





Abstract
Gliptins are accepted as a standard therapy for diabetes mellitus today. By inhibition of the enzyme dipeptidyl peptidase 4 (DPP4), gliptins prolong the GLP1-dependent insulin secretion in the pancreatic β-cells and thus support physiological blood glucose control. Various studies have now raised hope for an additional protective effect of pharmacological DPP4 inhibition in vascular diseases. Besides GLP1, especially, the inhibition of SDF1 cleavage has been shown to depict a relevant mechanism to enhance endothelial regeneration and reduce atherosclerosis progression via the SDF1–CXCR4 axis. Furthermore, several clinical trials have now shown an excellent safety profile of gliptin therapy in cardiovascular risk patients. In this review, we give a comprehensive overview on DPP4-dependent vascular functions and pathophysiological mechanisms with a detailed discussion of the underlying molecular mechanisms. We further analyse the role of pharmacological DPP4 inhibitors and their potential therapeutic impact on endothelial function and regeneration besides their effect during atherosclerosis development. Finally, we discuss presently available data from in vitro and in vivo studies with respect to the results of the recent clinical trials in diabetic and non-diabetic patients.
Introduction
Gliptins can pharmacologically inhibit the dipeptidyl peptidase 4 (DPP4). Their clinical use until today is restricted to the treatment of hyperglycaemia in type 2 diabetic (D.m.2) patients according to the ESC guidelines on diabetes and cardiovascular disease.1 While Sitagliptin was the first active substance authorized in 2007,2 various other gliptins have been approved within recent years.
By inhibiting DPP4, gliptins prolong insulin secretion from pancreatic β-cells via stabilization of GLP1/GIP.3 Due to their glucose-dependent effects on diabetes control, gliptins show beneficial effects on blood glucose and HbA1c without a relevant risk for hypoglycaemia or gain of body weight.2 Additionally assumed side effects like acute pancreatitis, higher probability of cardiovascular events, or the occurrence of heart failure are not increased in Sitagliptin- or Alogliptin-treated patients (TECOS and EXAMINE trials).4,5
Besides the primary positive gliptin effects, especially, preclinical data on DPP4 inhibitors could show several other beneficial effects like an enhanced regeneration of endothelial or myocardial injury6,7 or a reduction of atherosclerosis development.8,9 These positive gliptin side effects might depict a tremendous protective impact on vascular function, disease, and regeneration. Thus, several preclinical and clinical studies recently characterized gliptin-mediated therapeutic effects on the vascular system (Tables 1–3).
Impact of gliptin treatment on endothelial function
| Category | Active substance | Results |
|---|---|---|
| In vitro (aortic segments of C57Bl/6 mice)15 | Alogliptin | Alogliptin incubation-mediated dose-dependent relaxation of the aortic segments, whereas GLP1 pre-incubation had no effect. Endothelial denudation and addition of LNMMA reduced the effect of Alogliptin incubation |
| In vitro (aortic segments of ApoE-ko mice)9 | Des-Fluoro-Sitagliptin (DFS, 200 mg/kg/day) | Aortic segments of DFS-treated mice on high-fat diet showed an improved vascular function and significant higher eNOs phosphorylation after stimulation with acetylcholine |
| In vitro (Wistar rats)48 | Linagliptin (83 mg/kg) | The LPS-induced vascular dysfunction of rat aortic segments was significantly improved by Linagliptin treatment (e.g. reduced oxidative burst and suppressed attachment of human neutrophils on activated endothelial cells) |
| Clinical trial (10 volunteers; 10 days)28 | Alogliptin (25 mg/day) | Alogliptin significantly improved post-prandial endothelial function (increased FMD) and post-prandial lipaemia in healthy volunteers |
| Clinical trial (31 D.m.2 patients with insufficient blood glucose control, EDGE study; 12 weeks)49 | Sitagliptin (50 mg/day) | Sitagliptin treatment improved FMD, several blood parameters (e.g. cholesterol, HDL, lipids, GLP1, GIP, HbA1, and inflammatory markers), and the number of circulating CD34+ cells |
| Category | Active substance | Results |
|---|---|---|
| In vitro (aortic segments of C57Bl/6 mice)15 | Alogliptin | Alogliptin incubation-mediated dose-dependent relaxation of the aortic segments, whereas GLP1 pre-incubation had no effect. Endothelial denudation and addition of LNMMA reduced the effect of Alogliptin incubation |
| In vitro (aortic segments of ApoE-ko mice)9 | Des-Fluoro-Sitagliptin (DFS, 200 mg/kg/day) | Aortic segments of DFS-treated mice on high-fat diet showed an improved vascular function and significant higher eNOs phosphorylation after stimulation with acetylcholine |
| In vitro (Wistar rats)48 | Linagliptin (83 mg/kg) | The LPS-induced vascular dysfunction of rat aortic segments was significantly improved by Linagliptin treatment (e.g. reduced oxidative burst and suppressed attachment of human neutrophils on activated endothelial cells) |
| Clinical trial (10 volunteers; 10 days)28 | Alogliptin (25 mg/day) | Alogliptin significantly improved post-prandial endothelial function (increased FMD) and post-prandial lipaemia in healthy volunteers |
| Clinical trial (31 D.m.2 patients with insufficient blood glucose control, EDGE study; 12 weeks)49 | Sitagliptin (50 mg/day) | Sitagliptin treatment improved FMD, several blood parameters (e.g. cholesterol, HDL, lipids, GLP1, GIP, HbA1, and inflammatory markers), and the number of circulating CD34+ cells |
LNMMA, N-Monomethyl-L-arginine.
