





Abstract
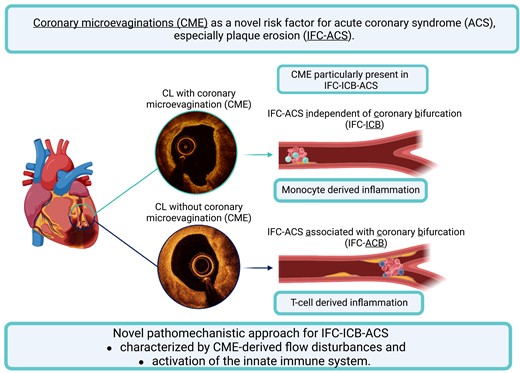
Coronary microevaginations (CMEs) represent an outward bulge of coronary plaques and have been introduced as a sign of adverse vascular remodelling following coronary device implantation. However, their role in atherosclerosis and plaque destabilization in the absence of coronary intervention is unknown. This study aimed to investigate CME as a novel feature of plaque vulnerability and to characterize its associated inflammatory cell–vessel–wall interactions.
A total of 557 patients from the translational OPTICO-ACS study programme underwent optical coherence tomography imaging of the culprit vessel and simultaneous immunophenotyping of the culprit lesion (CL). Two hundred and fifty-eight CLs had a ruptured fibrous cap (RFC) and one hundred had intact fibrous cap (IFC) acute coronary syndrome (ACS) as an underlying pathophysiology. CMEs were significantly more frequent in CL when compared with non-CL (25 vs. 4%, P < 0.001) and were more frequently observed in lesions with IFC-ACS when compared with RFC-ACS (55.0 vs. 12.7%, P < 0.001). CMEs were particularly prevalent in IFC-ACS-causing CLs independent of a coronary bifurcation (IFC-ICB) when compared with IFC-ACS with an association to a coronary bifurcation (IFC-ACB, 65.4 vs. 43.7%, P = 0.030). CME emerged as the strongest independent predictor of IFC-ICB (relative risk 3.36, 95% confidence interval 1.67–6.76, P = 0.001) by multivariable regression analysis. IFC-ICB demonstrated an enrichment of monocytes in both culprit blood analysis (culprit ratio: 1.1 ± 0.2 vs. 0.9 ± 0.2, P = 0.048) and aspirated culprit thrombi (326 ± 162 vs. 96 ± 87 cells/mm2, P = 0.017), while IFC-ACB confirmed the accumulation of CD4+ T cells, as recently described.
This study provides novel evidence for a pathophysiological involvement of CME in the development of IFC-ACS and provides first evidence for a distinct pathophysiological pathway for IFC-ICB, driven by CME-derived flow disturbances and inflammatory activation involving the innate immune system.
Registration of the study at clinicalTrials.gov (NCT03129503).
See the editorial comment for this article ‘Coronary ‘microevagination’ in intact fibrous cap acute coronary syndromes: another piece of the puzzle', by F. Prati and E. Arbustini, https://doi.org/10.1093/ehjci/jead277.
Introduction
Acute coronary syndromes (ACSs) remain the most devastating clinical manifestations of coronary artery disease (CAD) with an enormous global disease burden.1 It is well known that ACS arises from either plaques with ruptured fibrous caps (RFCs) or eroded coronary plaques with intact fibrous caps (IFCs),2–5 the latter being present in up to one-third of patients with ACS.2,6–10 Culprit lesions (CLs) with IFC are characterized by endothelial injury or the absence of the functional endothelial layer, i.e. endothelial denudation.11,12 The underlying pathophysiologic mechanism, leading to thrombus formation without plaque rupture, remains one of the most important unresolved questions within atherosclerosis research.4,13 Several studies recently observed a relationship between IFC-ACS, its CL, and the proximity of the CL to a coronary bifurcation,2,8 suggesting shear stress and local enrichment of adaptive immune cells and their effector molecules as potential pathophysiological mechanisms for IFC-ACS.8,14 However, with up to 40% of IFC-ACS lesions not being located at coronary bifurcations, further contributing factors for plaque destabilization, subsequently leading to IFC-ACS, were presumed.8
High-resolution intraluminal imaging techniques such as optical coherence tomography (OCT) allow for an in vivo characterization of the CL and the underlying anatomy of the coronary artery vessel wall.15 This enables a precise characterization of coronary plaque features and their respective susceptibility for vascular thrombosis in the context of ACS16,17—both directly at the ACS-causing culprit plaque and within the adjacent culprit vessel.
