





Abstract
Propionic acidaemia (PA) is an autosomal recessive disorder resulting from deficiency of propionyl-CoA carboxylase, a mitochondrial enzyme that metabolizes propionyl-CoA. Generally, patients with PA develop symptoms in the neonatal period due to protein intake through breastfeeding; however, late-onset PA with atypical symptoms, including cardiomyopathy, has been recently reported.
We present the case of a 25-year-old male with late-onset PA complicated by advanced heart failure (HF) due to isolated secondary dilated cardiomyopathy, who required left ventricular assist device (LVAD) implantation and finally underwent heart transplantation (HTx). Initially, the patient developed HF at the age of 16 and was diagnosed with mitochondrial cardiomyopathy. Due to refractory HF, he underwent an LVAD implantation and was scheduled for HTx. During the preoperative period for HTx, the patient suffered from sepsis due to the worsening of LVAD driveline exit-site infection complicated by overt metabolic acidosis, finally leading to the diagnosis of late-onset PA. After this diagnosis, adequate nutritional interventions were introduced, and the cardiac function was partially restored enough for him to be weaned-off LVAD; however, the patient became inotrope dependent and underwent HTx. The post-HTx course was uneventful with special nutritional management, and he has experienced no adverse metabolic events in the past 3 years.
Late-onset PA can cause isolated adult-onset cardiomyopathy, and LVAD or HTx should be considered when PA is complicated by advanced HF and is unresponsive to conventional medical therapies.
Learning points
Metabolic syndromes should be considered in the differential diagnosis of adult patients with isolated dilated cardiomyopathy.
Proper nutritional and medical interventions could ensure satisfactory myocardial recovery in patients with propionic acidaemia-related cardiomyopathy.
Left ventricular assist device implantation or heart transplantation should be considered in late-onset propionic acidaemia complicated by advanced heart failure refractory to conventional medical treatment.
Primary specialities involved other than cardiology
Cardiomyopathy secondary to inherited metabolic disorders.
Introduction
Propionic acidaemia (PA) is a rare autosomal recessive disorder caused by decreased activity of the mitochondrial enzyme propionyl-CoA carboxylase (PCC).1,2 The PCC metabolizes propionyl-CoA into methylmalonyl-CoA, which is converted to succinyl-CoA by methylmalonyl-CoA mutase. Metabolites derived from excess propionyl-CoA inhibit the glycine cleavage and urea cycle systems, leading to metabolic acidosis. Hyperglycinaemia and hyperammonaemia could cause neurological manifestations and developmental problems through their neurotoxicity, which causes acute encephalopathy.3,4 Cardiac diseases, such as cardiomyopathy and arrhythmias, are also common presentations of PA5; additionally, mild, late-onset PA could manifest as intellectual disability or delayed development during childhood, with or without metabolic acidosis.6 Furthermore, isolated adolescent-to-adult-onset dilated cardiomyopathy (DCM) due to PA with or without apparent metabolic disorders has been recently reported.7,8 These reports suggest that DCM can be the first symptom of PA even in previously healthy adults or adolescents.
Similarly, we present the case of a 25-year-old man with adolescent-onset isolated DCM, which was eventually diagnosed as PA following further investigations. Despite guideline-directed medical therapies, the patient’s condition gradually deteriorated, and he developed advanced heart failure (HF) requiring left ventricular assist device (LVAD) implantation followed by heart transplantation (HTx), within 6 years of his initial presentation (Timeline). To our knowledge, this is the first report of a patient with late-onset PA who received long-term LVAD therapy and a subsequent successful HTx.