Impact of gliptin treatment on endothelial function
| Category | Active substance | Results |
|---|---|---|
| In vitro (aortic segments of C57Bl/6 mice)15 | Alogliptin | Alogliptin incubation-mediated dose-dependent relaxation of the aortic segments, whereas GLP1 pre-incubation had no effect. Endothelial denudation and addition of LNMMA reduced the effect of Alogliptin incubation |
| In vitro (aortic segments of ApoE-ko mice)9 | Des-Fluoro-Sitagliptin (DFS, 200 mg/kg/day) | Aortic segments of DFS-treated mice on high-fat diet showed an improved vascular function and significant higher eNOs phosphorylation after stimulation with acetylcholine |
| In vitro (Wistar rats)48 | Linagliptin (83 mg/kg) | The LPS-induced vascular dysfunction of rat aortic segments was significantly improved by Linagliptin treatment (e.g. reduced oxidative burst and suppressed attachment of human neutrophils on activated endothelial cells) |
| Clinical trial (10 volunteers; 10 days)28 | Alogliptin (25 mg/day) | Alogliptin significantly improved post-prandial endothelial function (increased FMD) and post-prandial lipaemia in healthy volunteers |
| Clinical trial (31 D.m.2 patients with insufficient blood glucose control, EDGE study; 12 weeks)49 | Sitagliptin (50 mg/day) | Sitagliptin treatment improved FMD, several blood parameters (e.g. cholesterol, HDL, lipids, GLP1, GIP, HbA1, and inflammatory markers), and the number of circulating CD34+ cells |
| Category | Active substance | Results |
|---|---|---|
| In vitro (aortic segments of C57Bl/6 mice)15 | Alogliptin | Alogliptin incubation-mediated dose-dependent relaxation of the aortic segments, whereas GLP1 pre-incubation had no effect. Endothelial denudation and addition of LNMMA reduced the effect of Alogliptin incubation |
| In vitro (aortic segments of ApoE-ko mice)9 | Des-Fluoro-Sitagliptin (DFS, 200 mg/kg/day) | Aortic segments of DFS-treated mice on high-fat diet showed an improved vascular function and significant higher eNOs phosphorylation after stimulation with acetylcholine |
| In vitro (Wistar rats)48 | Linagliptin (83 mg/kg) | The LPS-induced vascular dysfunction of rat aortic segments was significantly improved by Linagliptin treatment (e.g. reduced oxidative burst and suppressed attachment of human neutrophils on activated endothelial cells) |
| Clinical trial (10 volunteers; 10 days)28 | Alogliptin (25 mg/day) | Alogliptin significantly improved post-prandial endothelial function (increased FMD) and post-prandial lipaemia in healthy volunteers |
| Clinical trial (31 D.m.2 patients with insufficient blood glucose control, EDGE study; 12 weeks)49 | Sitagliptin (50 mg/day) | Sitagliptin treatment improved FMD, several blood parameters (e.g. cholesterol, HDL, lipids, GLP1, GIP, HbA1, and inflammatory markers), and the number of circulating CD34+ cells |
LNMMA, N-Monomethyl-L-arginine.
Effects of pharmacological dipeptidyl peptidase 4 inhibition on endothelial regeneration
| Category | Active substance | Results |
|---|---|---|
| In vitro (HAoECs, scratch assay)30 | CXCL12 (SDF1) | CXCL12 stimulation enhanced migration of the CXCR4-expressing HAoECs into scratch area and AMD3100 reversed that effect. This proves the involvement of SDF1–CXCR4 signalling in endothelial regeneration |
| in vitro (human monocytes)38 | MCP1 (10 nmol/L) RANTES (100 ng/mL) | GLP1-pretreatment reduced monocyte-migration independent from the type of chemoattractant. This suggests that increased GLP1 levels can lead to reduced recruitment of inflammatory cells |
| In vivo (C57Bl/6 mice)6 | Sitagliptin (500 mg/kg/day) | DPP4 inhibition enhanced endothelial regeneration (via CXCR4/SDF1 pathway) and recruitment of ciPCs after electric endothelial injury. AMD3100 reversed the Sitagliptin effect |
| In vivo (CXCR4-ko, ApoE-ko, and DKO mice)30 | None | DKO mice showed an impaired re-endothelialization of carotid lesions after wire injury and an increased neointima formation compared with common ApoE-ko animals |
| Clinical trial (40 patients, D.m.2; 12 weeks)50 | Sitagliptin (50 mg/day) | Sitagliptin medication improved FMD, HbA1c, and increased adiponectin levels |
| Clinical trial (32 patients, D.m.2; 4 weeks)31 | Sitagliptin (100 mg/day) | Sitagliptin reduced DPP4 activity, increased number of ciPCs and SDF1 levels. MCP-1 levels decreased, VEGF levels remained unchanged |
| Clinical trial (76 patients with stable CAD and mild D.m.2 or impaired glucose tolerance; 1 year)35 | Sitagliptin (100 mg/day) | The Sitagliptin-treated patients showed less IMT progression, greater body-weight reduction, better 2 h values of blood glucose after OGT and improved HbA1c and LDL values |
| Category | Active substance | Results |
|---|---|---|
| In vitro (HAoECs, scratch assay)30 | CXCL12 (SDF1) | CXCL12 stimulation enhanced migration of the CXCR4-expressing HAoECs into scratch area and AMD3100 reversed that effect. This proves the involvement of SDF1–CXCR4 signalling in endothelial regeneration |
| in vitro (human monocytes)38 | MCP1 (10 nmol/L) RANTES (100 ng/mL) | GLP1-pretreatment reduced monocyte-migration independent from the type of chemoattractant. This suggests that increased GLP1 levels can lead to reduced recruitment of inflammatory cells |
| In vivo (C57Bl/6 mice)6 | Sitagliptin (500 mg/kg/day) | DPP4 inhibition enhanced endothelial regeneration (via CXCR4/SDF1 pathway) and recruitment of ciPCs after electric endothelial injury. AMD3100 reversed the Sitagliptin effect |
| In vivo (CXCR4-ko, ApoE-ko, and DKO mice)30 | None | DKO mice showed an impaired re-endothelialization of carotid lesions after wire injury and an increased neointima formation compared with common ApoE-ko animals |
| Clinical trial (40 patients, D.m.2; 12 weeks)50 | Sitagliptin (50 mg/day) | Sitagliptin medication improved FMD, HbA1c, and increased adiponectin levels |
| Clinical trial (32 patients, D.m.2; 4 weeks)31 | Sitagliptin (100 mg/day) | Sitagliptin reduced DPP4 activity, increased number of ciPCs and SDF1 levels. MCP-1 levels decreased, VEGF levels remained unchanged |
| Clinical trial (76 patients with stable CAD and mild D.m.2 or impaired glucose tolerance; 1 year)35 | Sitagliptin (100 mg/day) | The Sitagliptin-treated patients showed less IMT progression, greater body-weight reduction, better 2 h values of blood glucose after OGT and improved HbA1c and LDL values |
OGT, oral glucose tolerance test.
