





Abstract
RET polymorphisms have been involved in the clinical presentation and prognosis of multiple endocrine neoplasia type 2 (MEN2)-associated medullary thyroid carcinoma.
To investigate the effect of RET variants on the penetrance of pheochromocytoma (PHEO) in MEN2 patients. Methods: The RET variants L769L, S836S, and G691S/S904S were evaluated in a cohort of 153 MEN2 patients attending a tertiary teaching hospital. A comparison of RET variant frequencies between patients with and without PHEO was performed. Kaplan–Meier curves and Cox regression analysis were used to estimate the effect of RET variants on the age-dependent penetrance.
A total of 48 (31.4%) patients presented with MEN2-associated PHEOs. The mean age at diagnosis was 35.5±13.4 years, 60.4% of patients were women, and 92.8% had RET mutations at codon 634. The frequencies of RET polymorphisms were as follows: 20.1% L769L, 4.75% S836S, and 17.3% S904S/G691S. We did not observe any association between the frequencies of L769L, S836S, or S904S/G691S variants and PHEO development (all P>0.05). However, individuals carrying two RET polymorphic alleles had an increased estimated risk of PHEO (2.63; 95% CI, 1.4–5.0; P=0.004) and were younger at diagnosis when compared with those with one or no polymorphism (29.6±6.3 and 39.3±14.4 years respectively; P=0.006). Accordingly, additional analysis using Cox proportional hazard models demonstrated that the presence of two RET variants was associated with an increased risk for early PHEO development (hazard ratio, 5.99 (95% CI, 2.24–16.03); P<0.001).
RET polymorphic alleles have an additive effect on the estimated risk of age-related PHEO penetrance in MEN2 patients.
Introduction
Pheochromocytoma (PHEO) is a rare catecholamine-producing tumor that develops from the chromaffin cells of the adrenal medulla (1, 2). The tumor may occur sporadically (76–80%) or as part of inherited syndromes, such as multiple endocrine neoplasia type 2 (MEN2), von Hippel–Lindau disease, and neurofibromatosis type 1 (3, 4, 5). Mutations in the succinate dehydrogenase (SDH) gene complex (SDHB, SDHC, and SDHD) and, more recently, in the genes SDHA, SDHAF2, TMEM127, and MAX have also been linked to hereditary PHEO and paraglanglioma (6).
The MEN2 syndrome includes the following three clinically distinct forms: MEN2A, MEN2B, and familial medullary thyroid carcinoma (FMTC). Patients with MEN2A develop MTC, PHEO, and primary hyperparathyroidism (HPT). MEN2B patients have MTC, PHEO, ganglioneuromas of the digestive tract, mucosal neuromas, and skeletal abnormalities. In patients with FMTC, only the thyroid is affected. The RET proto-oncogene is the susceptibility gene for MEN2. The germline mutations in MEN2 are usually located either in the extracellular cysteine-rich region (exons 10 and 11) or in the intracellular tyrosine kinase domains (exons 13–16) of the RET protein (7).
Several studies have indicated a correlation between specific RET mutations and clinical presentation of MEN2. The presence of any mutation at codon 634 (exon 11) is associated with the presence of PHEO and HPT (8, 9, 10). By contrast, mutations at codons 768 and 804 are thus far associated with FMTC, whereas codon 918 mutations are MEN2B specific. Moreover, patients who harbor the C634R genotype present with more distant metastases than those who harbor C634W or C634Y mutations at the time of diagnosis, thus suggesting that even a change of specific amino acids may modify the natural development of the disease (11). A large study that included 92 carriers from 20 unrelated families worldwide showed that individuals who harbor the RET C634W mutation have a higher penetrance for PHEO (20% by the age of 30 years, 67% by age 50, and 92% by age 70) (12). Recently, risk profiles and penetrance estimations in MEN2A caused by germline RET exon 10 mutations were evaluated in a large multicenter study that included 340 subjects from 103 families. Frank-Raue et al. (13) observed that 50% penetrance was achieved by the age of 36 years for MTC, by 68 years for PHEO, and by 82 years for HPT. Based on genotype–phenotype correlation studies, the American Thyroid Association (ATA) developed recommendations for the age of prophylactic thyroidectomy in asymptomatic RET mutation carriers. The different mutations of the RET gene were classified into four risk categories based on the aggressiveness of the disease (A<B<C<D). Thus, the mutations associated with the MEN2B phenotype (ATA level D risk) are associated with the highest risk for early development of MTC, whereas in patients at ATA levels A and B (mutations in codons 768, 790, 791, 804, and 891 for level A and 609, 611, 618, 620, and 630 for level B), the risk for MTC is moderate (14).