Recently, outward bulges, called coronary microevaginations (CMEs; see Supplementary data online, Figure S1), have been identified as a novel coronary vascular remodelling phenomenon identified by OCT. CMEs trigger positive vessel wall remodelling due to evagination-based flow disturbances and have very recently been presented as risk factors for late stent thrombosis.18–23 The prevalence and characteristics of CME within the cascade of plaque destabilization mechanisms within the vessel wall irrespective of coronary device implantation remain incompletely understood. CME may be of special interest for the flow-dependent pathophysiologic mechanisms during IFC-ACS, especially in those cases where IFC-ACS occurs without a spatial interaction with a coronary bifurcation.
The aim of this subanalysis of the OPTICO-ACS study was to further analyse the relevance of CME in the context of different ACS-causing culprit pathologies.
Methods
Study design and patients
The entire study was performed as a pre-specified subanalysis within the prospective, multicentric OPTIcal COherence tomography in ACS (OPTICO-ACS) study programme, coordinated by the Charité—University Medicine Berlin and the Berlin Institute of Health (BIH), Germany. Details of the study design have recently been published elsewhere.8 Briefly, a total of 557 patients undergoing percutaneous coronary intervention (PCI) for ACS [including ST-segment elevation myocardial infarction (STE-ACS)24 or non-ST segment elevation myocardial infarction25] with a clearly identifiable CL in the absence of active inflammatory or malignant diseases were consecutively enrolled. Ethical approval was obtained from the local ethics committee of the Charité University Medicine (No. EA1/270/16), and the study was conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from all participating subjects and the study was registered at ClinicalTrials.gov (NCT03129503).
Acquisition and analysis of OCT images
The acquisition and analysis of OCT data were performed in accordance with the previously published methodology of the OPTICO-ACS trial.8 All OCTs were analysed for the underlying ACS-causing culprit pathology by two independent core labs, according to the pre-defined study-specific standard operating procedure using the Medis QIvus 3.0 (Medis Medical Imaging Service, Leiden, The Netherlands). IFC-ACS lesions were categorized as ‘associated with a coronary bifurcation’ (IFC-ACB), when the frame with its respective minimal lumen area (MLA) was situated within 3 mm of a side branch with a diameter >1 mm and as ‘independent of a coronary bifurcation’ (IFC-ICB) if the distance to a coronary bifurcation was over 3 mm or no bifurcation was present in the immediate surrounding, in accordance with previously published definitions.8 For the detection of CME, the following definition was predetermined: The presence of an outward bulge of intact endothelium in at least two adjacent frames of the coronary artery not caused by the merging of a side branch or created by two adjacent lumen protruding plaques. The inter-observer variability to identify incident CME was 95.0% (ĸ = 0.943). Further details of the OCT core lab image analysis are provided as Supplementary data online, Figure S2A: cases with disagreement on the type of ACS-causing CL or the presence of CME were reassessed to reach agreement between the two core labs with respect to all published data and definitions.8,15 Cases without consensus between the two core labs (n = 10, 2.3% of all OCT scans) were excluded from the analysis.
Biological sampling, immunophenotyping by flow cytometry, and thrombus analysis
Collection and analysis of biological samples have previously been published.8 Briefly, after receiving written patient consent and after diagnostic coronary angiography, 30 mL of blood were obtained directly from the ACS-causing CL (LOC) using an aspiration catheter system (Export Advance™; Medtronic, Minneapolis, MN, USA; Figure 1). Furthermore, 30 mL of systemic blood were collected directly from the arterial sheath (SYS). Extraction and fixation of thrombotic components were performed in 4% paraformaldehyde, after an initial dehydration in 30% sucrose solution (see Supplementary data online, Figure S2B), followed by immunofluorescence analysis. LOC and SYS blood was then promptly analysed using a flow cytometry-based immunophenotyping (see Supplementary data online, Figure S2C). To specifically focus on inflammatory properties directly at the ACS-causing CL, an intra-individual normalization was calculated using the SYS inflammatory level as a reference for pre-existent systemic inflammatory levels by calculation of a ‘culprit ratio’ (Figure 1).