Effects of pharmacological dipeptidyl peptidase 4 inhibition on endothelial regeneration
| Category | Active substance | Results |
|---|---|---|
| In vitro (HAoECs, scratch assay)30 | CXCL12 (SDF1) | CXCL12 stimulation enhanced migration of the CXCR4-expressing HAoECs into scratch area and AMD3100 reversed that effect. This proves the involvement of SDF1–CXCR4 signalling in endothelial regeneration |
| in vitro (human monocytes)38 | MCP1 (10 nmol/L) RANTES (100 ng/mL) | GLP1-pretreatment reduced monocyte-migration independent from the type of chemoattractant. This suggests that increased GLP1 levels can lead to reduced recruitment of inflammatory cells |
| In vivo (C57Bl/6 mice)6 | Sitagliptin (500 mg/kg/day) | DPP4 inhibition enhanced endothelial regeneration (via CXCR4/SDF1 pathway) and recruitment of ciPCs after electric endothelial injury. AMD3100 reversed the Sitagliptin effect |
| In vivo (CXCR4-ko, ApoE-ko, and DKO mice)30 | None | DKO mice showed an impaired re-endothelialization of carotid lesions after wire injury and an increased neointima formation compared with common ApoE-ko animals |
| Clinical trial (40 patients, D.m.2; 12 weeks)50 | Sitagliptin (50 mg/day) | Sitagliptin medication improved FMD, HbA1c, and increased adiponectin levels |
| Clinical trial (32 patients, D.m.2; 4 weeks)31 | Sitagliptin (100 mg/day) | Sitagliptin reduced DPP4 activity, increased number of ciPCs and SDF1 levels. MCP-1 levels decreased, VEGF levels remained unchanged |
| Clinical trial (76 patients with stable CAD and mild D.m.2 or impaired glucose tolerance; 1 year)35 | Sitagliptin (100 mg/day) | The Sitagliptin-treated patients showed less IMT progression, greater body-weight reduction, better 2 h values of blood glucose after OGT and improved HbA1c and LDL values |
| Category | Active substance | Results |
|---|---|---|
| In vitro (HAoECs, scratch assay)30 | CXCL12 (SDF1) | CXCL12 stimulation enhanced migration of the CXCR4-expressing HAoECs into scratch area and AMD3100 reversed that effect. This proves the involvement of SDF1–CXCR4 signalling in endothelial regeneration |
| in vitro (human monocytes)38 | MCP1 (10 nmol/L) RANTES (100 ng/mL) | GLP1-pretreatment reduced monocyte-migration independent from the type of chemoattractant. This suggests that increased GLP1 levels can lead to reduced recruitment of inflammatory cells |
| In vivo (C57Bl/6 mice)6 | Sitagliptin (500 mg/kg/day) | DPP4 inhibition enhanced endothelial regeneration (via CXCR4/SDF1 pathway) and recruitment of ciPCs after electric endothelial injury. AMD3100 reversed the Sitagliptin effect |
| In vivo (CXCR4-ko, ApoE-ko, and DKO mice)30 | None | DKO mice showed an impaired re-endothelialization of carotid lesions after wire injury and an increased neointima formation compared with common ApoE-ko animals |
| Clinical trial (40 patients, D.m.2; 12 weeks)50 | Sitagliptin (50 mg/day) | Sitagliptin medication improved FMD, HbA1c, and increased adiponectin levels |
| Clinical trial (32 patients, D.m.2; 4 weeks)31 | Sitagliptin (100 mg/day) | Sitagliptin reduced DPP4 activity, increased number of ciPCs and SDF1 levels. MCP-1 levels decreased, VEGF levels remained unchanged |
| Clinical trial (76 patients with stable CAD and mild D.m.2 or impaired glucose tolerance; 1 year)35 | Sitagliptin (100 mg/day) | The Sitagliptin-treated patients showed less IMT progression, greater body-weight reduction, better 2 h values of blood glucose after OGT and improved HbA1c and LDL values |
OGT, oral glucose tolerance test.
Impact of gliptin therapy on atherosclerosis development
| Category | Active substance | Results |
|---|---|---|
| In vitro (rat SMC)11 | Anagliptin (100 µM) | The Anagliptin-pretreated and LPS/DPP4-stimulated rat SMC showed a suppressed SMC proliferation and TNFα expression |
| In vitro (human mononuclear cells and human aortic endothelial cells)29 | Alogliptin (0.5/1/5 nM) | The addition of Alogliptin inhibited IL6- and IL-1β-expression in the LPS-stimulated cells |
| In vitro (THP-1 macrophages)39 | Sitagliptin (25 µM) and NVPDPP728 (270 µM) | DPP4 inhibition suppressed NLRP3, TLR4, and IL-1 β-expression in human macrophages |
| In vivo (ApoE-ko and db/db diabetic mice)13 | Vildagliptin (3 mg/kg/day) | Vildagliptin treatment suppressed atherosclerosis progression and foam cell formation in the mice. GLP1/GIP levels were increased and the positive effects could be reversed only partially by GLP1/GIP-receptor blockers |
| In vivo (ApoE-ko mice)11 | Anagliptin (0.3% in diet) | Anagliptin reduced atherosclerotic plaque burden and vascular lesions showed a reduced number of infiltrating monocytes/macrophages. Anagliptin treatment did not significantly alter ciPCs and SDF1 levels |
| In vivo (LDL-R-ko mice)36 | Alogliptin (40 mg/kg/day) | Treatment with Alogliptin improved metabolic indices and blood pressure. Abdominal fat tissue showed smaller adipocytes and a reduced macrophage content was observed in the aortic wall in high-fat diet fed mice |
| In vivo (ApoE-ko mice)8 | Sitagliptin (500 mg/kg/day) | Sitagliptin treatment significantly reduced the extent of atherosclerotic plaques. Monocytes differentiation was primed towards the reparative M2 subtype in mice on high-fat diet. Mechanistically, the Sitagliptin effect via the SDF1–CXCR4 pathway was proven by the addition of AMD3100, which reversed the Sitagliptin effect |
