | 9.7 | 10.3 | |
| 15 | 2 (80) | 1.0 (40) | 50 | 7.5 | 7.5 | 13.3 |
| 30-year Absolute risk of CVD (%) | Baseline LDL-C mmol/L (mg/dL) | LDL-C after reduction mmol/L (mg/dL) | Proportional risk reduction (%) | 30-year absolute risk (%) after 50% LDL-C reduction | ARR(%) | NNT | Guideline recommended treatment51 , 52 |
|---|---|---|---|---|---|---|---|
| 60 | 5 (200) | 2.5 (100) | 82.3 | 10.6 | 49.4 | 2 | Individualized (based on lifetime risk of ASCVD) |
| 60 | 4 (160) | 2.0 (80) | 75 | 15 | 45 | 2.2 | |
| 60 | 3 (120) | 1.5 (60) | 64.6 | 21.2 | 38.9 | 2.6 | |
| 60 | 2 (80) | 1.0 (40) | 50 | 30 | 30 | 3.3 | |
| 45 | 5 (200) | 2.5 (100) | 82.3 | 8 | 37 | 2.7 | Individualized (based on lifetime risk of ASCVD) |
| 45 | 4 (160) | 2.0 (80) | 75 | 11.3 | 33.8 | 3 | |
| 45 | 3 (120) | 1.5 (60) | 64.6 | 15.9 | 29.1 | 3.4 | |
| 45 | 2 (80) | 1.0 (40) | 50 | 22.5 | 22.5 | 4.4 | |
| 30 | 5 (200) | 2.5 (100) | 82.3 | 5.3 | 24.7 | 4 | Individualized (based on lifetime risk of ASCVD) |
| 30 | 4 (160) | 2.0 (80) | 75 | 7.5 | 22.5 | 4.4 | |
| 30 | 3 (120) | 1.5 (60) | 64.6 | 10.6 | 19.4 | 5.2 | |
| 30 | 2 (80) | 1.0 (40) | 50 | 15 | 15 | 6.7 | |
| 15 | 5 (200) | 2.5 (100) | 82.3 | 2.7 | 12.3 | 8.1 | Individualized (based on lifetime risk of ASCVD) |
| 15 | 4 (160) | 2.0 (80) | 75 | 3.8 | 11.3 | 8.9 | |
| 15 | 3 (120) | 1.5 (60) | 64.6 | 5.3 | 9.7 | 10.3 | |
| 15 | 2 (80) | 1.0 (40) | 50 | 7.5 | 7.5 | 13.3 |
Recommendations for treatment: expected clinical benefit per unit reduction in LDL-C. The proportional reduction in long-term risk is based on an expected 54% reduction in risk per millimole per litre reduction in LDL-C over 40 years of exposure as estimated from Mendelian randomization studies and is calculated as [(1−0.46 (absolute reduction in LDL-C in mmol/L))×100]. The absolute risk reduction is calculated as [ARR = Column 1 − Column 5], and the number needed to treat is calculated as [(1 ÷ ARR) × 100].
CVD, cardiovascular disease; ASCVD, atherosclerotic cardiovascular disease; ARR, absolute risk reduction; NNT, number needed to treat.
Low-density lipoprotein (LDL) as a causal factor for atherosclerotic cardiovascular disease: key implications
|
|
Low-density lipoprotein (LDL) as a causal factor for atherosclerotic cardiovascular disease: key implications
|
|
Recommendations for treatment
The evidence clearly indicates that reducing LDL-C by increasing the numbers of hepatic LDL receptors can reduce ASCVD (Figure 4). Because reducing LDL-C should have a constant effect on the risk of ASCVD per unit absolute reduction in LDL-C, and because LDL-C has both a causal and a cumulative effect on the risk of ASCVD, the proportional reduction in risk that a person can expect from reducing LDL-C will depend on their baseline LDL-C level, the absolute magnitude of the reduction in LDL-C, and the duration of therapy. Similarly, the absolute risk reduction that a person can expect from reducing LDL-C will depend on their baseline risk of ASCVD, baseline LDL-C level, the absolute magnitude of the reduction in LDL-C, and the duration of LDL-C-lowering therapy.