Timeline
| Date | Events |
|---|---|
| October 2012 to March 2013 | First episode of heart failure |
| Started on inotropes followed by intra-aortic balloon pump. Diagnosed with mitochondrial cardiomyopathy | |
| April 2014 to February 2015 | Second episode of heart failure |
| Listed for heart transplantation (HTx). Underwent left ventricular assist device (LVAD) implantation for bridge to transplantation. Developed Reye syndrome postoperatively. | |
| May 1–30, 2015 | Admission for LVAD driveline exit-site infection |
| Administration of antibiotics. | |
| June 2015 to October 2016 | Re-admission for LVAD driveline exit-site infection |
| Sepsis with metabolic crisis. Diagnosed with propionic acidaemia. Weaned-off LVAD with partial cardiac recovery. | |
| December 2016 to January 2019 | Third episode of heart failure |
| Started on inotropes. Received HTx on December 2018. | |
| Thereafter until November 2021 | Clinical course after HTx |
| Mild rejection episode 2 weeks post-HTx. No metabolic crisis. No HTx-related comorbidities. No restriction for physical activity. |
| Date | Events |
|---|---|
| October 2012 to March 2013 | First episode of heart failure |
| Started on inotropes followed by intra-aortic balloon pump. Diagnosed with mitochondrial cardiomyopathy | |
| April 2014 to February 2015 | Second episode of heart failure |
| Listed for heart transplantation (HTx). Underwent left ventricular assist device (LVAD) implantation for bridge to transplantation. Developed Reye syndrome postoperatively. | |
| May 1–30, 2015 | Admission for LVAD driveline exit-site infection |
| Administration of antibiotics. | |
| June 2015 to October 2016 | Re-admission for LVAD driveline exit-site infection |
| Sepsis with metabolic crisis. Diagnosed with propionic acidaemia. Weaned-off LVAD with partial cardiac recovery. | |
| December 2016 to January 2019 | Third episode of heart failure |
| Started on inotropes. Received HTx on December 2018. | |
| Thereafter until November 2021 | Clinical course after HTx |
| Mild rejection episode 2 weeks post-HTx. No metabolic crisis. No HTx-related comorbidities. No restriction for physical activity. |
| Date | Events |
|---|---|
| October 2012 to March 2013 | First episode of heart failure |
| Started on inotropes followed by intra-aortic balloon pump. Diagnosed with mitochondrial cardiomyopathy | |
| April 2014 to February 2015 | Second episode of heart failure |
| Listed for heart transplantation (HTx). Underwent left ventricular assist device (LVAD) implantation for bridge to transplantation. Developed Reye syndrome postoperatively. | |
| May 1–30, 2015 | Admission for LVAD driveline exit-site infection |
| Administration of antibiotics. | |
| June 2015 to October 2016 | Re-admission for LVAD driveline exit-site infection |
| Sepsis with metabolic crisis. Diagnosed with propionic acidaemia. Weaned-off LVAD with partial cardiac recovery. | |
| December 2016 to January 2019 | Third episode of heart failure |
| Started on inotropes. Received HTx on December 2018. | |
| Thereafter until November 2021 | Clinical course after HTx |
| Mild rejection episode 2 weeks post-HTx. No metabolic crisis. No HTx-related comorbidities. No restriction for physical activity. |
| Date | Events |
|---|---|
| October 2012 to March 2013 | First episode of heart failure |
| Started on inotropes followed by intra-aortic balloon pump. Diagnosed with mitochondrial cardiomyopathy | |
| April 2014 to February 2015 | Second episode of heart failure |
| Listed for heart transplantation (HTx). Underwent left ventricular assist device (LVAD) implantation for bridge to transplantation. Developed Reye syndrome postoperatively. | |
| May 1–30, 2015 | Admission for LVAD driveline exit-site infection |
| Administration of antibiotics. | |
| June 2015 to October 2016 | Re-admission for LVAD driveline exit-site infection |