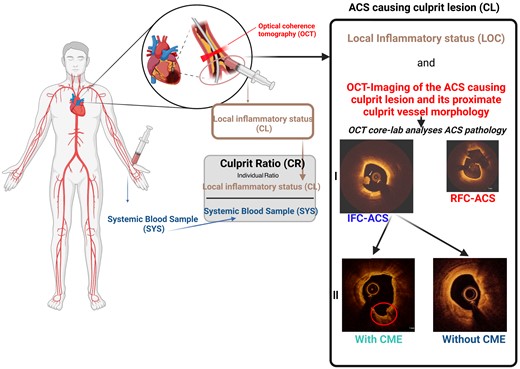
Assessment of inflammatory coronary vessel wall interactions by immunophenotyping. Each patient received simultaneous OCT imaging of the ACS-causing CL and local and systemic immunophenotyping used to calculate an intra-individual culprit ratio. ACS, acute coronary syndrome; CL, culprit lesion, IFC, intact fibrous cap; OCT, optical coherence tomography; RFC, ruptured fibrous cap.
Statistical analysis
Standard descriptive statistics were used to describe the distribution of parameters of interest for the entire study population and separately for CL vs. non-CL, and IFC-ACB vs. IFC-ICB, respectively. Values of flow cytometric measurements below the individual detection level were imputed by 10% of the minimum of valid data for the respective parameter. Group comparisons for categorical variables were performed by the χ2 test or the Fisher exact test in case of an expected cell frequency of 5 or less in cross tables. The Mann–Whitney U test was used for comparisons of continuous non-parametric variables. The association of the presence of IFC-ICB CL with CME and other established risk factors was determined by univariate and multivariable generalized linear models with robust error variances (Poisson distribution with a log link function26). Two-sided P-values <0.05 were considered statistically significant. The predictive value of CME in comparison with other established criteria of plaque vulnerability to characterize an IFC-ICB CL was assessed by receiver operating characteristic curve analysis and given as area under the curve (AUC). In addition, the AUC was estimated for multivariable models, including established cardiovascular risk factors and CME (see Table 1), and compared by the DeLong method.27 Statistical analyses were performed using IBM SPSS 25 (IBM, New York, USA), STATA 14,28 and GraphPad PRISM 7.00 (GraphPad Software Inc, CA, USA).
Predictive value of cardiovascular risk factors, established features of plaque vulnerability, and CME for the presence of IFC-ICB-induced ACS
| Risk factor | Univariate | Multivariate | ||||
|---|---|---|---|---|---|---|
| RR | 95% CI | P-value | RR | 95% CI | P-value | |
| Clinical risk factors | ||||||
| Age | 0.99 | 0.97–1.01 | 0.152 | 0.99 | 0.96–1.01 | 0.239 |
| Diabetes mellitus | 0.93 | 0.49–1.78 | 0.837 | 0.57 | 0.20–1.59 | 0.283 |
| LDL cholesterol at admission | 1.00 | 0.99–1.00 | 0.427 | 1.00 | 0.99–1.01 | 0.846 |
| hsCRP at admission | 1.00 | 1.00–1.01 | 0.367 | 1.00 | 0.99–1.01 | 0.694 |
| Presentation as STE-ACS | 1.13 | 0.67–1.89 | 0.650 | 0.93 | 0.47–1.84 | 0.832 |
| Established criteria of plaque vulnerability | ||||||
| MLA < 3.5 mm2 | 0.64 | 0.26–1.57 | 0.329 | 1.76 | 0.95–3.24 | 0.072 |
| Lipid arc circumferential extension >180° | 0.27 | 0.15–0.47 | <0.001 | 0.54 | 0.27–1.07 | 0.077 |
| Fibrous cap thickness <75 µm | 0.09 | 0.05–0.16 | <0.001 | 0.14 | 0.05–0.34 | <0.001 |
| Macrophages | 0.33 | 0.18–0.59 | <0.001 | 0.94 | 0.49–1.79 | 0.846 |
| CME | 5.77 | 3.44–9.70 | <0.001 | 3.36 | 1.67–6.76 | 0.001 |
| Risk factor | Univariate | Multivariate | ||||
|---|---|---|---|---|---|---|
| RR | 95% CI | P-value | RR | 95% CI | P-value | |
| Clinical risk factors | ||||||
| Age | 0.99 | 0.97–1.01 | 0.152 | 0.99 | 0.96–1.01 | 0.239 |
| Diabetes mellitus | 0.93 | 0.49–1.78 | 0.837 | 0.57 | 0.20–1.59 | 0.283 |
| LDL cholesterol at admission | 1.00 | 0.99–1.00 | 0.427 | 1.00 | 0.99–1.01 | 0.846 |
| hsCRP at admission | 1.00 | 1.00–1.01 | 0.367 | 1.00 | 0.99–1.01 | 0.694 |
| Presentation as STE-ACS | 1.13 | 0.67–1.89 | 0.650 | 0.93 | 0.47–1.84 | 0.832 |
| Established criteria of plaque vulnerability | ||||||
| MLA < 3.5 mm2 | 0.64 | 0.26–1.57 | 0.329 | 1.76 | 0.95–3.24 | 0.072 |
| Lipid arc circumferential extension >180° | 0.27 | 0.15–0.47 | <0.001 | 0.54 | 0.27–1.07 | 0.077 |
| Fibrous cap thickness <75 µm | 0.09 | 0.05–0.16 | <0.001 | 0.14 | 0.05–0.34 | <0.001 |
| Macrophages | 0.33 | 0.18–0.59 | <0.001 | 0.94 | 0.49–1.79 | 0.846 |
| CME | 5.77 | 3.44–9.70 | <0.001 | 3.36 | 1.67–6.76 | 0.001 |
ACS, acute coronary syndrome; CME, coronary microevagination; FC, fibrous cap; hsCRP, high-sensitive C-reactive protein; ICB, independent of coronary bifurcation; IFC, intact fibrous cap; LDL, low-density lipoprotein; MLA, minimum lumen area; STE-ACS, ST-elevation myocardial infarction.