In the last decade, several studies have focused on the potential role of neutral RET sequence variants in modifying the clinical course of MEN2-related MTC. Although controversial, an increased prevalence of the RET polymorphisms G691S, L769L, S836S, and S904S has been described in individuals with MTC (15, 16, 17, 18, 19). Other studies have shown that these variants could interfere in disease presentation (15, 16, 20, 21). Interestingly, recent findings suggest a potential additive effect of the different RET variants on the susceptibility and clinical course of sporadic MTC. Individuals harboring haplotypes with three or more RET polymorphic alleles have higher risks for MTC development and lymph node and distant metastases (22).
Conversely, the data regarding the role of RET variants in PHEO susceptibility or penetrance in MEN2 patients are limited. A small study that included 36 patients reported a 17% frequency of RET variants in patients with PHEO. Interestingly, patients carrying RET polymorphisms were older and had a tendency to have larger tumors when compared with those carrying the WT gene (23). A haplotype consisting of a unique combination of alleles at four RET variants within intron 1 was strongly associated with sporadic PHEO (24). The RET G9691S and S904S polymorphisms have also been observed in a patient with bilateral PHEO without any known PHEO-related mutations (25).
In this study, we aimed to investigate whether the RET neutral variants G691S, L769L, S836S, and S904S influence the penetrance or clinical presentation of PHEO in a large cohort of individuals with MEN2. These polymorphisms were selected based on their previous association with the clinical course of hereditary MTC. Although we did not observe any association between any single variant and PHEO development or clinical features, individuals who harbored two RET genetic variants presented with an age-related increased risk for developing PHEO.
Subjects and methods
Patients
Patients with a diagnosis of MEN2 who were admitted to the Endocrine Division at the Hospital de Clínicas de Porto Alegre (HCPA) were invited to participate in the study. Since 1997, our division has served as a reference center for molecular testing of RET germline mutations in Brazil; therefore, patients suspected of having MEN2 who were referred to us by other Brazilian centers for molecular investigation were invited to participate. Patients with a diagnosis of MEN2 who were followed at the University Hospital of the Ribeirão Preto School of Medicine, University of São Paulo (USP), were also included in the study. All the patients and their legal guardians had provided written consent according to the institutional Ethics Committee.
Our sample comprised 173 patients with MEN2 (48 with hereditary PHEO), including 159 patients (44 with PHEO) from our institution and 14 patients (four with PHEO) from the University Hospital of the Medical School of Ribeirão Preto (Ribeirão Preto, São Paulo). The MEN2 patients, belonging to 23 MEN2A and nine MEN2B unrelated families, were diagnosed using genetic screening. In this study, 20 patients who harbored mutations associated with FMTC (ATA level A risk, codons 891 and 768) were excluded.
The data collected for each individual included the clinical and histopathological characteristics of PHEO, the association of other endocrine neoplasias, the presence of affected family members, the presence of RET germline mutations, urinary fractionated metanephrines (HPLC), and diagnostic imaging (i.e. abdominal computed tomography and, in selected patients, whole-body metaiodobenzylguanidine scintigraphy).
Single-nucleotide polymorphism analysis in MEN2-associated PHEO
The following RET single-nucleotide polymorphisms (SNPs) were selected based on their previous association with the clinical course of MEN2: G691S (rs1799939, codon 691 of exon 11, GlyGGT→SerAGT), L769L (rs1800861, codon 769 of exon 13, LeuCTT→LeuCTG), S836S (rs1800862, codon 836 of exon 14, SerAGC→SerAGT), and S904S (rs1800863, codon 904 of exon 15, SerTCC→SerTCG). For genotyping, genomic DNA was prepared from peripheral blood leukocytes using a standard procedure. Genotype analysis was performed using Human Custom TaqMan SNP Genotyping Assays 40× (Applied Biosystems). Primer and probe sequences used for genotyping the RET variants were as follows: L769L (rs1800861): 5′-GGGTGGTTGACCTGCTTCAG-3′ (forward primer), 5′-CTGCTCTGTGCTGCATTTCAG-3′ (reverse primer), VIC-5′-AGGTCTCGAAGCTCA-3′, and FAM-5′-AGGTCTCGCAGCTCA-3′; S836S (rs1800862): 5′-GCGAGAGCCGCAAAGTG-3′ (forward primer), 5′-GTGAGGGCCCGCTCATC-3′ (reverse primer), VIC-5′-CAACTCCAGCTCCCTG-3′, and FAM-5′-CAACTCCAGTTCCCTG-3′; and S904S (rs1800863): 5′-GCTTGTCCCGAGATGTTTATGAAGA-3′ (forward primer), 5′-GGGCACCTGGCTCCT-3′ (reverse primer), VIC-5′-CTTCACGTAGGAATCC-3′, and FAM-5′-CTTCACGTACGAATCC-3′. The two variants G691S and S904S were in linkage disequilibrium (LD); therefore, only S904S was genotyped and the results are shown as G691S/S904S.