| Clinical trial (TRUST) (20 patients suffering coronary atherosclerosis and D.m.2; 48 weeks; ongoing)42 | Sitagliptin (50 mg/kg) | Patients suffering from CAD and D.m.2 were randomized to treatment with Sitagliptin or Placebo after successful PCI. Development of atherosclerosis is evaluated by IVUS (six centres, Japan) |
| Clinical trial (PROLOGUE) (463 patients suffering D.m.2; 12–24 months; not published)41 | Sitagliptin (50–100 mg/kg) | IMT is evaluated using ultrasound of the carotid artery (Sitagliptin vs. placebo) |
| Clinical trial (SPIKE) (282 hyperglycaemic D.m.2 patients, insufficient blood glucose control under insulin therapy; 2 years; ongoing, expected completion June 2016)40 | Sitagliptin (25 or 50 mg/kg) | This study randomizes patients to treatment with Sitagliptin or placebo. Blood chemistry is evaluated periodically, carotid IMT will be measured by ultrasound |
| Category | Active substance | Results |
|---|---|---|
| In vitro (rat SMC)11 | Anagliptin (100 µM) | The Anagliptin-pretreated and LPS/DPP4-stimulated rat SMC showed a suppressed SMC proliferation and TNFα expression |
| In vitro (human mononuclear cells and human aortic endothelial cells)29 | Alogliptin (0.5/1/5 nM) | The addition of Alogliptin inhibited IL6- and IL-1β-expression in the LPS-stimulated cells |
| In vitro (THP-1 macrophages)39 | Sitagliptin (25 µM) and NVPDPP728 (270 µM) | DPP4 inhibition suppressed NLRP3, TLR4, and IL-1 β-expression in human macrophages |
| In vivo (ApoE-ko and db/db diabetic mice)13 | Vildagliptin (3 mg/kg/day) | Vildagliptin treatment suppressed atherosclerosis progression and foam cell formation in the mice. GLP1/GIP levels were increased and the positive effects could be reversed only partially by GLP1/GIP-receptor blockers |
| In vivo (ApoE-ko mice)11 | Anagliptin (0.3% in diet) | Anagliptin reduced atherosclerotic plaque burden and vascular lesions showed a reduced number of infiltrating monocytes/macrophages. Anagliptin treatment did not significantly alter ciPCs and SDF1 levels |
| In vivo (LDL-R-ko mice)36 | Alogliptin (40 mg/kg/day) | Treatment with Alogliptin improved metabolic indices and blood pressure. Abdominal fat tissue showed smaller adipocytes and a reduced macrophage content was observed in the aortic wall in high-fat diet fed mice |
| In vivo (ApoE-ko mice)8 | Sitagliptin (500 mg/kg/day) | Sitagliptin treatment significantly reduced the extent of atherosclerotic plaques. Monocytes differentiation was primed towards the reparative M2 subtype in mice on high-fat diet. Mechanistically, the Sitagliptin effect via the SDF1–CXCR4 pathway was proven by the addition of AMD3100, which reversed the Sitagliptin effect |
| Clinical trial (TRUST) (20 patients suffering coronary atherosclerosis and D.m.2; 48 weeks; ongoing)42 | Sitagliptin (50 mg/kg) | Patients suffering from CAD and D.m.2 were randomized to treatment with Sitagliptin or Placebo after successful PCI. Development of atherosclerosis is evaluated by IVUS (six centres, Japan) |
| Clinical trial (PROLOGUE) (463 patients suffering D.m.2; 12–24 months; not published)41 | Sitagliptin (50–100 mg/kg) | IMT is evaluated using ultrasound of the carotid artery (Sitagliptin vs. placebo) |
| Clinical trial (SPIKE) (282 hyperglycaemic D.m.2 patients, insufficient blood glucose control under insulin therapy; 2 years; ongoing, expected completion June 2016)40 | Sitagliptin (25 or 50 mg/kg) | This study randomizes patients to treatment with Sitagliptin or placebo. Blood chemistry is evaluated periodically, carotid IMT will be measured by ultrasound |
Impact of gliptin therapy on atherosclerosis development
| Category | Active substance | Results |
|---|---|---|
| In vitro (rat SMC)11 | Anagliptin (100 µM) | The Anagliptin-pretreated and LPS/DPP4-stimulated rat SMC showed a suppressed SMC proliferation and TNFα expression |
| In vitro (human mononuclear cells and human aortic endothelial cells)29 | Alogliptin (0.5/1/5 nM) | The addition of Alogliptin inhibited IL6- and IL-1β-expression in the LPS-stimulated cells |
| In vitro (THP-1 macrophages)39 | Sitagliptin (25 µM) and NVPDPP728 (270 µM) | DPP4 inhibition suppressed NLRP3, TLR4, and IL-1 β-expression in human macrophages |
| In vivo (ApoE-ko and db/db diabetic mice)13 | Vildagliptin (3 mg/kg/day) | Vildagliptin treatment suppressed atherosclerosis progression and foam cell formation in the mice. GLP1/GIP levels were increased and the positive effects could be reversed only partially by GLP1/GIP-receptor blockers |
| In vivo (ApoE-ko mice)11 | Anagliptin (0.3% in diet) | Anagliptin reduced atherosclerotic plaque burden and vascular lesions showed a reduced number of infiltrating monocytes/macrophages. Anagliptin treatment did not significantly alter ciPCs and SDF1 levels |
| In vivo (LDL-R-ko mice)36 | Alogliptin (40 mg/kg/day) | Treatment with Alogliptin improved metabolic indices and blood pressure. Abdominal fat tissue showed smaller adipocytes and a reduced macrophage content was observed in the aortic wall in high-fat diet fed mice |
| In vivo (ApoE-ko mice)8 | Sitagliptin (500 mg/kg/day) | Sitagliptin treatment significantly reduced the extent of atherosclerotic plaques. Monocytes differentiation was primed towards the reparative M2 subtype in mice on high-fat diet. Mechanistically, the Sitagliptin effect via the SDF1–CXCR4 pathway was proven by the addition of AMD3100, which reversed the Sitagliptin effect |
