





Abstract
Prunus mongolica is an ecologically and economically important xerophytic tree native to Northwest China. Here, we report a high-quality, chromosome-level P. mongolica genome assembly integrating PacBio high-fidelity sequencing and Hi-C technology. The assembled genome was 233.17 Mb in size, with 98.89% assigned to eight pseudochromosomes. The genome had contig and scaffold N50s of 24.33 Mb and 26.54 Mb, respectively, a BUSCO completeness score of 98.76%, and CEGMA indicated that 98.47% of the assembled genome was reliably annotated. The genome contained a total of 88.54 Mb (37.97%) of repetitive sequences and 23,798 protein-coding genes. We found that P. mongolica experienced two whole-genome duplications, with the most recent event occurring ~3.57 million years ago. Phylogenetic and chromosome syntenic analyses revealed that P. mongolica was closely related to P. persica and P. dulcis. Furthermore, we identified a number of candidate genes involved in drought tolerance and fatty acid biosynthesis. These candidate genes are likely to prove useful in studies of drought tolerance and fatty acid biosynthesis in P. mongolica, and will provide important genetic resources for molecular breeding and improvement experiments in Prunus species. This high-quality reference genome will also accelerate the study of the adaptation of xerophytic plants to drought.
1. Introduction
Climate change-induced drought is responsible for substantial negative impacts to natural ecosystems, including altering ecosystem structure and function, and decreasing plant productivity, soil fertility, species richness, and plant cover.1 Nevertheless, many xeric plants have exhibited adaptability in the face of continued drought due to their unique adaptive traits.2 Xeric plants have historically been crucial for the maintenance of ecosystem health and agricultural development in arid regions. However, the distribution and population size of many xeric plant species have declined significantly due to human activities, such as grazing, tourism, and surface mining. This trend is expected to continue as economic and social development continues to reduce and degrade arid wildland habitats. Therefore, it is of critical importance to study and characterize the biological and genetic resources of xeric plants for breeding and other purposes.
The Mongolian almond (Prunus mongolica, Rosaceae) is a xeric, diploid (2n = 2x = 16)3 shrub distributed across the arid regions of western China, including inner and southern Mongolia, Gansu, and Ningxia, among others. P. mongolica is extremely drought resistant and grows primarily in arid hilly, mountainous, and desert regions where the annual rainfall ranges between 50 and 200 mm.4P. mongolica has evolved special genetic resources such as drought resistance, cold resistance, sand resistance, and barren resistance.5 Furthermore, P. mongolica is highly eco-functional, providing both biomass and medicinal compounds.6 The seeds of this species have high food and medicinal value, as the kernels are rich in oil (54.85%) and have been used to treat renal fibrosis.7 In China, P. mongolica has been listed as an endangered Tertiary relict plant and an endangered second-class protected plant, and has also been listed as ‘vulnerable’ by the International Union for the Conservation of Nature.
A thorough characterization of the P. mongolica genome would contribute to our understanding of the phylogeny of Rosaceae; the paleo-floristic, paleo-geographic, and climatic characteristics of the Tertiary Period; and the evolutionary and successional history of the flora endemic to arid central Asia. However, no previous genome-wide investigations of P. mongolica have been reported, likely because a high-quality chromosome-level gene map is unavailable. In this study, we assembled a chromosome-level P. mongolica genomic assembly using a combination of PacBio high-fidelity (HiFi) reads and Hi-C reads. Using this high-quality genome, we investigated the evolutionary history of P. mongolica, and identified a number of candidate genes involved in fatty acid biosynthesis and drought resistance. The genome assembly presented here will provide a valuable resource for the evolutionary study of xerophytic plants as well as for the successful breeding of this ecologically and economically important species.