Predictive value of cardiovascular risk factors, established features of plaque vulnerability, and CME for the presence of IFC-ICB-induced ACS
| Risk factor | Univariate | Multivariate | ||||
|---|---|---|---|---|---|---|
| RR | 95% CI | P-value | RR | 95% CI | P-value | |
| Clinical risk factors | ||||||
| Age | 0.99 | 0.97–1.01 | 0.152 | 0.99 | 0.96–1.01 | 0.239 |
| Diabetes mellitus | 0.93 | 0.49–1.78 | 0.837 | 0.57 | 0.20–1.59 | 0.283 |
| LDL cholesterol at admission | 1.00 | 0.99–1.00 | 0.427 | 1.00 | 0.99–1.01 | 0.846 |
| hsCRP at admission | 1.00 | 1.00–1.01 | 0.367 | 1.00 | 0.99–1.01 | 0.694 |
| Presentation as STE-ACS | 1.13 | 0.67–1.89 | 0.650 | 0.93 | 0.47–1.84 | 0.832 |
| Established criteria of plaque vulnerability | ||||||
| MLA < 3.5 mm2 | 0.64 | 0.26–1.57 | 0.329 | 1.76 | 0.95–3.24 | 0.072 |
| Lipid arc circumferential extension >180° | 0.27 | 0.15–0.47 | <0.001 | 0.54 | 0.27–1.07 | 0.077 |
| Fibrous cap thickness <75 µm | 0.09 | 0.05–0.16 | <0.001 | 0.14 | 0.05–0.34 | <0.001 |
| Macrophages | 0.33 | 0.18–0.59 | <0.001 | 0.94 | 0.49–1.79 | 0.846 |
| CME | 5.77 | 3.44–9.70 | <0.001 | 3.36 | 1.67–6.76 | 0.001 |
| Risk factor | Univariate | Multivariate | ||||
|---|---|---|---|---|---|---|
| RR | 95% CI | P-value | RR | 95% CI | P-value | |
| Clinical risk factors | ||||||
| Age | 0.99 | 0.97–1.01 | 0.152 | 0.99 | 0.96–1.01 | 0.239 |
| Diabetes mellitus | 0.93 | 0.49–1.78 | 0.837 | 0.57 | 0.20–1.59 | 0.283 |
| LDL cholesterol at admission | 1.00 | 0.99–1.00 | 0.427 | 1.00 | 0.99–1.01 | 0.846 |
| hsCRP at admission | 1.00 | 1.00–1.01 | 0.367 | 1.00 | 0.99–1.01 | 0.694 |
| Presentation as STE-ACS | 1.13 | 0.67–1.89 | 0.650 | 0.93 | 0.47–1.84 | 0.832 |
| Established criteria of plaque vulnerability | ||||||
| MLA < 3.5 mm2 | 0.64 | 0.26–1.57 | 0.329 | 1.76 | 0.95–3.24 | 0.072 |
| Lipid arc circumferential extension >180° | 0.27 | 0.15–0.47 | <0.001 | 0.54 | 0.27–1.07 | 0.077 |
| Fibrous cap thickness <75 µm | 0.09 | 0.05–0.16 | <0.001 | 0.14 | 0.05–0.34 | <0.001 |
| Macrophages | 0.33 | 0.18–0.59 | <0.001 | 0.94 | 0.49–1.79 | 0.846 |
| CME | 5.77 | 3.44–9.70 | <0.001 | 3.36 | 1.67–6.76 | 0.001 |
ACS, acute coronary syndrome; CME, coronary microevagination; FC, fibrous cap; hsCRP, high-sensitive C-reactive protein; ICB, independent of coronary bifurcation; IFC, intact fibrous cap; LDL, low-density lipoprotein; MLA, minimum lumen area; STE-ACS, ST-elevation myocardial infarction.