| Clinical trial (TRUST) (20 patients suffering coronary atherosclerosis and D.m.2; 48 weeks; ongoing)42 | Sitagliptin (50 mg/kg) | Patients suffering from CAD and D.m.2 were randomized to treatment with Sitagliptin or Placebo after successful PCI. Development of atherosclerosis is evaluated by IVUS (six centres, Japan) |
| Clinical trial (PROLOGUE) (463 patients suffering D.m.2; 12–24 months; not published)41 | Sitagliptin (50–100 mg/kg) | IMT is evaluated using ultrasound of the carotid artery (Sitagliptin vs. placebo) |
| Clinical trial (SPIKE) (282 hyperglycaemic D.m.2 patients, insufficient blood glucose control under insulin therapy; 2 years; ongoing, expected completion June 2016)40 | Sitagliptin (25 or 50 mg/kg) | This study randomizes patients to treatment with Sitagliptin or placebo. Blood chemistry is evaluated periodically, carotid IMT will be measured by ultrasound |
| Category | Active substance | Results |
|---|---|---|
| In vitro (rat SMC)11 | Anagliptin (100 µM) | The Anagliptin-pretreated and LPS/DPP4-stimulated rat SMC showed a suppressed SMC proliferation and TNFα expression |
| In vitro (human mononuclear cells and human aortic endothelial cells)29 | Alogliptin (0.5/1/5 nM) | The addition of Alogliptin inhibited IL6- and IL-1β-expression in the LPS-stimulated cells |
| In vitro (THP-1 macrophages)39 | Sitagliptin (25 µM) and NVPDPP728 (270 µM) | DPP4 inhibition suppressed NLRP3, TLR4, and IL-1 β-expression in human macrophages |
| In vivo (ApoE-ko and db/db diabetic mice)13 | Vildagliptin (3 mg/kg/day) | Vildagliptin treatment suppressed atherosclerosis progression and foam cell formation in the mice. GLP1/GIP levels were increased and the positive effects could be reversed only partially by GLP1/GIP-receptor blockers |
| In vivo (ApoE-ko mice)11 | Anagliptin (0.3% in diet) | Anagliptin reduced atherosclerotic plaque burden and vascular lesions showed a reduced number of infiltrating monocytes/macrophages. Anagliptin treatment did not significantly alter ciPCs and SDF1 levels |
| In vivo (LDL-R-ko mice)36 | Alogliptin (40 mg/kg/day) | Treatment with Alogliptin improved metabolic indices and blood pressure. Abdominal fat tissue showed smaller adipocytes and a reduced macrophage content was observed in the aortic wall in high-fat diet fed mice |
| In vivo (ApoE-ko mice)8 | Sitagliptin (500 mg/kg/day) | Sitagliptin treatment significantly reduced the extent of atherosclerotic plaques. Monocytes differentiation was primed towards the reparative M2 subtype in mice on high-fat diet. Mechanistically, the Sitagliptin effect via the SDF1–CXCR4 pathway was proven by the addition of AMD3100, which reversed the Sitagliptin effect |
| Clinical trial (TRUST) (20 patients suffering coronary atherosclerosis and D.m.2; 48 weeks; ongoing)42 | Sitagliptin (50 mg/kg) | Patients suffering from CAD and D.m.2 were randomized to treatment with Sitagliptin or Placebo after successful PCI. Development of atherosclerosis is evaluated by IVUS (six centres, Japan) |
| Clinical trial (PROLOGUE) (463 patients suffering D.m.2; 12–24 months; not published)41 | Sitagliptin (50–100 mg/kg) | IMT is evaluated using ultrasound of the carotid artery (Sitagliptin vs. placebo) |
| Clinical trial (SPIKE) (282 hyperglycaemic D.m.2 patients, insufficient blood glucose control under insulin therapy; 2 years; ongoing, expected completion June 2016)40 | Sitagliptin (25 or 50 mg/kg) | This study randomizes patients to treatment with Sitagliptin or placebo. Blood chemistry is evaluated periodically, carotid IMT will be measured by ultrasound |
Effects of dipeptidyl peptidase 4
The DPP4 (or CD26) is a member of the prolyl-oligopeptidase superfamily, which comprises three enzyme subtypes (DPP8, DPP9, and FAP) and two non-enzymes (DPP10 and DPP4-like-peptide 6)10 besides the DPP4. Regarding vascular function and regeneration, DPP4 is well analysed and involved in numerous enzymatic and few non-enzymatic signalling pathways. The enzymatic activity of DPP4 in the vascular system is pathophysiologically most relevant in insulin/glucose metabolism, recruitment of circulating progenitor cells (ciPCs) to injured tissues, development of atherosclerosis, and vascular inflammation.6,8,11 Besides SDF1 and GLP1/GIP, DPP4 is involved in the cleavage of N-terminal dipeptides from macrophage-derived cytokine (MDC, CCL22), Eotaxin (CCL11), regulated on activation, normal T cell expressed and secreted (RANTES) (CCL5), inducible T-cell-α-chemoattractant [Interferon-inducible T-cell α chemoattractant (ITAC), CXCL11], interferon γ-inducible-protein [Interferon γ-induced protein 10 (IP10), CXCL10], Monokine induced by gamma interferon (MIG) (CXCL9) among others that are all involved in vascular pathology12 (Figure 1).
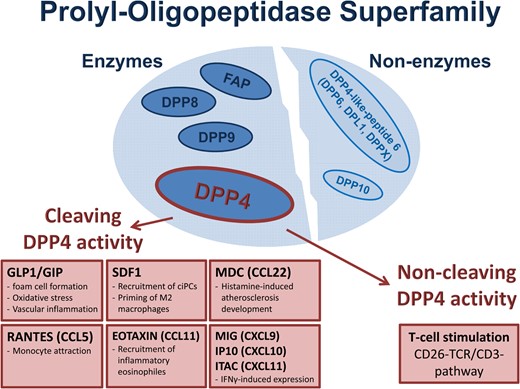
The prolyl-oligopeptidase superfamily: enzymatic and non-enzymatic members. Enzymatic function of dipeptidyl peptidase 4 includes cleavage of the incretins GLP1/GIP as well as SDF1 and several other cytokines (MIG, IP10, ITAC, macrophage-derived cytokine, Eotaxin, and RANTES). Non-enzymatic effects of dipeptidyl peptidase 4 are based on T-cell activation via the T-cell receptor complex stimulation.