Tables 2–4 provide estimates of the potential clinical benefit that can be achieved by lowering plasma LDL-C concentration based on a person’s baseline risk of ASCVD, baseline LDL-C, and the duration of lipid-lowering therapy, expressed as both expected proportional and absolute risk reductions. Physicians can use this information to discuss with their patients about the potential benefits of lowering LDL-C based on the causal effect of LDL on the risk of ASCVD.
Impact of other exposures on the casual effect of LDL on ASCVD
In addition to LDL, multiple other exposures have been reported to be associated with the risk of ASCVD including elevated systolic blood pressure, diabetes, and tobacco smoking.1 Both the Mendelian randomization studies and the meta-analyses of randomized trials demonstrate that changes in LDL-C have a very consistent proportional effect on the risk of ASCVD across different levels of various risk factors. As a result, a given absolute reduction in LDL-C is associated with the same proportional reduction in the risk of ASCVD, regardless of the presence or absence of other risk factors. However, because persons with more risk factors have a higher absolute rate of ASCVD in comparison with persons with fewer risk factors, the constant proportional reduction in risk per millimole per litre reduction in LDL-C will translate into greater absolute risk reductions for persons with increasing numbers of risk factors or among persons who are at a higher risk of cardiovascular disease more generally.
Finally, an important area for future research is to identify persons who are most likely to benefit from LDL-C-lowering therapies. The probability that LDL will be retained within the arterial intima leading to the development and growth of atherosclerotic plaque increases with increasing concentration of circulating LDL particles.6 Because retention of LDL particles is a probabilistic event, one would expect, therefore, that people with similar LDL-C level will have a distribution of underlying atherosclerotic plaque burdens. Genetic factors may influence whether a person is more or less likely to retain LDL-C within the arterial intima or influence the degree to which retention of LDL particles triggers the inflammatory process or oxidative changes that influence the rate of plaque growth and the propensity for plaque disruption.
Indeed, many of the genetic variants most strongly associated with ASCVD in GWAS are in genes that encode for components of the arterial wall, which may modify susceptibility to retaining LDL or modify responses to the accumulated LDL within the artery wall.27 This hypothesis is consistent with the results of a meta-analysis of statin trials that reported that persons with the highest tertile of a genetic ASCVD risk score derived a greater proportional reduction in the risk of cardiovascular events per millimole per litre reduction in LDL-C than did persons with the lowest tertile of the genetic risk score.53 The genetic score used in this study may have included variants that lead to a greater susceptibility to retain LDL particles. Identifying genetic and other factors that influence the likelihood of retaining LDL particles within the intima is an active area of ongoing research and may eventually help to personalize the prevention of cardiovascular disease by identifying persons who are most vulnerable to the deleterious effects of LDL-C and therefore are most likely to benefit from therapies that lower LDL-C.
Conclusions
Considered together, the strong and consistent evidence from the genetic studies, prospective epidemiologic cohort studies, Mendelian randomization studies, and randomized intervention trials discussed here, supported by mechanistic evidence to be presented in the second Consensus Statement on LDL causality, establishes that LDL is not merely a biomarker of increased risk but a causal factor in the pathophysiology of ASCVD. The key implications for this conclusion are presented in Box 1.
Funding
Travel and meeting logistics were supported by unrestricted educational grants from MSD and Amgen to the European Atherosclerosis Society. These companies were not present at the Consensus Panel meetings, had no role in the design or content of the manuscript, and had no right to approve or disapprove the final document. Funding to pay the Open Access publication charges for this article was provided by the European Atherosclerosis Society.