Results
Baseline characteristics
After OCT assessment of their ACS-causing CL pathology, followed by a comprehensive OCT Corelab analysis, 358 patients with either RFC-ACS or IFC-ACS as CL morphology were included in the final analysis of this pre-specified substudy (for details, see Supplementary data online, Figure S3). In addition, 521 non-CLs (NCLs) located within the culprit vessels were identified and subjected to OCT analysis following the same criteria as stated above for the ACS-causing culprit plaques. IFC- and RFC-ACS lesions were identified as underlying ACS pathology in 100 (27.9%) and 258 (72.1%) patients, respectively. Within the subgroup of patients presenting with IFC-ACS (n = 100), 48 (48%) lesions were IFC-ACS CL associated with a coronary bifurcation (IFC-ACB).
Supplementary data online, Table S1 summarizes the clinical baseline and laboratory data for the entire study cohort. Patients were predominantly men (74%, n = 318), with no sex or gender-based differences present, with a mean age of 63 ± 11.9 years and mostly presented with STE-ACS (62%, n = 269; see Supplementary data online, Table S1). The prevalence of cardiovascular risk factors was high with arterial hypertension and Type 2 diabetes observed in 335 (78%) and 89 (21%) patients, respectively (see Supplementary data online, Table S1). Within the subgroup of patients presenting with IFC-ACS (n = 100), 48 (48%) lesions were IFC-ACB, while 52% of IFC-ACS cases were independent (>3 mm away from a bifurcation or no bifurcation present) from a coronary bifurcation (IFC-ICB).
CME in ACS-culprit vessels
The incidence of CME was significantly higher (P < 0.001, Figure 2A) in CL (n = 88; 24.6%) when compared with NCL (n = 20; 3.8%). This was similar to the frequency of other established features of plaque vulnerability, including higher presence of thin-cap-fibroatheromas, smaller fibrous plaque cap thickness, higher maximum lipid arc, smaller MLA, and an accumulation of macrophages (all: P < 0.001, see Supplementary data online, Table S2).
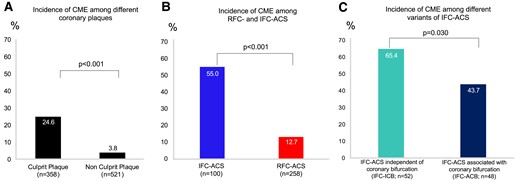
The presence of CMEs on coronary plaques. (A) Incidental finding of CME on ACS-causing culprit (n = 358) and non-culprit lesions (n = 521). (B) Incidence of CME within ACS-causing CL with ruptured (n = 258, RFC-ACS) and intact fibrous cap (n = 100, IFC-ACS). (C) Incidence of CME within IFC-ACS independent of a coronary bifurcation (n = 52, IFC-ICB) and IFC-ACS associated with a coronary bifurcation (n = 48, IFC-ACB). ACB, associated with coronary bifurcation; ACS, acute coronary syndrome; AUC, area under the curve; CME, coronary microevagination; ICB, independent of coronary bifurcation; IFC, intact fibrous cap.
Among culprit plaques, there was also a four-fold higher prevalence of CME in IFC-ACS when compared with RFC-ACS (55 vs. 13%, P < 0.001, Figure 2B and Supplementary data online, Table S3). Likewise, the maximum CME size (3.5 ± 2.5% of total lumen area) was higher in IFC-ACS when compared with RFC-ACS (2.0 ± 2.3%, P < 0.001; Supplementary data online, Table S3). CMEs were not associated with pre-dilation using an undersized balloon prior to OCT imaging in all ACS CLs (52% of non-CME CL, 42% of CME CL received pre-dilation, P = 0.116).
Importantly, with respect to the proximity of IFC-ACS-causing CL to flow-altering coronary bifurcations, CMEs were significantly more frequent (n = 34; 65%; P = 0.030) in IFC-ICB CL when compared with IFC-ACB (n = 21; 44%; Figure 2C). A comparison of established plaque vulnerability characteristics revealed no differences with respect to MLA<3.5 mm2, fibrous cap (FC) thickness <75 μm, max lipid arc >180°, as well as the prevalence of OCT-detected macrophages among IFC-ACB and IFC-ICB (see Supplementary data online, Table S4).