The reactions were conducted in a 96-well plate in a total of 5-ml reaction volume using 2 ng genomic DNA, TaqMan Genotyping Master Mix 1× (Applied Biosystems), and Custom TaqMan Genotyping Assay 1×. The plates were positioned in a real-time PCR thermal cycler (7500 Fast Real PCR System, Life Technologies) and heated for 10 min at 95 °C, followed by 45 cycles of 95 °C for 15 s and 62 °C for 1 min. Fluorescence data from each plate were analyzed using automated allele-calling software (SDS 2.1, Life Technologies).
Statistical analysis
The results are expressed as the mean±s.d. values. Clinical and oncological characteristics and SNPs were compared using the χ2-test for qualitative variables and Student's t-test for quantitative variables. The Hardy–Weinberg equilibrium for each SNP was assessed using the χ2-test.
The haplotypes were constructed based on the combination of allelic variants, and their frequencies were inferred using the phase 2.1 program, which implements a Bayesian statistical method. We also used the phase 2.1 program to compare the distributions of different RET haplotypes between MEN2 patients with and without PHEO by employing permutation analyses of 1000 random replicates.
Poisson regression analysis was performed to evaluate the estimated risk of PHEO, using PHEO as the dependent variable and age, RET mutations, and number of RET SNPs as independent variables. The age-dependent penetrance for PHEO was estimated using Kaplan–Meier curves, and comparisons between curves were performed using the log-rank test. We performed a Cox regression to assess the effect of the number of RET variants on the time of a specified event, i.e. presence of PHEO.
Statistical Package for the Social Sciences 18.0 Software (PASW, Inc., Chicago, IL, USA) was used for all the analyses. P<0.05 was considered as statistically significant.
Results
RET polymorphisms in MEN2 patients
The molecular data for families with MEN2 are given in Table 1. Of the 32 independent families with MEN2 analyzed, 20 families were classified as MEN2A, three as MEN2A associated with cutaneous lichen amyloidosis (CLA), and nine as MEN2B. All but three MEN2A kindred had a mutation at RET codon 634 in exon 11, the most prevalent mutation (92.8% of cases). All MEN2B individuals presented with the characteristic phenotype and mutation at codon 918, exon 16, resulting in the substitution of a methionine residue by threonine (M918T).
Clinical presentation and RET germline mutations in individuals with multiple endocrine neoplasia type 2.
| Phenotype | No. of families | RET mutation | ATA risk level | Affected individuals | CCH | MTC | PHEO | HPT |
|---|---|---|---|---|---|---|---|---|
| MEN2A | 9 | C634Y | C | 70 | 1 | 64 | 20 | 14 |
| 6 | C634R | C | 16 | 1 | 11 | 7 | 4 | |
| 1 | C634W | C | 3 | 2 | 2 | 2 | ||
| 1 | C634S | C | 1 | 1 | 1 | 0 | ||
| 1 | C620R | B | 3 | 3 | 1 | 0 | ||
| 2 | C618R | B | 7 | 7 | 3 | 0 | ||
| 1 | C634R | C | 2 | 2 | 0 | 0 | ||
| MEN2A+CLA | 1 | C634Y | C | 31 | 27 | 5 | 6 | |
| 1 | C634W | C | 6 | 1 | 5 | 3 | 0 | |
| MEN2B | 9 | M918T | D | 14 | 14 | 6 | – | |
| Total | 32 | 153a | 3 | 136 | 48 | 26 |
| Phenotype | No. of families | RET mutation | ATA risk level | Affected individuals | CCH | MTC | PHEO | HPT |
|---|---|---|---|---|---|---|---|---|
| MEN2A | 9 | C634Y | C | 70 | 1 | 64 | 20 | 14 |
| 6 | C634R | C | 16 | 1 | 11 | 7 | 4 | |
| 1 | C634W | C | 3 | 2 | 2 | 2 | ||
| 1 | C634S | C | 1 | 1 | 1 | 0 | ||
| 1 | C620R | B | 3 | 3 | 1 | 0 | ||
| 2 | C618R | B | 7 | 7 | 3 | 0 | ||
| 1 | C634R | C | 2 | 2 | 0 | 0 | ||
| MEN2A+CLA | 1 | C634Y | C | 31 | 27 | 5 | 6 | |
| 1 | C634W | C | 6 | 1 | 5 | 3 | 0 | |
| MEN2B | 9 | M918T | D | 14 | 14 | 6 | – | |
| Total | 32 | 153a | 3 | 136 | 48 | 26 |
MTC, hereditary medullary carcinoma; PHEO, pheochromocytoma; HPT, hyperparathyroidism; CCH, C cell hyperplasia.
Fourteen individuals are still awaiting MTC surgery.