| Sepsis with metabolic crisis. Diagnosed with propionic acidaemia. Weaned-off LVAD with partial cardiac recovery. | |
| December 2016 to January 2019 | Third episode of heart failure |
| Started on inotropes. Received HTx on December 2018. | |
| Thereafter until November 2021 | Clinical course after HTx |
| Mild rejection episode 2 weeks post-HTx. No metabolic crisis. No HTx-related comorbidities. No restriction for physical activity. |
Case presentation
A previously healthy 16-year-old adolescent man was referred to our institution with acute HF. Upon admission, he was classified as having New York Heart Association Class IV HF and was started on 4.4 μg/kg/min of dobutamine and 0.14 μg/kg/min of milrinone. Physical examination revealed that the patient had relatively low blood pressure and tachycardia (blood pressure, 94/62 mmHg; heart rate, 105 b.p.m.) combined with cold extremities, implying severe low cardiac output. Echocardiography revealed an enlarged left ventricle with severely reduced left ventricular contraction [left ventricular diastolic dimension (LVDd), 82 mm; left ventricular ejection fraction (LVEF), 19%], and brain natriuretic peptide (BNP) increased to 1012.9 pg/mL at admission (Figure 1A). Furthermore, high-intensity lesions in the basal ganglia were observed on T2-weighted-FLAIR magnetic resonance images (MRI; Figure 1B). Based on morphological abnormalities of the mitochondria on electron microscopic imaging of the biopsied right ventricular myocardium and the basal ganglia lesions observed on MRI, the patient was diagnosed with mitochondrial cardiomyopathy (Figure 1D and E). Similarly, a reduced enzyme activity of respiratory chain Complex I was confirmed via a biochemical analysis of the patient’s skeletal muscle. After intensive treatments, including 3 weeks of an intra-aortic balloon pump, 3 months of continuous inotrope infusion, and up-titration of cardioprotective medications, the HF resolved, and the basal ganglia lesions disappeared (Figure 1C). No episode of metabolic acidosis was observed during the initial 6-month-long admission, and the patient was discharged with medications for HF, as indicated by the current guidelines. Details regarding echocardiogram, BNP value, and HF medications at discharge (including β-blockers, angiotensin-converting enzyme inhibitors, and mineralocorticoid receptor antagonists) are demonstrated in Figure 2. Thirteen months after discharge, at 18 years of age, the patient was admitted again to our institution on account of gradually deteriorating HF. His BNP value, which had once decreased to <100 pg/mL, increased to >500 pg/mL, and despite aggressive treatment for HF, including multiple inotropic infusions (2.5 μg/kg/min of dobutamine and 0.25 μg/kg/min of milrinone), the patient’s condition did not improve. He finally underwent LVAD implantation (Jarvik 2000; Jarvik Heart Inc., New York, NY, USA) for a bridge to HTx. Immediately after LVAD implantation, the patient developed severe, acute liver injury with metabolic acidosis, which was diagnosed as Reye syndrome induced by aspirin administration; the metabolic acidosis and liver injury gradually resolved after 2 weeks of multidisciplinary treatments, such as mechanical ventilation, plasmapheresis, and continuous haemodialysis with conventional postoperative care. Regarding medications, carvedilol was changed to bisoprolol, and imidapril was changed to enalapril (Figure 2). He was finally discharged home with LVAD support, 8 months after LVAD implantation. At discharge, ventricular reverse remodelling was observed (LVDd, 42 mm; LVEF, 19%), and his BNP was within normal limits (16.8 pg/mL; Figure 2).
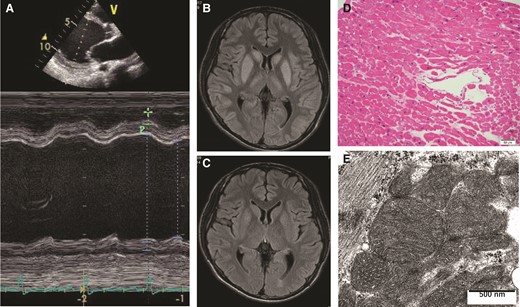
An echocardiographic image (A), magnetic resonance images of the brain (B, C), a histopathological image of the myocardium (D, haematoxylin-eosin stain), and an electron microscopic image of the myocardium (E), at the time of the first admission for heart failure.