CME distinguish ICB- from ACB-IFC-ACS-causing culprit lesions
By multivariable regression analysis, the presence of CME was significantly associated with the presence of IFC-ICB [relative risk (RR) = 3.36, 95% confidence interval (CI) 1.67–6.76, P = 0.001; Table 1] and outranged cardiovascular risk factors and other established factors of plaque vulnerability,29 such as MLA <3.5 mm2 (RR = 1.76, 95% CI 0.95–3.24, P = 0.072), lipid arc circumferential >180° (RR = 0.54, 95% CI 0.27–1.07, P = 0.077), the presence of macrophages (RR = 0.94, 95% CI 0.49–1.79, P = 0.846), as well as CL with a FC thickness <75 μm showing an inverse correlation indicating that IFC-ICB are associated with thicker FCs (RR = 0.14, 95% CI 0.05–0.34, P < 0.001). Likewise, CME showed the highest predictive value (AUC = 0.777) for IFC-ICB lesions when compared with other OCT-derived criteria of plaque vulnerability (Figure 3A). Finally, adding CME to a model representing established OCT-derived plaque criteria and selected clinical parameters (Figure 3B) resulted in a further significant increase in the AUC (AUC +0.08; P = 0.001) for the accuracy to predict IFC-ICB lesions.
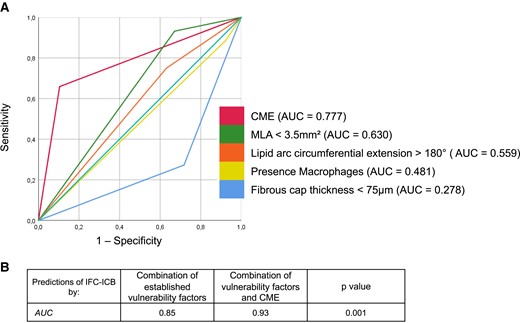
(A) The presence of CME in comparison with other established criteria of plaque vulnerability to characterize IFC-ICB-causing culprit lesions. (B) Added accuracy of CME to established criteria of plaque vulnerability to predict IFC-ICB-ACS. ACB, associated with a coronary bifurcation; ACS, acute coronary syndrome; AUC, area under the curve; CME, coronary microevagination; ICB, independent of coronary bifurcation; IFC, intact fibrous cap; MLA, minimal lumen area.
Differential inflammatory microenvironment at an IFC-ICB CL
The inflammatory microenvironment of IFC-ACB CL was characterized by a significantly higher concentration of a subset of CD3+CD4+ T cells (0.9 ± 0.2 vs. 1.1 ± 0.2, P = 0.034; Figure 4) when compared with IFC-ICB CL. In contrast, IFC-ICB CL was characterized by a more pronounced activation of the innate immune system with a selective local enrichment of monocytes (1.1 ± 0.2 vs. 0.9 ± 0.2, P = 0.048; Figure 4), especially the subgroup of CD14+CD16− classical monocytes (1.0 ± 0.2 vs. 0.9 ± 0.2, P = 0.034; Figure 4). Other immune cells were comparable between IFC-ACB and IFC-ICB irrespective of their proximity to a coronary bifurcation (Figure 4). Importantly, the observed monocyte enrichment in IFC-ICB CL was also replicable by investigating thrombotic material aspirated from IFC-ICB CL (326 ± 162 vs. 96 ± 87 cells/mm2; P = 0.017; Figure 5 and see Supplementary data online, Table S5).

Culprit lesion ratio (CR) of different immune cells in IFC-ACS among their proximity to coronary bifurcations. All values are expressed as a median (IQR). ACB, associated with coronary bifurcation; CD, cluster of differentiation; CL, culprit lesion; ICB, independent of coronary bifurcation; IFC-ACS, Intact fibrous cap acute coronary syndrome; IQR, interquartile range; NK, natural killer; SD, standard deviation.
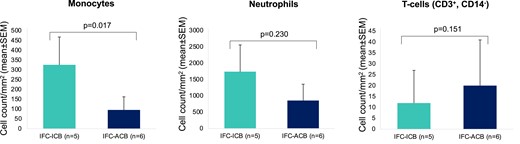
Cell counts of different immune cells as derived from coronary thrombi aspirated in IFC-ACS, associated with a coronary bifurcation (IFC-ACB) or independent of a coronary bifurcation (IFC-ICB). ACB, associated with a coronary bifurcation; CD, cluster of differentiation; CL, culprit lesion; ICB, independent of coronary bifurcation; IFC-ACS, intact fibrous cap acute coronary syndrome; SD, standard deviation.