Clinical presentation and RET germline mutations in individuals with multiple endocrine neoplasia type 2.
| Phenotype | No. of families | RET mutation | ATA risk level | Affected individuals | CCH | MTC | PHEO | HPT |
|---|---|---|---|---|---|---|---|---|
| MEN2A | 9 | C634Y | C | 70 | 1 | 64 | 20 | 14 |
| 6 | C634R | C | 16 | 1 | 11 | 7 | 4 | |
| 1 | C634W | C | 3 | 2 | 2 | 2 | ||
| 1 | C634S | C | 1 | 1 | 1 | 0 | ||
| 1 | C620R | B | 3 | 3 | 1 | 0 | ||
| 2 | C618R | B | 7 | 7 | 3 | 0 | ||
| 1 | C634R | C | 2 | 2 | 0 | 0 | ||
| MEN2A+CLA | 1 | C634Y | C | 31 | 27 | 5 | 6 | |
| 1 | C634W | C | 6 | 1 | 5 | 3 | 0 | |
| MEN2B | 9 | M918T | D | 14 | 14 | 6 | – | |
| Total | 32 | 153a | 3 | 136 | 48 | 26 |
| Phenotype | No. of families | RET mutation | ATA risk level | Affected individuals | CCH | MTC | PHEO | HPT |
|---|---|---|---|---|---|---|---|---|
| MEN2A | 9 | C634Y | C | 70 | 1 | 64 | 20 | 14 |
| 6 | C634R | C | 16 | 1 | 11 | 7 | 4 | |
| 1 | C634W | C | 3 | 2 | 2 | 2 | ||
| 1 | C634S | C | 1 | 1 | 1 | 0 | ||
| 1 | C620R | B | 3 | 3 | 1 | 0 | ||
| 2 | C618R | B | 7 | 7 | 3 | 0 | ||
| 1 | C634R | C | 2 | 2 | 0 | 0 | ||
| MEN2A+CLA | 1 | C634Y | C | 31 | 27 | 5 | 6 | |
| 1 | C634W | C | 6 | 1 | 5 | 3 | 0 | |
| MEN2B | 9 | M918T | D | 14 | 14 | 6 | – | |
| Total | 32 | 153a | 3 | 136 | 48 | 26 |
MTC, hereditary medullary carcinoma; PHEO, pheochromocytoma; HPT, hyperparathyroidism; CCH, C cell hyperplasia.
Fourteen individuals are still awaiting MTC surgery.
The frequencies of RET polymorphisms were as follows: 20.1% L769L, 4.75% S836S, and 17.3% S904S/G691S. The observed SNP frequencies were similar to those reported in the literature (17, 21). We had previously evaluated the frequency of RET polymorphisms in a group of 308 cancer unaffected volunteers attending the blood donation facility of our institution (22). The frequencies of RET variants in the control subjects were as follows: 20.8% L769L, 4.2% S836S, and 21.7% S904S and did not differ from those observed in MEN2A patients (all P>0.05). Confirming previous studies, the two variants G691S and S904S were in LD; therefore, to avoid the inclusion of redundant information, the results were grouped and referred to as G691S/S904S (16, 17, 26). All genotypes analyzed were in Hardy–Weinberg equilibrium (P>0.20).
Clinical and oncological features of MEN2-associated PHEO
The frequency of PHEO in our sample was 31.4% (48 out of 153 patients). The mean age at the time of diagnosis was 35.5±13.4 years and 60.4% were women. Bilateral PHEO occurred synchronously or metachronously in 29.2% of patients. The mean age at the time of MTC diagnosis was significantly different between patients with and without PHEO (31.4±13.1 vs 21.7±14.7 years respectively; P=0.0001; Table 2). Out of 48 individuals with PHEO, MTC was diagnosed before PHEO in 27 patients (56.2%) and after PHEO in three patients (6.2%). The identified RET germline mutations in PHEO patients were as follows: 10.4% C634W, 52% C634Y, 14.6% C634R, 2.1% C634S, 6.3% C618R, 2.1% C620R, and 12.5% M918T.
Clinical features and genotype of MEN2 patients according to the presence of pheochromocytoma.
| Patients | Total | Pheo | P | |
|---|---|---|---|---|
| + | − | |||
| n | 153 | 48 | 105 | |
| Sex, female (%) | 55.6 | 60.4 | 53.3 | 0.52 |
| Age (years)a | 25.2±14.9 | 31.4±13.1 | 21.7±14.7 | 0.0001 |
| Age (years)b | – | 35.5±13.4 | – | – |
| L769L (%)c | 34.1 | 46.3 | 28.7 | 0.06 |
| S836S (%)c | 9.6 | 12.2 | 8.5 | 0.53 |
| G691S/S904S (%)c | 33.3 | 31.7 | 34 | 0.94 |
| Patients | Total | Pheo | P | |
|---|---|---|---|---|
| + | − | |||
| n | 153 | 48 | 105 | |
| Sex, female (%) | 55.6 | 60.4 | 53.3 | 0.52 |
| Age (years)a | 25.2±14.9 | 31.4±13.1 | 21.7±14.7 | 0.0001 |
| Age (years)b | – | 35.5±13.4 | – | – |
| L769L (%)c | 34.1 | 46.3 | 28.7 | 0.06 |
| S836S (%)c | 9.6 | 12.2 | 8.5 | 0.53 |
| G691S/S904S (%)c | 33.3 | 31.7 | 34 | 0.94 |
Pheo, pheochromocytoma.