The incretins GLP1 and GIP are linked to the other cytokines mentioned above by their physiologic way of inactivation via the exopeptidase DPP4. The DPP4 is expressed on membranes of various cell types (especially, leukocytes, endothelial, and epithelial cells) and freely available in the serum. While gliptins have the ability to inhibit DPP4 activity, they might improve vasoregenerative effects mediated by SDF1 and GLP1/GIP among multiple other protective signalling pathways.
Vascular function of dipeptidyl peptidase 4 substrates
In addition to incretin function in glucose metabolism, GLP1/GIP shows several beneficial effects, like a reduced atherosclerosis progression in Vildagliptin-treated non-diabetic and diabetic ApoE-ko mice. These mice showed slower progression of atherosclerotic lesions when compared with the placebo group. Furthermore, foam cell formation from peritoneal macrophages was also reduced which further supports the anti-atherosclerotic GLP/GIP-mediated effect.13 Other beneficial incretin effects are an improved vascular function, reduced oxidative stress, and attenuated vascular inflammation in diabetic patients treated with (post-prandial) GLP1 infusions14 (Figure 2). However, as treatment with the incretin-antagonist Exendin was not able to completely inhibit Vildagliptin-mediated vascular effects, other incretin-independent but DPP4-dependent pathways of gliptin therapy seem to be involved in the regulation of vascular homeostasis. Furthermore, Shah et al.15 were able to confirm this assumption while treating aortic segments of C57Bl/6 mice with Alogliptin. Gliptins improved vascular function and NO production via phosphorylation of eNOS and Akt in these tissues independent from a pretreatment with Exendin.
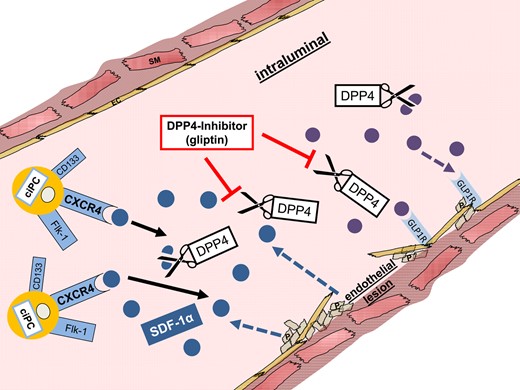
Endothelial effects of pharmacological dipeptidyl peptidase 4 inhibition. Activated platelets and smooth muscle and endothelial cells secret SDF1. SDF1 binds to the CXCR4 receptor of circulating progenitor cells and mediates their recruitment to the injured vascular wall. GLP1 binds to the GLP1 receptor on the vascular wall and improves endothelial function. Gliptins can inhibit cleavage of SDF1 and GLP1 and thus facilitate an improved vascular recovery.
After endothelial injury, the chemokine SDF1 is highly expressed in vascular walls, atherosclerotic plaques, and attached platelets.16 When active, SDF1 mediates vascular regeneration, e.g. via recruitment of ciPCs.6 Additionally, Borghini et al.17 could show that high SDF1 protein expression is associated with a decreased risk of myocardial infarction and an improved endothelial function in humans. Another SDF1-mediated mechanism during vascular regeneration is the recruitment of bone marrow-derived smooth muscle progenitors and platelets to sites of vascular injury. This further underlines the crucial role of the SDF1–CXCR4 signalling in vascular remodelling.18
Macrophage-derived cytokine is another molecule involved in the development of atherosclerosis and highly expressed by macrophages and dendritic cells. Histamine is a key mediator in the development of atherosclerosis that can induces MDC expression in human monocytes. Histamine decarboxylase (HDC) catalyses the metabolization of l-histidine to histamine. Likewise, knockout of HDC led to a significant reduction of atherosclerotic plaque burden, which further confirmed the histamine dependency of MDC-mediated atherosclerosis development.19
Eotaxin mediates recruitment of eosinophilic granulocytes to various sites of inflammation. Regarding atherosclerosis, Eotaxin is especially upregulated on smooth muscle cells (SMCs) in atherosclerotic plaques, which shows its close involvement in vascular inflammation.20
Another important chemokine is RANTES, a pro-atherogenic chemokine involved in T-cell migration and monocyte adhesion to atherosclerotic plaques. von Hundelshausen et al.21 demonstrated that especially activated platelets secret RANTES that can induce monocyte adhesion on inflammatory and atherosclerotic vascular tissue. Besides, D.m.2 patients overexpress RANTES in the serum, which may partly explain the accelerated progression of atherosclerosis in diabetes.
Interferon γ is a further relevant factor in progression of atherosclerosis and induces expression of the DPP4 substrates I-TAC, IP-10, and MIG.22 These molecules are highly expressed by macrophages, endothelial cells, and SMCs isolated from human atherosclerotic plaques. Their CXC-motif receptor 3 (CXCR3) is mainly expressed on the surface of T-lymphocytes. I-TAC is highly secreted by endothelial cells and downregulates CXCR3 on activated T cells in neovessels of atherosclerotic plaques. I-TAC might, therefore, be responsible for T-lymphocyte recruitment to atherosclerotic plaques by inducing their arrest and facilitating their diapedesis. Additionally, IP-10 improves T-cell retention after binding to CXCR3 without negatively affecting the receptor expression in all three plaque-associated cells.23 Like in MDC, Eotaxin, and RANTES, inhibition of I-TAC, IP-10, and MIG cleavage may have adverse vascular effects and thus compensate GLP1/GIP- and SDF1-mediated vasoprotection improved by gliptin therapy.6,8
Besides the soluble form of DPP4/CD26, a cell-bound, non-cleaving variant of CD26 is co-localized to the T-cell receptor (TCR)/CD3 complex. In this non-cleaving form, CD26 serves as a receptor for adenosine deaminase (ADA) on T-lymphocytes and plays a certain role in T-cell activation. The inhibition of T-cell activation by adenosine, proven by Dong et al., explains the important role of ADA in the CD26–TCR/CD3 pathway. In absence of ADA, adenosine was able to suppress IL-2 expression of T cells significantly.24 This effect can be explained by activation of intracellular tyrosine kinases once CD26 is (co-)stimulated by ADA.25 After the initiation of the CD26–TCR/CD3 pathway, these tyrosine kinases induce expression of the pro-inflammatory IL-2.24 Due to the fact that T cells can mediate pro-atherogenic as well as anti-atherogenic effects depending on the underlying T-cell subtype, further specifications are necessary to address potential therapeutic targets in T-cell regulation during the development of atherosclerosis.26 Nevertheless, T-cell activation via the CD26–TCR/CD3 pathway is a clear proof for the relevance of the non-cleaving function of DPP4 in vascular inflammation and diseases (Figure 3).