Discussion
This pre-specified subanalysis of the OPTICO-ACS study demonstrated for the first time the relevance of CME in ACS patients. Furthermore, this study highlights CME as a novel independent plaque feature in IFC-ICB, a subtype of IFC-ACS without association to coronary bifurcations, and without any known evidence for its underlying pathophysiology yet. This study extends previous reports on the OCT-based observation of CME with a particular focus on their relation to coronary bifurcations and a differential participation of the innate and adaptive immune system in IFC-ICB and IFC-ACB lesions.8 Therefore, this study advances our pathophysiological understanding of IFC-ACS and promotes the concept that IFC-ACS comprises a variety of different pathophysiological causes, finally ending in an eroded CL (Graphical Abstract).
CMEs and their impact on increasing plaque vulnerability
Evaginations as assessed by OCT have recently been described as a unique feature of plaque remodelling after treating CAD with different coronary devices.18–22 These evaginations after PCI have been associated with local flow disturbances in previous 3D remodelling studies, possibly explaining their association with positive vessel remodelling and very late stent thrombosis.18,19,30 This subanalysis of the OPTICO-ACS trial demonstrates for the first time a significant role of CME also in the absence of device implantation, with a quarter of all ACS patients showing CME within the culprit plaque. The progression of atherosclerosis and subsequent plaque destabilization seem to have similar key mechanisms to those following device implantation, namely the processes of adverse vessel remodelling.11 Since the OPTICO-ACS study programme uses high-resolution intraluminal OCT imaging, a precise identification of the individual ACS triggering underlying pathophysiology was possible and allowed for the depiction of the complex pictures of different ACS phenotypes and the role of CME therein.
Role of coronary flow disturbances in the pathophysiology of IFC-ACS
The pathogenesis of IFC-ACS remains one of the main unresolved issues in atherosclerosis research,4 with large proportions of past research focusing on the pathogenesis of the most frequent ACS pathophysiology, RFC-ACS. However, with increasing incidences of IFC-ACS, possibly due to the modern era of intensive lipid-lowering therapies,13 the exact pathomechanism of IFC-ACS has shifted into the centre of interest in atherosclerotic research.4
Recent large trials have suggested that IFC-ACS lesions frequently occur in close proximity to coronary bifurcations,2,8 hence emphasizing the role of shear stress and subsequent flow alterations to be causal for the development of IFC-ACS.8,14 Nonetheless, with this pathomechanism, we can only explain the occurrence of IFC-ACS in association with coronary bifurcations. However, with ∼40% of IFC-ACS occurring independently of coronary bifurcations,2,8 other factors causing endothelial injury come into play.11,12,31 The clear accumulation of CME in IFC-ACS and their increased prevalence in the subgroup of IFC-ICB support the hypothesis that CME by themselves induce flow alterations, possibly resulting in adverse vessel remodelling. In support of this hypothesis, CMEs were identified as the strongest predictor of IFC-ICB, surpassing established factors of plaque vulnerability.29 This suggests that the epiphenomenon of CME as identified by intracoronary imaging methods may help us to detect a coronary plaque prone to erosion at an early stage. Of note, this would certainly need to be confirmed by further prospective longitudinal studies, potentially allowing for an investigation into how CMEs develop as this cannot be concluded within this study design.