Age, at the time of diagnosis of MTC, expressed as the mean±s.d.
Age, at the time of diagnosis of Pheo, expressed as the mean±s.d.
Data available for 135 patients.
Clinical features and genotype of MEN2 patients according to the presence of pheochromocytoma.
| Patients | Total | Pheo | P | |
|---|---|---|---|---|
| + | − | |||
| n | 153 | 48 | 105 | |
| Sex, female (%) | 55.6 | 60.4 | 53.3 | 0.52 |
| Age (years)a | 25.2±14.9 | 31.4±13.1 | 21.7±14.7 | 0.0001 |
| Age (years)b | – | 35.5±13.4 | – | – |
| L769L (%)c | 34.1 | 46.3 | 28.7 | 0.06 |
| S836S (%)c | 9.6 | 12.2 | 8.5 | 0.53 |
| G691S/S904S (%)c | 33.3 | 31.7 | 34 | 0.94 |
| Patients | Total | Pheo | P | |
|---|---|---|---|---|
| + | − | |||
| n | 153 | 48 | 105 | |
| Sex, female (%) | 55.6 | 60.4 | 53.3 | 0.52 |
| Age (years)a | 25.2±14.9 | 31.4±13.1 | 21.7±14.7 | 0.0001 |
| Age (years)b | – | 35.5±13.4 | – | – |
| L769L (%)c | 34.1 | 46.3 | 28.7 | 0.06 |
| S836S (%)c | 9.6 | 12.2 | 8.5 | 0.53 |
| G691S/S904S (%)c | 33.3 | 31.7 | 34 | 0.94 |
Pheo, pheochromocytoma.
Age, at the time of diagnosis of MTC, expressed as the mean±s.d.
Age, at the time of diagnosis of Pheo, expressed as the mean±s.d.
Data available for 135 patients.
We did not observe any association between the frequencies of the L769L, S836S, or S904S/G691S variants and MEN2-associated PHEO (all P>0.05; Table 3).
Frequency of RET haplotypes in MEN2 patients.
| Haplotypes | Presence/absence of | Polymorphic alleles (n) | Haplotypes (%) | |||
|---|---|---|---|---|---|---|
| L769L (T>G) | S836S (C>T) | S904S (C>G) | PHEOa (−) | PHEOa (+) | ||
| Hpt1 | − | − | − | None | 68.0 | 57.0 |
| Hpt2 | + | − | − | 1 | 11.7 | 22.0 |
| Hpt3 | − | + | − | 1 | 0.64 | 0.60 |
| Hpt4 | − | − | + | 1 | 15.0 | 15.4 |
| Hpt5 | + | + | − | 2 | 4.25 | 4.66 |
| Hpt6 | + | − | + | 2 | 0.35 | 0.58 |
| Hpt7 | − | + | + | 2 | – | – |
| Hpt8 | + | + | + | 3 | – | – |
| Haplotypes | Presence/absence of | Polymorphic alleles (n) | Haplotypes (%) | |||
|---|---|---|---|---|---|---|
| L769L (T>G) | S836S (C>T) | S904S (C>G) | PHEOa (−) | PHEOa (+) | ||
| Hpt1 | − | − | − | None | 68.0 | 57.0 |
| Hpt2 | + | − | − | 1 | 11.7 | 22.0 |
| Hpt3 | − | + | − | 1 | 0.64 | 0.60 |
| Hpt4 | − | − | + | 1 | 15.0 | 15.4 |
| Hpt5 | + | + | − | 2 | 4.25 | 4.66 |
| Hpt6 | + | − | + | 2 | 0.35 | 0.58 |
| Hpt7 | − | + | + | 2 | – | – |
| Hpt8 | + | + | + | 3 | – | – |
Hpt, haplotype; P=0.16. The P value for the comparisons of haplotype frequencies between patients, PHEO (−) and PHEO (+), was calculated using the permutation test (1000 replications).
Frequency calculated based on the number of chromosomes (n=270).
Frequency of RET haplotypes in MEN2 patients.