2. Materials and methods
2.1. Plant materials and DNA extraction
Fresh leaves of mature P. mongolica specimens were collected from Yinchuan Botanical Garden (106°10’36’’E,38°25’19’’N), Ningxia Province, northwestern China (Fig. 1). Leaf samples were immediately frozen in liquid nitrogen and stored for further analysis. For whole-genome sequencing, total genomic DNA was extracted from fresh leaves using a modified CTAB method.8 After extraction, the concentration, integrity, and purity of the DNA were determined using a NanoDrop spectrophotometer (A260/A280 = 1.8, A260/A230 = 2.0–2.2) and a Qubit.

Morphological features of Prunus mongolica. (A) Whole plant in flowers, (B) flower, (C) Young fruit, (D) Ripe fruit and branches.
2.2. Genome sequencing
An Illumina genomic library was constructed according to Illumina’s standard protocol. Paired-end (PE) reads (2 × 150 bp) sequenced on an Illumina NovaSeq 6000 platform were used for genomic survey and assessment. To produce CCS reads (HiFi) for contig assembly, the genomes were sequenced using a PacBio Sequel II platform (Pacific Biosciences). For Hi-C sequencing, the chromosomal structure was crosslinked with formaldehyde, and the genomic DNA was digested using HindIII.9 After a Hi-C library with a 300–700 bp insert size was constructed, the concentration and insert size were detected using a Qubit2.0 and an Agilent 2100, and the effective concentration was quantified by qPCR. After the libraries were qualified, high-throughput sequencing was performed with an Illumina NovaSeq 6000 platform, with a PE150 reading length.10 To aid in gene annotation and phylogenomic analyses, fresh leaves, roots, shoots, flowers, and fruits from the same P. mongolica specimen were collected for RNA sequencing (RNA-seq). High-quality RNA-seq libraries were prepared and sequenced with an Illumina NovaSeq 6000 platform. RNA-seq reads were filtered using Trimmomatic11 (version 0.36), with default parameters. Low-quality sequencing reads were filtered out and were excluded from further analyses. All sequencing services were provided by Biomarker Technologies Co., Ltd. (Beijing, China).
2.3. Genome survey and assembly
Genome size and complexity were estimated based on the k-mer distribution of Illumina short reads. GenomeScope12 (version 2.0) was used to count the distribution of 21-mers, with default parameters. A de novo assembly of the PacBio HiFi reads was constructed with Hifiasm13 (version 0.16), and redundant sequences were filtered out with purge_dups.14 CEGMA15 (version 2.5) and BUSCO16 (version 4) were used to assess the completeness of the genome and gene annotation.
2.4. Chromosome assembly using Hi-C
To generate high-quality Hi-C reads for chromosome-level assembly, adapter sequences and low-quality PE reads were removed. The resultant high-quality Hi-C data were truncated at putative Hi-C junctions and mapped to contigs using BWA17 (version 0.7.10-r789). HiC-Pro18 (version 2.10.0) was used to filter valid reads, and only uniquely mapped PE reads were selected for further analyses. Genome sequences were clustered and ordered onto chromosomes using LACHESIS, with the following parameters: Cluster_min_re_sites = 100, Cluster_max_link_density = 2, order_min_n_res_in_trunk = 15, order_min_n_res_in_shreds = 1.5.
2.5. Protein-coding gene prediction
We integrated three approaches to annotate protein-coding genes in the genome: de novo prediction, homology search, and transcript-based assembly. The de novo gene models were predicted using two ab initio gene-prediction software tools, Augustus19 (version 3.1.0) and SNAP20 (2006-07-28). For the homology-based approach, GeMoMa21 (version 1.7) was used to construct a reference gene model using data from several related species (P. dulcis, P. armeniaca, P. mume, and P. persica). For the transcript-based prediction, RNA-seq data were mapped to the reference genome using Hisat22 (version 2.1.0) and assembled by Stringtie23 (version 2.1.4). GeneMarkS-T24 (version 5.1) was used to predict genes based on the assembled transcripts. PASA25 (version 2.4.1) was used to predict genes based on a combination of full-length PacBio transcripts and unigenes assembled by Trinity26 (version 2.11). Gene models from these different approaches were combined using EVM27 (version 1.1.1) and updated using PASA.