Conflict of interest: J.B. has received research grants from Amgen, AstraZeneca, NovoNordisk, Pfizer and Regeneron/Sanofi and honoraria for consultancy and lectures from Amgen, AstraZeneca, Eli Lilly, Merck, Novo-Nordisk, Pfizer, and Regeneron/Sanofi. E.B. has received honoraria from AstraZeneca, Amgen, Genfit, MSD, Sanofi-Regeneron, Unilever, Danone, Aegerion, Chiesi, Rottapharm, Lilly and research grants from Amgen, Danone and Aegerion. A.L.C. has received research grants to his institution from Amgen, Astra-Zeneca, Merck, Regeneron/Sanofi, and Sigma Tau, and honoraria for advisory boards, consultancy or speaker bureau from Abbot, Aegerion, Amgen, AstraZeneca, Eli Lilly, Genzyme, Merck/MSD,Mylan, Pfizer, Rottapharm and Sanofi-Regeneron. M.J.C. has received research grants from MSD, Kowa, Pfizer, and Randox and honoraria for consultancy/speaker activities from Amgen, Kowa, Merck, Sanofi, Servier, Unilever, and Regeneron. S.F. has the following disclosures for the last 12 months: Compensated consultant and advisory activities with Merck, Kowa, Sanofi, Amgen, Amarin, and Aegerion. B.A.F. has received research grants from Merck, Amgen and Esperion Therapeutics and received honoraria for lectures, consulting and/or advisory board membership from Merck, Amgen, Esperion, Ionis, and the American College of Cardiology. I.G. has received speaker fees from MSD and Pfizer relating to cardiovascular risk estimation and lipid guidelines, and consultancy/speaker fee from Amgen. H.N.G. has received research grants from Merck, Sanofi-Regeneron, and Amgen. He consults for Merck, Sanofi, Regeneron, Lilly, Kowa, Resverlogix, Boehringer Ingelheim. R.A.H. has received research grants from Aegerion, Amgen, The Medicines Company, Pfizer, and Sanofi. He consults for Amgen, Aegerion, Boston Heart Diagnostics, Gemphire, Lilly, and Sanofi. J.D.H reports honoraria/research grants from Aegerion, Alnylam, Catabasis, Lilly, Merck, Pfizer, Novartis, Regeneron, Sanofi. R.M.K is a Member, Merck Global Atherosclerosis Advisory Board. U.L. has received honoraria for lectures and/or consulting from Amgen, Medicines Company, Astra Zeneca, MSD, Berlin Chemie, Bayer, Abbott, and Sanofi. U.L. aufs has received honoraria for board membership, consultancy, and lectures from Amgen, MSD, Sanofi, and Servier. L.M. has received honoraria for consultancy and lectures from Amgen, Danone, Kowa, Merck, and Sanofi-Regeneron. S.J.N. has received research support from Amgen, AstraZeneca, Anthera, Cerenis, Novartis, Eli Lilly, Esperion, Resverlogix, Sanofi-Regeneron, InfraReDx. and LipoScience and is a consultant for Amgen, AstraZeneca, Boehringer Ingelheim, CSL Behring, Eli Lilly, Merck, Takeda, Pfizer, Roche, Sanofi-Regeneron, Kowa. and Novartis. B.G.N. reports consultancies and honoraria for lectures from AstraZeneca, Sanofi, Regeneron, Aegerion, Fresenius, B Braun, Kaneka, Amgen. C.J.P. has received research support from Roche, MSD and honoraria from MSD, Sanofi/Regeneron, Amgen and Pfizer. F.J.R. has received grants/research support from Amgen and Sanofi and has received speaker fees or honoraria for consultation from AstraZeneca, Merck, Amgen, and Sanofi. K.K.R. has received research grants from Amgen, Sanofi-Regeneron and Pfizer and honoraria for lectures, advisory boards or as a steering committee member from Aegerion, Amgen, Sanofi-Regeneron, Pfizer, AstraZeneca, Cerenis, ISIS Pharma, Medco, Resverlogix, Kowa, Novartis, Cipla, Lilly, Algorithm, Takeda, Boehringer Ingelheim, MSD. Esperion, and AbbieVie. H.S. has received research grants from AstraZeneca, MSD, Bayer Vital, sanofi-aventis, and Pfizer and honoraria for speaker fees from AstraZeneca, MSD, Genzyme, sanofi-aventis, and Synlab. He has consulted for MSD and AstraZeneca. M.R.T. has received speaker fees from Amgen, Astra Zeneca, Chiesi Pharma and Eli Lilly and speaker fees and research support from Amgen, Sanofi Aventis and Novo Nordisk. She has consulted for AstraZeneca. L.T. has received research funding and/or honoraria for advisory boards, consultancy or speaker bureau from Abbott Mylan, Actelion, Aegerion, Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Daiichi-Sankyo, GlaxoSmithKline, Menarini, Merck, Novartis, Pfizer, Sanofi-Regeneron, Servier and Synageva. G.F.W. has received research support from Amgen and Sanofi-Regeneron. O.W. has received honoraria for lectures or consultancy from Sanofi, Amgen, MSD, and Astra-Zeneca. B.v.S, and J.K.S. report no disclosures.