| Haplotypes | Presence/absence of | Polymorphic alleles (n) | Haplotypes (%) | |||
|---|---|---|---|---|---|---|
| L769L (T>G) | S836S (C>T) | S904S (C>G) | PHEOa (−) | PHEOa (+) | ||
| Hpt1 | − | − | − | None | 68.0 | 57.0 |
| Hpt2 | + | − | − | 1 | 11.7 | 22.0 |
| Hpt3 | − | + | − | 1 | 0.64 | 0.60 |
| Hpt4 | − | − | + | 1 | 15.0 | 15.4 |
| Hpt5 | + | + | − | 2 | 4.25 | 4.66 |
| Hpt6 | + | − | + | 2 | 0.35 | 0.58 |
| Hpt7 | − | + | + | 2 | – | – |
| Hpt8 | + | + | + | 3 | – | – |
| Haplotypes | Presence/absence of | Polymorphic alleles (n) | Haplotypes (%) | |||
|---|---|---|---|---|---|---|
| L769L (T>G) | S836S (C>T) | S904S (C>G) | PHEOa (−) | PHEOa (+) | ||
| Hpt1 | − | − | − | None | 68.0 | 57.0 |
| Hpt2 | + | − | − | 1 | 11.7 | 22.0 |
| Hpt3 | − | + | − | 1 | 0.64 | 0.60 |
| Hpt4 | − | − | + | 1 | 15.0 | 15.4 |
| Hpt5 | + | + | − | 2 | 4.25 | 4.66 |
| Hpt6 | + | − | + | 2 | 0.35 | 0.58 |
| Hpt7 | − | + | + | 2 | – | – |
| Hpt8 | + | + | + | 3 | – | – |
Hpt, haplotype; P=0.16. The P value for the comparisons of haplotype frequencies between patients, PHEO (−) and PHEO (+), was calculated using the permutation test (1000 replications).
Frequency calculated based on the number of chromosomes (n=270).
Additive effect of RET polymorphisms on clinical presentation of PHEO
We examined the additive effect of RET risk alleles on clinical features of PHEO in MEN2 individuals. The distributions of RET risk alleles were as follows: 53 (39.3%) individuals harbored no risk allele, 60 (44.4%) harbored one risk allele, and 22 (16.3%) harbored two RET risk alleles. We used a Bayesian statistical method to estimate the frequency of the different haplotypes produced by combinations of the L769L, S836S, and S904S/G691S polymorphisms. In total, six haplotypes were inferred (Table 3), and permutation analyses showed that the distributions of these six haplotypes were not significantly different between patients with and without PHEO (P=0.16; Table 4). Interestingly, no patient harbored all three RET polymorphisms.
Additive effect of RET polymorphic alleles on pheochromocytoma susceptibility (n=135). The independent variables included in the multiple regression analyses were age, RET germline mutations (codons 618, 620, 634, and 918), and number of RET polymorphisms.
| Risk alleles | PHEO (+)a | PHEO (−)b | OR (95% CI) | P |
|---|---|---|---|---|
| None | 14 (34.1) | 39 (41.5) | 1 | |
| One | 17 (41.5) | 43 (45.7) | 1.3 (0.8–2.3) | 0.365 |
| Two | 10 (24.4) | 12 (12.8) | 2.63 (1.4–5.0) | 0.004 |
| Risk alleles | PHEO (+)a | PHEO (−)b | OR (95% CI) | P |
|---|---|---|---|---|
| None | 14 (34.1) | 39 (41.5) | 1 | |
| One | 17 (41.5) | 43 (45.7) | 1.3 (0.8–2.3) | 0.365 |
| Two | 10 (24.4) | 12 (12.8) | 2.63 (1.4–5.0) | 0.004 |
Data available for 41 patients with PHEO.
Data available for 94 patients without PHEO.
Additive effect of RET polymorphic alleles on pheochromocytoma susceptibility (n=135). The independent variables included in the multiple regression analyses were age, RET germline mutations (codons 618, 620, 634, and 918), and number of RET polymorphisms.
| Risk alleles | PHEO (+)a | PHEO (−)b | OR (95% CI) | P |
|---|---|---|---|---|
| None | 14 (34.1) | 39 (41.5) | 1 | |
| One | 17 (41.5) | 43 (45.7) | 1.3 (0.8–2.3) | 0.365 |
| Two | 10 (24.4) | 12 (12.8) | 2.63 (1.4–5.0) | 0.004 |
| Risk alleles | PHEO (+)a | PHEO (−)b | OR (95% CI) | P |
|---|---|---|---|---|
| None | 14 (34.1) | 39 (41.5) | 1 | |
| One | 17 (41.5) | 43 (45.7) | 1.3 (0.8–2.3) | 0.365 |
| Two | 10 (24.4) | 12 (12.8) | 2.63 (1.4–5.0) | 0.004 |
Data available for 41 patients with PHEO.
Data available for 94 patients without PHEO.
Patients were subsequently assembled according to the number of genetic variants into three groups: zero, one, or two genetic variants. First, we evaluated whether the presence of multiple RET variants would increase the estimated risk for PHEO development. Poisson regression analysis showed that the presence of two RET risk alleles was associated with a 2.63-fold increased risk for PHEO development (Table 4). We also observed a significant difference in age at the time of diagnosis of PHEO between patients without or with one variant and patients who harbored two genetic variants (39.3±14.4 and 29.6±6.3 years respectively; P=0.006), suggesting an age-related effect on PHEO penetrance. To test this hypothesis, we used the Kaplan–Meier model. As gene dysfunction is present from birth, we assumed that the individual age at the time of diagnosis would indicate the period of exposure. Kaplan–Meier estimates of cumulative PHEO diagnosis yielded distinct curves for patients harboring no polymorphism or one or three RET polymorphisms (P=0.003; Fig. 1). In patients with two polymorphisms, one polymorphism, or no polymorphisms, 50% penetrance was achieved by the ages of 28.9, 39, and 47.7 years respectively (P=0.003; Fig. 1).