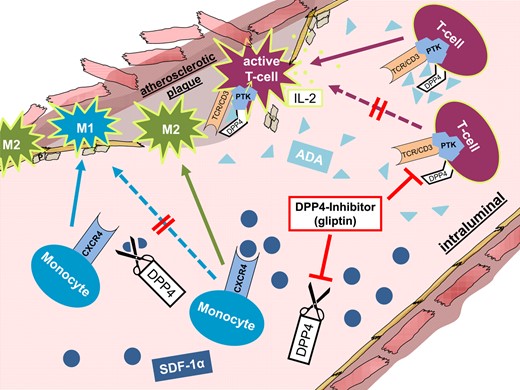
Anti-atherosclerotic effects of pharmacological dipeptidyl peptidase 4 inhibition. After binding to the monocyte CXCR4 receptor, SDF1 can prime their differentiation towards a regenerative M2 phenotype. In contrast, adenosine deaminase can couple to the dipeptidyl peptidase 4 on the surface of circulating T cells and thus activate these cells via a protein tyrosine kinase. Inhibition of dipeptidyl peptidase 4 in atherosclerosis can lead to increased numbers of reparative M2 macrophages in the atherosclerotic plaque area and inhibit inflammatory T-cell accumulation.
Impact of gliptin therapy
Effects on vascular function
Several studies have evaluated modulation of endothelial function by gliptin therapy. Shah et al.15 showed Alogliptin-mediated and GLP1-independent vasodilatation of aortic segments, caused by NO-upregulation and activation of the potassium channels in the vessels. Besides, Matsubara et al. showed an improved vascular function mediated by Sitagliptin in aortic segments from ApoE-ko mice. The aortic rings were stimulated with acetylcholine and showed an increase in eNOS phosphorylation, which further confirmed the positive impact of gliptin treatment on endothelial function.9
In clinical trials, endothelial function was evaluated by non-invasive vascular ultrasound or peripheral arterial tone measurements. The latter method evaluated vascular function and reactive hyperaemia using the peripheral arterial tonometry index (RHI). Sitagliptin significantly improved the RHI in treated patients.27 Correspondingly, flow-mediated dilatation of the brachial artery was significantly improved in Sitagliptin-treated patients. Noda et al.28 showed similar results measuring flow-mediated dilatation (FMD) in Alogliptin-medicated patients.
Besides the fact that gliptins may reduce expression of cytokines like TNFα, NFκB, IL1, and IL6,11,29 these clinical trials lack detailed analyses on the underlying molecular mechanisms. Thus, further studies are needed to decrypt the beneficial gliptin effects in endothelial dysfunction.
Effects on progenitor cell mobilization and recruitment on vascular healing
Only few working groups exclusively focused on the influence of gliptins on endothelial regeneration. However, few studies show that DPP4 inhibitors have the potential to accelerate endothelial regeneration after arterial vascular injury. Noels et al.30 could nicely show an accelerated endothelial cell growth and improved wound healing mediated by SDF1. Data from clinical trials further support a beneficial impact of gliptins on endothelial healing. Fadini et al. analysed blood samples from D.m.2 patients after a 4-week period of Sitagliptin treatment. Dipeptidyl peptidase 4 inhibition resulted in increased plasma levels of SDF1 and increased numbers of ciPCs, which can mediate vascular protective effects.31 Several other working groups were able to confirm the positive effect of DPP4 inhibition on progenitor cell mobilization.32 To study cell-based endothelial regeneration, various indirect (thermal and electric) or direct (wire injury, balloon angioplasty) injury models have been established.6,30,33 While electric injury is an excellent method to induce endothelial injuries of a defined length, wire injury and balloon angioplasty appear to injure the vessel in a more superficial way without affecting the medial and adventitial layer. All these models represent adequate methods to study different mechanisms of endothelial recovery, e.g. via recruitment of ciPCs6 or potential anti-apoptotic effects as well as their corresponding adverse proliferation of SMCs.11,30
Brenner et al. induced endothelial injuries in the common carotid artery of C57Bl/6 mice. Sitagliptin enhanced endothelial regeneration by improving mural progenitor cell recruitment. Sitagliptin inhibited cleavage of SDF1, which could thus bind to the CXCR4 receptor on ciPCs. AMD3100 abrogated the protective gliptin-mediated effect. Likewise, Noels et al.30 could prove the necessity of the SDF1–CXCR4 signalling for endothelial regeneration in an endothelial CXCR4-ko mouse model (Figure 2).