References
Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, Wang H, Liu T, Wasserman SM, Sever PS, Pedersen TR. FOURIER Steering Committee and Investigators. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med 2017; doi: 10.1056/NEJMoa1615664.
Author notes
These authors contributed equally as senior authors.
The opinions expressed in this article are not necessarily those of the Editors of the European Heart Journal or of the European Society of Cardiology.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments
Based on this vast body of evidence, we disagree with the suggestion by Sloop and St. Cyr that our conclusion that LDL causes ASCVD is non-falsifiable, and therefore non-scientific. Indeed, the causality of LDL has been directly tested in numerous randomized trials that have evaluated the effect of random allocation to multiple different LDL lowering therapies or placebo on the risk of developing incident cardiovascular events. The suggestion that the “limited success of cholesterol-lowering therapy in numerous prospective randomized controlled studies, some of which show significant decreases in serum LDL cholesterol but no improvement in outcome” refutes the causal effect of LDL on the risk of ASCVD is not a quantitatively literate argument. Instead, a synthesis of the totality of the evidence from randomized trials evaluating therapies that lower LDL by multiple different mechanisms provides overwhelming quantitative evidence that LDL causes ASCVD, that lowering LDL reduces the risk of ASCVD, and that this effect is dose dependent and proportional to the absolute change in plasma LDL cholesterol concentration (1,2).
Furthermore, the suggestion by Sloop and St. Cyr that “proving” LDL causes ASCVD would require a randomized trial evaluating “lifetime therapy with a drug effective enough to completely eliminate serum LDL cholesterol” is, ironically for them, a non-falsifiable, and therefore non-scientific, straw-man because such a trial would be impossible to conduct and therefore will never be performed. Fortunately, however, Nature has conducted this trial for us. Numerous genetic mutations that increase lifetime exposure to LDL cholesterol are associated with a markedly increased lifetime risk of ASCVD, while mutations that lead to lifetime exposure to lower LDL are associated with a corresponding markedly lower lifetime risk of ASCVD (3). Indeed, naturally random allocation to nearly all genetic variants that lead to lifetime exposure to lower LDL are associated with a corresponding dose-dependent lower lifetime risk of ASCVD (4).
The remarkable concordance between the results of Nature’s randomized genetic trials and the results of the randomized trials evaluating multiple different LDL lowering therapies provides powerful and unbiased confirmation that LDL causes ASCVD, and that lowering LDL reduces the risk of cardiovascular events (5-8). To conclude otherwise would require Sloop and St. Cyr to count Mother Nature herself among the LDL “apologists”.
Authors:
Brian A Ference
Henry N Ginsberg
M John Chapman
Alberico L Catapano
References
1. Cholesterol Treatment Trialists’ (CTT) Collaboration, Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, Peto R, Barnes EH, Keech A, Simes J, Collins R. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170 000 participants in 26 randomised trials. Lancet 2010;376:1670–1681.