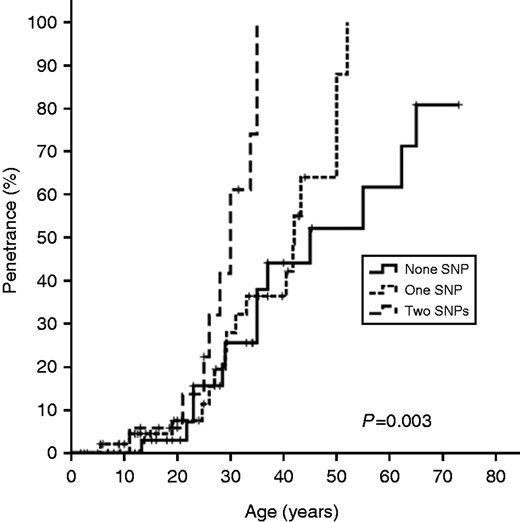
Kaplan–Meier estimates for the proportion of MEN2 patients who developed pheochromocytoma. The log-rank test was used to compare curves.
The Cox proportional hazard survival test was used to assess whether the observed effect of multiple variant alleles on PHEO penetrance was independent of the specific germline mutation. Patients who harbored two RET variants exhibited an increased risk for early PHEO development regardless of the RET mutation (hazard ratio (HR), 5.99 (95% CI, 2.24–16.03); Table 5).
Additive effect of RET polymorphic alleles on pheochromocytoma susceptibility (n=135). The independent variables included in the analysis of Cox regression were numbers of RET polymorphic alleles and RET germline mutations (codons 618, 620, 634, and 918).
| Risk alleles | b | P | HR | CI |
|---|---|---|---|---|
| None | – | – | 1 | – |
| One | 0.40 | 0.31 | 1.49 | (0.68–3.27) |
| Two | 1.79 | 0.000 | 5.99 | (2.24–16.03) |
| Risk alleles | b | P | HR | CI |
|---|---|---|---|---|
| None | – | – | 1 | – |
| One | 0.40 | 0.31 | 1.49 | (0.68–3.27) |
| Two | 1.79 | 0.000 | 5.99 | (2.24–16.03) |
b, Regression coefficient; HR, hazard ratio.
Additive effect of RET polymorphic alleles on pheochromocytoma susceptibility (n=135). The independent variables included in the analysis of Cox regression were numbers of RET polymorphic alleles and RET germline mutations (codons 618, 620, 634, and 918).
| Risk alleles | b | P | HR | CI |
|---|---|---|---|---|
| None | – | – | 1 | – |
| One | 0.40 | 0.31 | 1.49 | (0.68–3.27) |
| Two | 1.79 | 0.000 | 5.99 | (2.24–16.03) |
| Risk alleles | b | P | HR | CI |
|---|---|---|---|---|
| None | – | – | 1 | – |
| One | 0.40 | 0.31 | 1.49 | (0.68–3.27) |
| Two | 1.79 | 0.000 | 5.99 | (2.24–16.03) |
b, Regression coefficient; HR, hazard ratio.
Discussion
PHEO is present in ∼50% of patients with MEN2 syndrome. RET mutations at codon 634 or 918 are associated with the presence of PHEO; however, the reasons for incomplete penetrance have not been established. In this study, we demonstrate that patients carrying multiple RET variants have an increased risk for development of PHEO. Individuals carrying two RET polymorphisms were younger than those carrying one or no polymorphism at the time of diagnosis, which suggests an age-related effect on PHEO penetrance.
The relationship between specific RET proto-oncogene mutations and the presence or absence of endocrine neoplasias in MEN2 syndrome has been demonstrated. Mutations at codon 634 (ATA level B) are associated with the development of PHEO in ∼29–50% of MEN2A patients (14, 27). PHEO is also present in ∼50% of patients harboring the MEN2B-specific RET codon 918 mutation (ATA level D) (7). By contrast, PHEO is rarely present in patients with RET mutations in exon 13 (codons 768, 790, and 791), exon 14 (codons 804 and 844), or exon 15 (codon 891) (all ATA level A risk mutations). PHEO is usually diagnosed after MTC. Machens et al. (27) studied a cohort of MEN2 patients with a mean observation period of 27 years and observed that PHEO developed in 28, 21, and 3% of RET carriers in the highest-risk category (mutations in codon 918), high-risk category (mutations in codons 634, 630, 609, 611, 618, and 620), and least-high-risk category (mutations in codons 768, 790, 791, 804, and 891) respectively. The time of diagnosis of PHEO differed significantly according to the RET risk category with means of 26.4 years (highest risk), 34.9 years (high risk), and 46.5 years (least high risk). Despite the cumulative knowledge available regarding molecular mechanisms, the reasons why some patients do not develop PHEO remain unknown.