However, progenitor cell-based pharmacological improvement of endothelial cell proliferation might also induce neointimal hyperplasia.18 In contrast, Terawaki et al.34 showed Linagliptin to be an appropriate medication to reduce neointima formation. In line with these results, CXCR4 deficiency led to an increased neointima formation after wire-induced endothelial injury. This impaired vascular healing was based on the interruption of the underlying SDF1–CXCR4 pathway. Furthermore, non-invasive ultrasound analyses in mice as well as in clinical trials confirmed beneficial effects of gliptin treatment on neointima formation.30,35
Effects on atherosclerosis progression
Various preclinical studies have shown a reduction of atherosclerosis development in gliptin-treated animals independent from an improved glucose control (Figure 3). This might be based on enhanced metabolic indices like attenuated insulin resistance and improved β-cell function, reduced systolic blood pressure, and decreased TNFα- and IL-6 levels.36 Importantly, post-prandial lipaemia was also significantly lower under treatment with Alogliptin.28 Likewise, a meta-analysis on blood-lipid levels indicated a decrease in DPP4-inhibitor-treated patients.37
Besides a reduction of plaque burden, gliptin treatment further led to a stabilization of atherosclerotic lesions in ApoE-ko mice, potentially based on an altered plaque macrophage content.38 Interestingly, Vildagliptin medication led to an attenuated progression of atherosclerotic disease, less foam cell formation in peritoneal macrophages, reduced macrophage content in atherosclerotic plaques, but no changes in cholesterol levels.36 Increased GLP1/GIP concentrations, however, may only partially explain these positive effects. In fact, DPP4-dependent atherosclerosis progression appears to occur also via various other signalling pathways.8,13 Another member of the gliptin family, Anagliptin, achieved comparable results in ApoE-ko mice comprising significantly reduced serum cholesterol, plaque burden, and macrophage content in the vascular walls. A decreased monocyte-based inflammatory reaction led to this attenuated atherosclerosis development. Lipopolysaccharide (LPS)-stimulated THP1 cells showed a reduced TNFα expression after Anagliptin incubation. Additionally, Anagliptin-pretreated SMCs incubated with recombinant human DPP4 showed an inhibition of SMC proliferation.11 In line with the anti-inflammatory impact of gliptins shown by Ta et al. (inhibition of IL-6 and IL-1β expression in human mononuclear cells),29 Dai et al.39 could demonstrate a reduction of NLRP3 inflammasome and IL-1β expression in human macrophages. Hence, reduced vascular inflammation mediated by pharmacological DPP4 inhibition depicts an important factor for the inhibition of atherosclerosis development.
Recent data by Brenner et al. further proves the anti-atherosclerotic effect of gliptin treatment in ApoE-ko mice and decrypts another mechanism for the initiation of atherosclerosis. Gliptin-treated mice showed a significantly lower development of atherosclerotic plaques in aortae than the placebo group. Administration of AMD3100 reversed the positive gliptin effect, which proved that Sitagliptin-induced inhibition of atherosclerotic plaque formation at least in part works via an enhanced SDF1–CXCR4 signalling.
Fluorescence-activated cell sorting analyses regarding macrophage content and differentiation of inflammatory M1 and reparative M2 macrophage subtypes in aortic walls of the ApoE-ko mice demonstrated a Sitagliptin-mediated increase in mural protective M2 macrophages. In vitro analyses confirmed a gliptin-based priming of monocyte differentiation into this macrophage subtype.8
Due to these promising preclinical results, DPP4 inhibitors may depict an auspicious option for the treatment and prevention of atherosclerosis. However, currently only few clinical trials especially focus on intima-media thickness (IMT) and atherosclerosis development. Up to now, these studies are still ongoing or final analyses not yet completed. All these clinical trials (PROLOGUE, SPIKE, and TRUST) enrol D.m.2 patients who did not receive gliptin treatment for the last 3 months. In the PROLOGUE and SPIKE study, the patients sustained no cardiac events before.40,41,42 In contrast, the TRUST study enrols stable coronary artery disease (CAD) patients who underwent successful percutaneous coronary intervention (PCI).42 First results will probably be available by the end of this year.
Future prospects
Currently, gliptin therapy is restricted to glycaemic control in D.m.2 patients. However, based on the studies presented above, DPP4 inhibitors seem to represent a promising therapeutic approach for the treatment of vascular diseases in addition to glucose control. In vitro and preclinical models have shown beneficial effects on vascular function, endothelial regeneration, and inhibition of atherosclerosis via various signalling pathways in detail (Figure 4). Furthermore, DPP4 inhibitors are a well-tolerated class of drugs in diabetic and non-diabetic patients after acute coronary syndrome.43
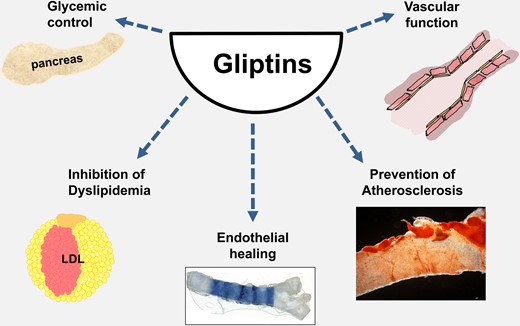
Potential therapeutic implications of pharmacological dipeptidyl peptidase 4 inhibition. Dipeptidyl peptidase 4 inhibitors help to control blood glucose in patients. Besides, present in vitro and preclinical data suggest a potential positive effect on vascular function, dyslipidaemia, endothelial healing, and prevention of atherosclerosis. However, long-term clinical data are still pending.
Although gliptins have convincingly shown a positive biological effect in preclinical models, recently published clinical trials using DPP4 inhibitors are less encouraging (e.g. EXAMINE, GUARD, and TECOS trials4,5,44). However, as already suggested by Zhong et al.,45 presently available data do not yet facilitate a final judgement on the impact of gliptin therapy in cardiovascular disease of diabetic patients. Besides the results of the ongoing clinical trials PROLOGUE, TRUST, or SPIKE,40,41,42 future trials also in non-diabetic patients will have to be taken into account.
Other enzymatic members of the prolyl-oligopeptidase superfamily
In contrast to DPP4, the function of DPP8/9 is not yet clear. Due to their ‘DPP4-like’ expression pattern, their actual relevance in cell signalling may still be underestimated.46 Furthermore, their homology and their common substrates with DPP4 might identify DPP8/9 as another important therapeutic target for dipeptidyl peptidase inhibition. Regarding gliptin use, Huan et al.47 demonstrated that inhibition of DPP8/9 was clearly dependent on gliptin-type and -dosage. These findings underline the potential relevance of DPP8/9 in future mechanistic gliptin studies.
Funding
The Else Kröner-Fresenius-Stiftung (2012_A192) kindly supported this work.
Conflict of interest: W.-M.F. and C.B. received honorarium from MSD for scientific presentations and advisory board participation, respectively.

{kind=link}
{kind=link}
{kind=link}
{kind=link}