2.6. Functional annotation
Functional annotation of P. mongolica protein-coding genes was performed by a BLASTP against several public databases, including EggNOG28 (5.0, http://eggnog5.embl.de/#/app/home. de/download/eggnog_5.0/), gene ontology29 (GO, 20200615, http://geneontology.org), eukaryotic orthologous groups30 (KOG, 20110125), Pfam31 (version 33.1), TrEMBL,32 Non-Redundant33 (NR, 202009, ftp://ftp.ncbi.nlm.nih.gov/blast/db), SwissProt32 (202005, http://ftp.ebi.ac.uk/pub/databases/swissprot), and Kyoto Encyclopedia of Genes and Genomes34 (KEGG, 20191220), with an E-value cut-off of 1.0 × 10−5.
2.7. Identification of repetitive elements
RepeatModeler 235 (version 2.0.1) was used to customize a de novo genomic repeat library which could automatically execute two de novo repeat-finding programs: RECON36 (version 1.0.8) and RepeatScout37 (version 1.0.6). To classify the predicted results, RepeatClassifier35 was used to search the Dfam38 (version 3.5) database. Full-length long terminal repeat retrotransposons (fl-LTR-RTs) were identified using both LTRharvest39 (version 1.5.10) (-minlenltr 100 -maxlenltr 40000 -mintsd 4 -maxtsd 6 -motif TGCA -motifmis 1 -similar 85 -vic 10 -seed 20 -seqids yes) and LTR_FINDER40 (version 1.07) (-D 40000 -d 100 -L 9000 -l 50 -p 20 -C -M 0.9). The high-quality intact fl-LTR-RTs and the non-redundant LTR library were then produced using LTR_retriever41 (version 2.9.0). The sequences flanking both sides of LTRs were extracted and compared using MAFFT42 (version 7.205), and the distance was calculated using the Kimura model in EMBOSS43 (version 6.6.0). The integration times (million years ago, Mya) of intact LTRs were estimated using the following equation: T = K/2r, where K is the number of nucleotide substitutions per site between each LTR pair, and r is the nucleotide substitution rate (7 × 10–9 substitutions per site per year44). A non-redundant, species-specific TE library was constructed by combining the de novo TE sequence library with data from the Repbase (version 19.06), REXdb (version 3.0) and Dfam (version 3.2) databases. Final P. mongolica genomic TE sequences were identified and classified by homology search against the library using RepeatMasker45 (version 4.10). Tandem repeats were annotated using Tandem Repeats Finder46 (version 409) and the MIcroSAtellite identification tool47 (version 2.1).
2.8. Identification of pseudogenes and non-coding RNA genes
In this study, we identified several non-coding RNA molecules, including miRNA, rRNA, tRNA, snoRNA, and snRNA. tRNAscan-SE48 (version 1.3.1) was used to predict tRNA, using ‘eukaryote’ parameters. Barrnap49 (version 0.9) was used to predict rRNA. miRBase50 (release 21) was used to predict miRNA. Both snoRNA and snRNA genes were predicted using INFERNAL51 (version 1.1) to search the Rfam52 (release 14.5) database. GenBlastA53 (version 1.0.4) and GeneWise54 (version 2.4.1) were used to predict pseudogenes.
2.9. Reconstruction of the phylogenetic tree
OrthoFinder55 (version 2.5.1) was used to identify homologous genes in P. mongolica and 10 other species. The protein sequences of the single-copy orthologs were aligned with MAFFT43 (version 7.205), with default parameters. The unique gene families were identified and annotated using the Pfam56 (version 33.1) database. GO and KEGG enrichment analyses were carried out using clusterProfiler57 (version 3.6.0). To estimate the best substitution models, ModelFinder58 was implemented in IQ-TREE59 (version 1.6.11). To construct the maximum-likelihood tree, RAxML was used with the best-fit substitution model, with 1,000 bootstrap replicates. Divergence time was estimated using the MCMCTreeR60 (version 1.1) program in the Phylogenetic Analysis by Maximum Likelihood software61 (PAML, version 4.9i), using three secondary calibration points [P. mongolica vs Amborella trichopoda (179–199.1), P. dulcis vs P. armeniaca (3.8–10.2), and P. dulcis vs Fragaria nilgerrensis (44.5–89.3)] obtained from the TimeTree database (http://www.timetree.org/). The graphical phylogenetic tree was constructed using MCMCTreeR.