Distinct immunological characteristics of IFC-ACS due to the proximity to coronary bifurcations
Using a unique translational study design to investigate the microenvironment of an ACS-causing CL, we recently revealed a flow-dependent activation and accumulation of the adaptive immune system in IFC-ACS.8 In fact, an accumulation, activation, and endothelial adhesion of CD8+ T cells was initiated and maintained by altered flow conditions,8,32 giving a first solid pathophysiological explanation for the genesis of IFC-ACS located close to coronary bifurcations. This substudy adjusts this recent finding by revealing clear inflammatory differences among IFC-ACB and IFC-ICB. In fact, the present study fills a gap in our pathophysiologic understanding by identifying a local accumulation of monocytes in IFC-ICB, where a high incidence of flow-mediating CME was observed. Monocytes are an integral part of the processes of vessel inflammation and especially during positive vessel remodelling33,34 and therefore may explain the local enrichment observed in IFC-ICB. Consequently, CME may alter coronary blood flow and ultimately lead to the activation of a coronary plaque, resulting in IFC-ACS, even in the absence of a coronary bifurcation (IFC-ICB). Therefore, future studies are needed to establish this alternative pathophysiological pathway of IFC-ACS and to identify potential targets for personalized treatment algorithms, including anti-inflammatory therapies or interventional plaque-sealing strategies.35,36
Limitations
There are several limitations in this study, which should be considered. First, while patients with known inflammatory or malignant diseases, or acute clinical or laboratory signs of infections were excluded from the study, the possibility cannot be ruled out that subjects with inapparent comorbidities may have been included in the study and hence influenced the results. Secondly, even though thrombus aspiration was performed in the immediate proximity of the CL, the aspirated material may not exclusively represent the microenvironment of the CL. Since local blood was withdrawn only at the time of ACS, it cannot be definitely determined whether local immune cell accumulations are the consequence or cause of the plaque morphology. To further answer this question, a study design with multiple time points for local blood withdrawal would be required. Since OCT was not performed in the left main disease, coronary bypass grafts, CL in very distal locations, and cases where the CL was unclear, the results of the study may not be applicable to these patient populations. Thirdly, since two independent core labs evaluated each case, exclusions due to unattainable agreements between core labs may have introduced a selection bias. Lastly, with pre-dilatation with an undersized balloon being necessary in some patients, the possibility that the integrity of the culprit plaque was affected cannot be ruled out; however, a statistical work-up showed no significant accumulation of CME on CLs experiencing prior pre-dilation. In order to prove a causal relationship between inflammatory cell accumulation and CME density, a different study design would be required. Prospective studies are needed to confirm CME as a source or marker of plaque vulnerability in IFC-ICB.
Conclusion
This dedicated substudy from the translational OPTICO-ACS study programme identified for the first time a potential role of CME in the pathogenesis of IFC-ICB, where CME-induced coronary flow alterations and monocyte-derived inflammation may contribute to advanced adverse plaque remodelling. Moreover, the occurrence of CME in the absence of previous stenting holds great potential for CME as a novel marker of plaque vulnerability, specifically during plaque destabilization with subsequent IFC-ACS.
Supplementary data
Supplementary data are available at European Heart Journal - Cardiovascular Imaging online.
Acknowledgements
The authors acknowledge the support of the catheterization staff at all three sites of Charité University Medicine Berlin. Furthermore, the authors express their appreciation to Sabine Knüppel, Andrea Heuberger, MD, and Joachim Weber, MD. Finally, the authors thank the staff of the BIH, including Anne Gössinger (study coordinator), Kristin Simon, and Katrin Haug (study nurses). All figures are original, with the graphical abstract and Figure 1 created with www.Biorender.com and Figure 4 created using Graph Pad Prism 9 (GraphPad Software Inc).
Funding
The OPTICO-ACS trial was funded by a CRU grant from the Berlin Institute of Health (BIH, Berlin, Germany), as well as from the Deutsches Zentrum für Herzkreislaufforschung (“German Centre for Cardiovascular Research”; DZHK; FKZ: 81Z2100202, Berlin., Germany) to D.M.L. Additionally, an education grant from Abbott Vascular (Chicago, Illinois, USA) was received for providing OCT imaging to D.M.L. N.K. was supported by the Deutsche Stiftung für Herzforschung (Project: F/39/17, Frankfurt am Main, Germany). T.G. was part of the Berlin Institute of Health (BIH, Berlin Germany) Junior Clinician Scientist Program. T.G. received a research fellowship by the German Cardiac Society (#DGK12/2020, Düsseldorf, Germany), a Postdoc-Start-up Grant by the German Centre for Cardiovascular Research (DZHK, Berlin, Germany), and a Vascular Biomedicine Grant by the Berlin Center for Translational Vascular Biomedicine (BIH, Charité, MDC, Berlin, Germany).
Data availability
The data underlying this article will be shared on reasonable request with the corresponding author.
References
Author notes
Conflict of interest: D.M.L. received lecture honoraria from Amgen, Abbott Vascular, AstraZeneca, and Novo Nordisk. M.J. received consulting fees from Biotronik, TriCares, Veryan, and Shockwave, and is in the Steering Committee of Biotronik and Edwards Lifesciences. T.D.T. received payment honoraria from Novartis, AstraZeneca, Berlin Chemie, Abbott, NeoVasc, and Amgen. U.L. reports lecture and advisory honorary from Abbott. All other authors report no conflicts of interest.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}