Sequential changes in echocardiographic findings (left ventricular diastolic dimension and ejection fraction), brain natriuretic peptide, and medications before receiving heart transplantation. Basic cardioprotective medications including beta-blockers, angiotensin-converting enzyme inhibitors, and mineralocorticoid receptor antagonists, were administered before left ventricular assist device implantation. After successful left ventricular assist device implantation, ventricular reverse remodelling occurred, which deteriorated with the development of metabolic crisis. ACEI, angiotensin-converting enzyme inhibitor; BNP, brain natriuretic peptide; EF, ejection fraction; LVAD, left ventricular assist device; LVDd, left ventricular diastolic dimension; MR, mineralocorticoid receptor
Two months after the second discharge, the patient was admitted for the third time to our institution due to an LVAD driveline exit-site infection. Despite various medical and surgical therapies, the infection worsened and finally involved the LVAD pump; he finally developed multiple drug-resistant Pseudomonas aeruginosa (MDRP) sepsis due to pump pocket infection. Despite treatment for sepsis, the patient’s overall condition deteriorated rapidly; he became weak and developed anorexia, recurrent vomiting, and tachypnea. Blood examinations revealed metabolic acidosis with hyperammonaemia, and mechanical ventilation was required. Along with the patient’s general condition, his cardiac function rapidly deteriorated (LVDd, 72 mm; LVEF, 10%) and his BNP increased to 886.2 pg/mL (Figure 2), despite substantial LVAD support. A metabolic screening was performed because the symptoms were characteristic of an inherited metabolic disorder, and PA was diagnosed from the typical results obtained from metabolic screening (Table 1). Briefly, a diagnosis of PA is made when there are elevated concentrations of propionylcarnitine (C3), glycine, and alanine in the blood. Increased urinary excretion of 3-hydroxypropionate, propionylglycine, and methylcitrate are also characteristic of PA. Based on the diagnosis of PA, glucose was intravenously administered to avoid catabolism, and colistin was administered for MDRP sepsis, resulting in a gradual improvement of the patient’s heart function and general condition. He was weaned from mechanical ventilation, and partial left ventricular reverse remodelling occurred (LVDd, 52 mm), enough for him to be weaned-off LVAD (Figure 2). Further examinations for PA revealed genetic mutations in the PCCA gene (PCCA c.923dupT, PCCA c.229C > T), and the patient’s PCC enzymatic activity, quantified by ultra-high-performance liquid chromatography tandem mass spectrometry, was low (2.24%; Table 2).9 After he was weaned-off the LVAD, bisoprolol was uptitrated from 2.5 to 15 mg to enhance his myocardial recovery. Angiotensin II receptor antagonists were also added for cardioprotection (Figure 2).
Acute-phase metabolic screening
| Acylcarnitine profile in dried blood spot | |
| Propionylcarnitine (C3) | 21.24 nmol/mL |
| Free carnitine (C0) | 23.31 nmol/mL |