2.10. Analyses of gene-family expansion and contraction
Based on the identified gene families, the phylogenetic tree, and the predicted divergence times, we used Computational Analysis of gene Family Evolution62 (CAFE, version 4.2) to analyse gene-family expansion and contraction. In CAFE, a random birth and death model is used to study gene gain or loss in gene families across a specified phylogenetic tree. Then, a conditional P-value is calculated for each gene family, and families with conditional P-values < 0.05 are considered to have an accelerated rate of gene gain or loss. The expanded and contracted gene families in P. mongolica (P-value ≤ 0.05) were subjected to KEGG pathway enrichment analysis. This method utilized hypergeometric test algorithms, and the Q-value (False Discovery Rate, FDR) was calculated to adjust the P-value using the R package q-value (https://github.com/StoreyLab/qvalue).
2.11. Positively selected genes
Based on the phylogenetic tree, we selected five closely related species (P. armeniaca, P. mume, P. dulcis, P. mongolica, and P. persica) to estimate the ratio (ω) of non-synonymous (Ka) to synonymous (Ks) nucleotide substitutions using PAML63 (version 4.9e). ClusterProfiler57 (version 3.6.0) was used to perform GO and KEGG enrichment analyses on the positively selected genes of P. mongolica.
2.12. Synteny analysis and whole-genome duplication
Synteny analysis was performed using Diamond64 (version 0.9.29.130). MCScanX65 was used to identify collinear blocks. The synonymous mutation rate (Ks) and fourfold synonymous third-codon transversion rate (4DTv) are commonly used to identify whole-genome duplications (WGDs). Here, wgd software66 (version 1.1.1) was used with a custom script (https://github.com/JinfengChen/Scripts) to identify WGD events in P. mongolica.
2.13. Identification of fatty acid biosynthesis and drought resistance genes
Drought resistance, fatty acid biosynthesis, and triglyceride biosynthesis genes in the P. mongolica genome were identified by homologous search against the Arabidopsis thaliana genome. The candidate genes were further filtered by checking their Enzyme Commission number.
3. Results and discussion
3.1. High-quality P. mongolica genome assembly
Prior to assembly, a total of 18.18 Gb of Illumina data were generated (Supplementary Table S1). In total, 1,574,319,910 k-mers (k = 21) were identified, and a major peak was observed at a k-depth of 33 (Supplementary Fig. S1). After removing low-frequency k-mers, the P. mongolica genome size was estimated to be 226.47 Mb, with a high level of heterozygosity (1.61%) and a large percentage of repeat sequences (39.01%). To assemble this highly complex genome, we obtained a total of 9.59 Gb (×42.34 genome coverage) of HiFi long reads, with a maximum read length of 42,695 bp and an average read length of 15,176 bp (Supplementary Tables S1 and S2). Using Hifiasm stitching, the total length of the genomic contig sequence was found to be 271.26 Mb, with a contig N50 of 24.33 Mb. After removal of haplotigs and overlaps, the total length of the genomic contig sequence was 233.17 Mb, with a contig N50 of 24.33 Mb (Supplementary Table S2). To validate the quality of the Hifiasm stitching, we first mapped the Illumina reads back to the assembly and obtained an overall mapping rate of 98.36%, 10-fold minimum genome coverage of 97.54%, and an average sequencing depth of ×73 (Supplementary Table S3). Furthermore, we obtained a BUSCO completeness score of 98.76% and CEGMA indicated that 98.47% of the assembled P. mongolica genome was reliably annotated (Supplementary Tables S4 and S5). Taken together, these results suggest that our P. mongolica genome assembly was high quality and complete.