2. Silverman MG, Ference BA, Im K, Wiviott SD, Giugliano RP, Grundy SM, Braunwald E, Sabatine MS. Association between lowering LDL-C and cardiovascular risk reduction among different therapeutic interventions: a systematic review and meta-analysis. JAMA 2016;316:1289-1297.
3. Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006;354:1264–1272.
4. Ference BA, Yoo W, Alesh I, Mahajan N, Mirowska KK, Mewada A, Kahn J, Afonso L, Williams KA Sr, Flack JM. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a Mendelian randomization analysis. J Am Coll Cardiol 2012;60:2631-2639.
5. Ference BA, Majeed F, Penumetcha R, Flack JM, Brook RD. Effect of naturally random allocation to lower low-density lipoprotein cholesterol on the risk of coronary heart disease mediated by polymorphisms in NPC1L1, HMGCR, or both: a 2 x 2 factorial Mendelian randomization study. J Am Coll Cardiol 2015;65:1552–1261.
6. Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, Theroux P, Darius H, Lewis BS, Ophuis TO, Jukema JW, De Ferrari GM, Ruzyllo W, De Lucca P, Im K, Bohula EA, Reist C, Wiviott SD, Tershakovec AM, Musliner TA, Braunwald E, Califf RM; IMPROVE-IT Investigators. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med 2015;372:2387-2397
7. Ference BA, Robinson JG, Brook RD, Catapano AL, Chapman MJ, Neff DR, Voros S, Giugliano RP, Davey Smith G, Fazio S, Sabatine MS. Variation in PCSK9 and HMGCR and Risk of Cardiovascular Disease and Diabetes. N Engl J Med. 2016; 375:2144-2153.
8. Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, Wang H, Liu T, Wasserman SM, Sever PS, Pedersen TR. FOURIER Steering Committee and Investigators. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med 2017; doi: 10.1056/NEJMoa1615664.
The statement that the key events in the initiation of ASCVD are the retention and accumulation of cholesterol-rich apoB-containing lipoproteins within the arterial intima at sites of predilection for plaque formation is also too vague to be scientific. Apparently, the authors have not seriously considered that LDL is retained throughout the arterial tree bound to proteoglycans within diffuse intimal thickening (1). Thus, some other factor must be present to create sites of predilection. The authors may also be unaware that fatty streaks, which are rich in cholesterol derived from apoB-containing lipoproteins, routinely resolve without sequelae (2). Even mainstream atherogenesis theorists acknowledge that at least some fatty streaks do not progress to atherosclerotic plaques. The inability to predict which fatty streaks will progress makes the idea that the accumulation of apoB-containing lipoproteins is a key event in atherogenesis non-falsifiable and nonscientific. The authors should make a testable theory which explains how LDL can be atherogenic in one place and not another.
It is apparent that LDL cholesterol alone does not cause atherosclerotic cardiovascular disease. There is no other conclusion possible given the limited success of cholesterol-lowering therapy in numerous prospective randomized controlled studies, some of which show significant decreases in serum LDL cholesterol but no improvement in outcome (3). Elevated LDL merely contributes to the disease by increasing blood viscosity, especially at low rates of shear. Elevated LDL is merely one of several true risk factors for atherosclerosis which increase blood viscosity and therefore increase the risk of developing a mural thrombus. The circumstance which causes or inevitably leads to atherosclerosis should only be present at sites of predilection. That circumstance is the organizing mural thrombus, as originally proposed by the Welsh pathologist J.B. Duguid in the mid twentieth century.
References
1. Sloop GD. A critical analysis of the role of cholesterol in atherosclerosis. Atherosclerosis 1999; 142(2): 265-8.
2. Sloop GD, Perret RS, Brahney JS, Oalmann M. A description of two morphologic patterns of aortic fatty streaks, and a hypothesis of their pathogenesis. Atherosclerosis 1998; 141: 153-160.
3. DeBruff R. Cholesterol paradox: a correlate does not a surrogate make. Evid Based Med 2017; 22:15-19.