| Acylcarnitine profile in serum | |
| Propionylcarnitine (C3) | 28.55 nmol/mL |
| Free carnitine (C0) | 6.20 nmol/mL |
| Urinary analysis | |
| 3-Hydroxypropionate | 518.10; ref <1.10 |
| Propionylglycine | 9.02; ref <0.00 |
| Methylcitrate | 25.02; ref <1.10 |
| 3-Hydroxybutyrate | 1582.43; ref <3.70 |
| Acetoacetate | 130.05; ref <0.00 |
| 3-Hydroxyisovalerate | 9.33; ref <2.30 |
| 2-Hydroxyisovalerate | 4.67; ref <0.00 |
| Acylcarnitine profile in dried blood spot | |
| Propionylcarnitine (C3) | 21.24 nmol/mL |
| Free carnitine (C0) | 23.31 nmol/mL |
| Acylcarnitine profile in serum | |
| Propionylcarnitine (C3) | 28.55 nmol/mL |
| Free carnitine (C0) | 6.20 nmol/mL |
| Urinary analysis | |
| 3-Hydroxypropionate | 518.10; ref <1.10 |
| Propionylglycine | 9.02; ref <0.00 |
| Methylcitrate | 25.02; ref <1.10 |
| 3-Hydroxybutyrate | 1582.43; ref <3.70 |
| Acetoacetate | 130.05; ref <0.00 |
| 3-Hydroxyisovalerate | 9.33; ref <2.30 |
| 2-Hydroxyisovalerate | 4.67; ref <0.00 |
Acute-phase metabolic screening
| Acylcarnitine profile in dried blood spot | |
| Propionylcarnitine (C3) | 21.24 nmol/mL |
| Free carnitine (C0) | 23.31 nmol/mL |
| Acylcarnitine profile in serum | |
| Propionylcarnitine (C3) | 28.55 nmol/mL |
| Free carnitine (C0) | 6.20 nmol/mL |
| Urinary analysis | |
| 3-Hydroxypropionate | 518.10; ref <1.10 |
| Propionylglycine | 9.02; ref <0.00 |
| Methylcitrate | 25.02; ref <1.10 |
| 3-Hydroxybutyrate | 1582.43; ref <3.70 |
| Acetoacetate | 130.05; ref <0.00 |
| 3-Hydroxyisovalerate | 9.33; ref <2.30 |
| 2-Hydroxyisovalerate | 4.67; ref <0.00 |
| Acylcarnitine profile in dried blood spot | |
| Propionylcarnitine (C3) | 21.24 nmol/mL |
| Free carnitine (C0) | 23.31 nmol/mL |
| Acylcarnitine profile in serum | |
| Propionylcarnitine (C3) | 28.55 nmol/mL |
| Free carnitine (C0) | 6.20 nmol/mL |
| Urinary analysis | |
| 3-Hydroxypropionate | 518.10; ref <1.10 |
| Propionylglycine | 9.02; ref <0.00 |
| Methylcitrate | 25.02; ref <1.10 |
| 3-Hydroxybutyrate | 1582.43; ref <3.70 |
| Acetoacetate | 130.05; ref <0.00 |
| 3-Hydroxyisovalerate | 9.33; ref <2.30 |
| 2-Hydroxyisovalerate | 4.67; ref <0.00 |
Amino acid, enzyme activity, and genetic analysis
| Amino acid profile | |
| Glycine | 516.2 nmol/mL |
| Alanine | 567.0 nmol/mL |
| PCC enzyme activity | |
| 2.24% | |
| Genetic analysis | |
| PCCA c.229C > T | PCCA c.923dupT |
| * | * |
 |  |
| Amino acid profile | |
| Glycine | 516.2 nmol/mL |
| Alanine | 567.0 nmol/mL |
| PCC enzyme activity | |
| 2.24% | |
| Genetic analysis | |
| PCCA c.229C > T | PCCA c.923dupT |
| * | * |
| |
PCCA, propionyl-CoA carboxylase alpha chain.
Amino acid, enzyme activity, and genetic analysis
| Amino acid profile | |
| Glycine | 516.2 nmol/mL |
| Alanine | 567.0 nmol/mL |
| PCC enzyme activity | |
| 2.24% | |
| Genetic analysis | |
| PCCA c.229C > T | PCCA c.923dupT |
| * | * |
| |
| Amino acid profile | |
| Glycine | 516.2 nmol/mL |
| Alanine | 567.0 nmol/mL |
| PCC enzyme activity | |
| 2.24% | |
| Genetic analysis | |
| PCCA c.229C > T | PCCA c.923dupT |
| * | * |
| |
PCCA, propionyl-CoA carboxylase alpha chain.