To reconstruct a chromosome-level assembly, we generated 31.49 Gb (×139.04 genome coverage) of Hi-C clean reads (Supplementary Table S1) and anchored the contigs onto pseudochromosomes using the Hi-C scaffolding approach. Ultimately, 227,864,786 bp of sequences (97.73% of the entire assembly) were assigned to eight pseudochromosomes (Fig. 2A), corresponding to the haploid chromosome number of P. mongolica. The lengths of the pseudochromosomes ranged from 18,973,745 to 47,189,407 bp (Supplementary Table S6). The corresponding heat map revealed that the eight chromosomal groupings were clearly distinguishable and that all pseudochromosomes exhibited a well-organized diagonal pattern of intra-chromosomal interactions (Fig. 2B and Supplementary Fig. S2), suggesting a high-quality Hi-C-assisted genome assembly. The length of sequences whose order and orientation could be determined was 225,330,101 bp, accounting for 98.89% of the total length of mapped chromosome sequences. The final genome assembly contained very few gaps, with an average of seven gaps per pseudochromosome. The contig and scaffold N50 values were 24,328,480 bp and 26,540,977 bp, respectively (Table 1 and Supplemental Table S7). Taken together, these statistics verified that our genome assembly was precise, complete, and of high quality at the chromosome scale.
Global statistics of Prunus mongolica genome assembly and annotation
| Parameter | Size or number |
|---|---|
| Estimate of genome size (survey), Mb | 226,470,058 |
| Assembled genome size, bp | 233,169,053 |
| Total length of contigs, bp | 233,168,353 |
| Total number of contigs | 91 |
| N50 of contigs, bp | 24,328,480 |
| Largest contig, bp | 30,055,353 |
| Total length of scaffolds, bp | 233,169,053 |
| Total number of scaffolds | 84 |
| N50 of scaffolds, bp | 26,540,977 |
| Largest scaffold, bp | 47,189,607 |
| GC content, % | 38.20 |
| Complete CEGMA, % | 98.47 |
| Complete BUSCOs, % | 98.76 |
| Total length of repeat, bp | 88,539,904 |
| Repeat density, % | 37.97 |
| Long terminal repeat (LTR) density, % | 17.92 |
| Microsatellite repeat density, % | 0.94 |
| Number of protein-coding genes | 23,798 |
| Number of annotated genes | 23,702 |
| Number of rRNA | 832 |
| Number of tRNA | 625 |
| Number of miRNAs | 217 |
| Number of snRNAs | 283 |
| Number of snoRNAs | 491 |
| Number of pseudogenes | 161 |
| Parameter | Size or number |
|---|---|
| Estimate of genome size (survey), Mb | 226,470,058 |
| Assembled genome size, bp | 233,169,053 |
| Total length of contigs, bp | 233,168,353 |
| Total number of contigs | 91 |
| N50 of contigs, bp | 24,328,480 |
| Largest contig, bp | 30,055,353 |
| Total length of scaffolds, bp | 233,169,053 |
| Total number of scaffolds | 84 |
| N50 of scaffolds, bp | 26,540,977 |
| Largest scaffold, bp | 47,189,607 |
| GC content, % | 38.20 |
| Complete CEGMA, % | 98.47 |
| Complete BUSCOs, % | 98.76 |
| Total length of repeat, bp | 88,539,904 |
| Repeat density, % | 37.97 |
| Long terminal repeat (LTR) density, % | 17.92 |
| Microsatellite repeat density, % | 0.94 |
| Number of protein-coding genes | 23,798 |
| Number of annotated genes | 23,702 |
| Number of rRNA | 832 |
| Number of tRNA | 625 |
| Number of miRNAs | 217 |