Several studies have investigated the effect of neutral RET sequence variants on the disease presentation of MEN2-related MTC (15, 16, 17, 20, 21). However, we found only one study that evaluated the potential role of RET variants on the penetrance or clinical course of MEN2-associated PHEO. Fernandez et al. (28) investigated the effect of RET polymorphisms/haplotypes as modifier loci for MEN2 and analyzed the correlation with the type of RET mutation in a series of 114 Spanish probands but found no association of any RET sequence variants and the presence of PHEO. In this study, we evaluated the RET polymorphisms L769L, S836S, and G691S/S904S in a large cohort of MEN2 patients followed at our institution. We did not detect any significant association between a single RET genetic variant and the development of PHEO (all P>0.05).
Several lines of evidence indicate that tumorigenesis in humans is a multistep process (29). Considering that two or more distinct genetic changes may be necessary for tumor development and progression, it is reasonable to consider the analysis of combined effects of polymorphisms rather than the effect of a single genetic variant. Thus, a potential additive effect of gene polymorphisms on the development of several diseases, including susceptibility for cancer, has attracted much attention in recent studies (30, 31, 32, 33). In prostate cancer, the gene–gene interaction of vascular endothelial growth factor (VEGF(VEGFA)) and thrombospondin 1 (TSP1(THBS1)) polymorphisms increased the estimated risk of an aggressive phenotype. Interestingly, a significant effect of gene dosage on the number of potential high-risk genotypes was observed (31). An additive effect of gene polymorphisms on tumor necrosis factor α (TNFα) and nuclear factor of κ light chain gene enhancer inactivated B cells (NFκB) has been demonstrated for susceptibility of esophageal squamous cell carcinoma in a population of northern India. Patients with one or more risk allele presented with a 1.67-fold increased risk for this malignancy (33).
We have recently investigated the effect of multiple RET polymorphic alleles on the susceptibility and tumor aggressiveness of sporadic MTC. Individuals harboring haplotypes with three or more polymorphic alleles showed a 3.79-fold increased risk for MTC development. Remarkably, we also found an association between the number of RET risk alleles and advanced disease at the time of diagnosis, indicating that the analysis of RET haplotypes may provide a higher predictive value than a study of single polymorphisms (22). In this study, we investigated the effect of multiple RET variants on MEN2-associated PHEO penetrance. We observed an association between the number of RET risk alleles and PHEO susceptibility and age-related penetrance. Individuals harboring two RET polymorphic alleles not only presented with a 2.6-fold increase in the estimated risk of PHEO (95% CI, 1.4–5.0; P=0.004) but also were younger at the time of diagnosis when compared with those with one or no polymorphism (29.6±6.3 and 39.3±14.4 years respectively; P=0.006). Accordingly, the Cox proportional hazard survival model identified the presence of two polymorphic alleles as an independent risk factor for PHEO development in MEN2 patients (HR, 5.99 (95% CI, 2.24–16.03); P<0.0001).
The molecular mechanism by which these RET polymorphisms may affect the development and evolution of MTC has not been established. Quantitative studies of mRNA levels in tumor tissues showed similar levels of RET expression in individuals with or without these polymorphisms (17). It has been suggested that base exchange in the DNA molecule could create a new alternative splicing site, leading to erroneous ligand or microRNA binding; altered mRNA structure, stability, or copy number; or altered synthesis of a truncated protein (34). It is also conceivable that these neutral variants are in LD with an as yet unknown functional variant (21). Indeed, we have recently shown that the RET 3′-UTR variants rs76759170 and rs3026785 are in strong LD with the L769L (|D′|=−1; r2=0.16) and S836S (|D′|=−1; r2=0.989) variants (Ceolin L, Siqueira DR, Romitti M, Ferreira CV & Maia AL 2013, unpublished observations). These biological interactions may influence the occurrence of somatic mutations or alternatively may indirectly reflect other polymorphisms that are in LD with these genotyped polymorphisms and thus contribute to tumorigenesis.
In conclusion, our results indicate a synergistic effect of RET risk alleles on the susceptibility and age-related penetrance in MEN2-associated PHEO. However, considering that the development of human tumors is affected by the individual's genetic background, it is essential to evaluate the role of RET polymorphisms in other populations.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil), Fundação de Amparo Pesquisa do Estado do Rio Grande do Sul (FAPERGS, Brazil), Fundo de Incentivo à Pesquisa do Hospital de Clínicas de Porto Alegre (FIPE, Brazil), and Programa de Apoio a Núcleos de Excelência (PRONEX, Brazil).
References
Author notes
(A L Maia is now at Serviço de Endocrinologia, Hospital de Clnicas de Porto Alegre, Rua Ramiro Barcelos 2350, 90035-003 Porto Alegre, Rio Grande do Sul, Brazil).

{kind=link}