The patient was again discharged from the hospital; however, his cardiac function gradually deteriorated while on oral HF medications (LVDd, 72 mm; LVEF, 31%); therefore, the patient was admitted and became inotrope dependent (Figure 2). After 2 years of waiting with inotropic support, the patient underwent HTx. At the time of HTx, additional prophylactic nutritional and medical support was introduced to avoid metabolic crisis (Table 3). Postoperatively, glucose was intravenously administered for 3 days to maintain blood glucose levels between 120 and 200 mg/dL, and carnitine was intravenously administered at a daily dose of 3000 mg to facilitate organic acid metabolism, followed by oral carnitine at the same dose after extubation. Protein intake was restricted to between 0.5 and 1.0 g daily. Standard triple-drug immunosuppression therapy using tacrolimus, mycophenolate mofetil, and steroids was administered without induction therapy. Moderate acute cellular rejection (International Heart and Lung Transplantation grade 2R) was observed at 2 weeks post-HTx; otherwise, the clinical course was uneventful. Acute cellular rejection was resolved by administering 50 mg of prednisone daily for 3 days, and trough levels of tacrolimus was strictly maintained at no <10 ng/mL until 1 year after HTx. Because of mild renal dysfunction, immunosuppression therapy was converted to everolimus with reduced tacrolimus, 2 years after HTx. Currently, it has been 3 years since HTx, and the patient’s physical activity is unrestricted with no incidence of metabolic crises. The patient is currently stable and on a normal diet.
Prophylactic nutritional and medical managements at heart transplantation
| Glucose supplementation | Intravenously administered to maintain blood glucose levels between 120 and 200 mg/dL until third postoperative day |
| Carnitine supplementation | Intravenously administered 3000 mg daily, followed by oral administration 3000 mg daily after extubation |
| Protein restriction | Protein intake is restricted between 0.5 and 1.0 g/kg/day (regular hospital renal diet is served) |
| Glucose supplementation | Intravenously administered to maintain blood glucose levels between 120 and 200 mg/dL until third postoperative day |
| Carnitine supplementation | Intravenously administered 3000 mg daily, followed by oral administration 3000 mg daily after extubation |
| Protein restriction | Protein intake is restricted between 0.5 and 1.0 g/kg/day (regular hospital renal diet is served) |
Prophylactic nutritional and medical managements at heart transplantation
| Glucose supplementation | Intravenously administered to maintain blood glucose levels between 120 and 200 mg/dL until third postoperative day |
| Carnitine supplementation | Intravenously administered 3000 mg daily, followed by oral administration 3000 mg daily after extubation |
| Protein restriction | Protein intake is restricted between 0.5 and 1.0 g/kg/day (regular hospital renal diet is served) |
| Glucose supplementation | Intravenously administered to maintain blood glucose levels between 120 and 200 mg/dL until third postoperative day |
| Carnitine supplementation | Intravenously administered 3000 mg daily, followed by oral administration 3000 mg daily after extubation |
| Protein restriction | Protein intake is restricted between 0.5 and 1.0 g/kg/day (regular hospital renal diet is served) |
Discussion
Herein, we presented a case of adolescent-onset DCM secondary to PA, which was initially treated with conventional HF medical therapies and ultimately resolved after HTx, 6 years after the patient’s initial presentation. This case highlights important clinical implications of the management of isolated adult-onset cardiomyopathy due to metabolic diseases, such as PA. First, late-onset PA may form a substantial disease group in isolated adult-onset DCM. Second, patients with cardiac diseases secondary to PA can recover when appropriate conventional medical therapies are administered. Third, LVAD and HTx are promising therapeutic alternatives for patients with advanced HF due to PA-associated DCM in whom conventional medical therapies are ineffective. Appropriate nutritional management enables these patients to safely undergo invasive interventions without developing metabolic crises.
In addition to previously reported cases of adult-onset DCM secondary to PA, a recent clinical study confirmed the clinical implications of PA in adult DCM. Riemersma et al.8 conducted metabolic screening in 36 adults with DCM and found one with PA from the derivation cohort. This result was validated in a cohort of 157 adults with DCM, wherein one patient with PA was identified. Similar to these previous reports, our report supports the hypothesis that some adults with DCM have PA and that aggressive metabolic screening should be performed in patients with idiopathic DCM. Since metabolic crisis without appropriate intervention can be fatal in patients with PA, misdiagnosis or diagnostic delays should be avoided. In our case, appropriate nutritional management was not initially provided as the original diagnosis was mitochondrial cardiomyopathy, and the patient developed metabolic acidosis and sepsis following LVAD implantation. However, once the correct diagnosis (PA) was established and appropriate nutritional management was administered, the patient safely underwent HTx surgery. Without appropriate nutritional management, HTx surgery may also induce a metabolic crisis as it is invasive and induces catabolism.