| Number of snRNAs | 283 |
| Number of snoRNAs | 491 |
| Number of pseudogenes | 161 |
Global statistics of Prunus mongolica genome assembly and annotation
| Parameter | Size or number |
|---|---|
| Estimate of genome size (survey), Mb | 226,470,058 |
| Assembled genome size, bp | 233,169,053 |
| Total length of contigs, bp | 233,168,353 |
| Total number of contigs | 91 |
| N50 of contigs, bp | 24,328,480 |
| Largest contig, bp | 30,055,353 |
| Total length of scaffolds, bp | 233,169,053 |
| Total number of scaffolds | 84 |
| N50 of scaffolds, bp | 26,540,977 |
| Largest scaffold, bp | 47,189,607 |
| GC content, % | 38.20 |
| Complete CEGMA, % | 98.47 |
| Complete BUSCOs, % | 98.76 |
| Total length of repeat, bp | 88,539,904 |
| Repeat density, % | 37.97 |
| Long terminal repeat (LTR) density, % | 17.92 |
| Microsatellite repeat density, % | 0.94 |
| Number of protein-coding genes | 23,798 |
| Number of annotated genes | 23,702 |
| Number of rRNA | 832 |
| Number of tRNA | 625 |
| Number of miRNAs | 217 |
| Number of snRNAs | 283 |
| Number of snoRNAs | 491 |
| Number of pseudogenes | 161 |
| Parameter | Size or number |
|---|---|
| Estimate of genome size (survey), Mb | 226,470,058 |
| Assembled genome size, bp | 233,169,053 |
| Total length of contigs, bp | 233,168,353 |
| Total number of contigs | 91 |
| N50 of contigs, bp | 24,328,480 |
| Largest contig, bp | 30,055,353 |
| Total length of scaffolds, bp | 233,169,053 |
| Total number of scaffolds | 84 |
| N50 of scaffolds, bp | 26,540,977 |
| Largest scaffold, bp | 47,189,607 |
| GC content, % | 38.20 |
| Complete CEGMA, % | 98.47 |
| Complete BUSCOs, % | 98.76 |
| Total length of repeat, bp | 88,539,904 |
| Repeat density, % | 37.97 |
| Long terminal repeat (LTR) density, % | 17.92 |
| Microsatellite repeat density, % | 0.94 |
| Number of protein-coding genes | 23,798 |
| Number of annotated genes | 23,702 |
| Number of rRNA | 832 |
| Number of tRNA | 625 |
| Number of miRNAs | 217 |
| Number of snRNAs | 283 |
| Number of snoRNAs | 491 |
| Number of pseudogenes | 161 |
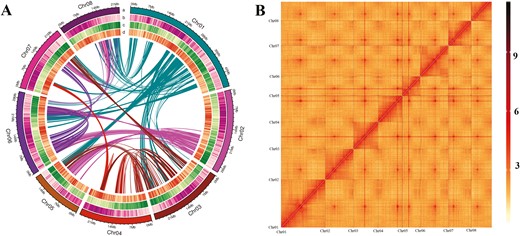
Overview of Prunus mongolica and its genome. (A) Landscape of the P. mongolica genome. The tracks from outer to inner circles indicate the following: a, chromosome ideograms; b, TE (transposable elements) density; c, gene density; d, GC content. (B) Heatmap showing Hi-C interactions at a resolution of 300 kb.
3.2. Annotation of the P. mongolica genome
We annotated a total of 88,539,904 bp of repetitive sequences, accounting for 37.96% of the entire genome (Supplementary Table S8). Transposable elements (TEs) were the most abundant class of repeats, spanning 59,900,478 bp or 25.68% of the genome. Long terminal repeat-retrotransposons (LTR-RTs) were the next most abundant class of repeats (27,352,348 bp or 11.72% of the genome), including Ty1-Copia superfamily retrotransposons (4.84%), Ty3-Gypsy superfamily retrotransposons (6.53%), and other LTR elements (0.36%). A total of 166,321 tandem repeats were also identified, accounting for 14,174,342 bp or 6.08% of the genome. In addition, we identified 28,806,220 bp (12.35% of the genome) of unknown repetitive sequences, which may be specific to P. mongolica.