The potential for cardiac recovery may be an important characteristic of PA-related DCM. Laemmle et al.10 summarized four previously reported cases of late-onset PA, including their patient, and rapid recovery from HF was observed in three cases. In their patient, the LVEF increased substantially from 25 to 40% within 10 days of treatment initiation, and cardiac function was maintained for at least 4 months. Furthermore, in other previous reports, cardiac recovery was observed in both adult and paediatric patients with DCM secondary to PA, after liver transplantation.11,12 In our patient, as shown in Figure 2, LVDd, LVEF, and BNP values drastically changed throughout the clinical course until HTx was performed. In addition to HF medication and LVAD support, cardiac function appeared to be greatly influenced by the patient’s metabolic condition. These previous reports and the current case provide important evidence regarding the clinical management of patients with PA-associated DCM. Appropriate medical and nutritional interventions may play a key role in preventing metabolic crises and ensuring myocardial recovery.
Although appropriate medical and nutritional interventions are effective treatment strategies for patients with PA, some patients have refractory HF who requires mechanical support or HTx. Two adults with DCM secondary to PA reportedly underwent HTx; however, to our knowledge, this is the first case report of a bridge to HTx with long-term durable LVAD support in a patient with late-onset PA.7,13 Since the indication criteria for both LVAD and HTx in patients with isolated DCM secondary to PA are similar to those of patients with idiopathic DCM, such a bridge is crucial if patients’ conditions are complicated by systemic symptoms, such as neurological disorders. In our case, despite severe cardiac disease requiring LVAD and HTx, the patient demonstrated no neurological symptoms, including developmental disorders, that could contraindicate LVAD and HTx. Regarding these symptomatic discrepancies, ammonia and glycine, which have been reported to trigger neurotoxicity when elevated,3,14 were normal in our patient and might have been the reason for the absence of neurological manifestations. Therefore, the presence or absence of neurological symptoms is crucial to receiving advanced HF therapies (LVAD and HTx). However, acute metabolic crises could occur even in patients with PA with isolated DCM, especially during catabolic events and the perioperative period of invasive surgeries. We administered intravenous glucose and carnitine to this patient during the perioperative and early postoperative periods, which resulted in an uneventful clinical course.
In conclusion, since isolated DCM secondary to PA is extremely rare in adults with HF, a diagnostic delay or misdiagnosis may occur. As described herein, our patient experienced unusual episodes of liver injury with metabolic acidosis during the perioperative period of LVAD implantation and another episode of metabolic acidosis due to sepsis from LVAD driveline exit-site infection. These events are associated with PA and could have been avoided if the correct diagnosis had been established during the patient’s initial admission. Metabolic syndromes should be included in the differential diagnosis of adults with isolated DCM, as metabolic crises can be fatal, and appropriate nutritional and medical interventions in patients with PA-related cardiomyopathy could result in myocardial recovery.
Lead author biography

Osamu Seguchi is a board-certified cardiologist and works for advanced heart failure therapies including mechanical circulatory support and heart transplantation. One of his specific interests is in the diagnosis and treatment for rare cardiomyopathy secondary to systemic disease such as muscular dystrophy, metabolic disorder, and others.
Supplementary material
Supplementary material is available at European Heart Journal – Case Reports.
Acknowledgements
The authors thank Dr Yasuhiro Maeda from Nagoya City University for his role in quantifying enzyme activity of PCC. They also thank Dr Kohei Moribayashi, Dr Tetsufumi Motokawa, and Dr Aki Shionoiri for their roles in patient care.
Slide sets: A fully edited slide set detailing this case and suitable for local presentation is available online as Supplementary data.
Consent: The authors confirm that written consent for submission and publication of this case report, including images and associated text, has been obtained from the patient, in line with COPE guidance.
Conflict of interest: None declared.
Funding: None declared.
Data availability
The data underlying this article are available in the article and it will be provided on request.

{kind=link}
{kind=link}
{kind=link}
Comments