By combining transcriptome-based, homology-based, and ab initio methods, a total of 23,798 protein-coding genes were predicted, of which 23,573 (99.05%) were located on the eight pseudochromosomes (Table 1, Supplementary Table S9, and Supplementary Fig. S3A). The average gene length was 3,448.46 bp with 5.67 exons per gene, with an average exon length of 1,793.77 bp (Supplementary Table S10 and Supplementary Fig. S3B–D). Besides the protein-coding genes, we also identified 2,448 small RNAs and 161 pseudogenes. Of the small RNAs, we identified 625 tRNAs, 832 rRNAs, 217 microRNAs, 283 snRNAs, and 491 snoRNAs (Table 1). A BUSCO completeness score of 98.88% was obtained at the gene model level, indicating the high completeness of gene annotation (Supplementary Table S4). Taken together, these statistics indicate the high accuracy of gene prediction across the P. mongolica genome.
Searching these genes against the Pfam, Swiss-Prot, TrEMBL, EggNOG (Supplementary Fig. S4A), and NR (Supplementary Table S11) databases resulted in the functional annotation of 88.62%, 80.99%, 99.58%, 84.71%, and 99.07% genes, respectively. We further annotated these genes using the KOG, GO (Supplementary Fig. S4B), and KEGG databases (Supplementary Table S11).
3.3. Evolutionary history of P. mongolica
To investigate the evolutionary history of P. mongolica, we compared the P. mongolica genome to eight closely related Rosaceae plants (P. persica, P. avium, P. mume, P. dulcis, P. armeniaca, Malus domestica, Rosa chinensis, and F. nilgerrensis) and two outgroups (A. trichopoda and Vitis vinifera) (Supplemental Table S12). Among these 11 plant species, 33,254 orthologous families and 1,552 single-copy families were identified. Based on the single-copy genes, a maximum likelihood-based phylogenetic analysis was conducted, revealing that the most recent common ancestor of the 11 species contained 23,287 gene families and 1,538 high-quality single-copy orthologous genes (Fig. 3C). The results further indicated that P. mongolica is most closely related to P. persica and P. dulcis. Molecular dating using A. trichopoda and V. vinifera for fossil calibration indicated that P. mongolica-P. dulcis emerged 2–6 Mya and that P. mongolica-P. persica emerged 2–5 Mya.
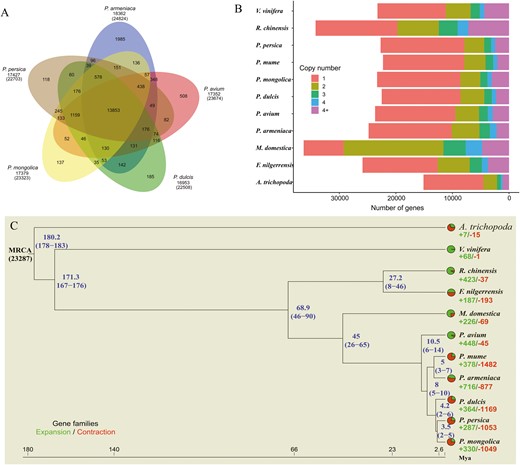
(A) Venn diagram of gene families cluster in five prunus species. (B and C) phylogenetic analysis, divergence time estimates, and the number of gene copies and their distribution among 11 plant species. The tree was constructed on the basis of 1,538 single-copy truly orthologous genes. Divergence times (Mya) are indicated by the blue numbers beside the branch nodes. The numbers of gene-family expansion and contraction events are indicated by green and red numbers, respectively, on each species branch. Pie charts show the proportions of gene families that underwent expansion or contraction. MRCA, most recent common ancestor.
Among the 11 species, all protein-coding genes were clustered into 28,283 orthogroups based on sequence homology. A total of 8,272 gene families were shared by all 11 species, and 25 P. mongolica-specific gene families were found (Supplementary Fig. S5 and Supplementary Table S13). Comparison of gene families among the five Prunus species revealed that 13,853 gene families were shared among P. mongolica, P. persica, P. avium, P. mume, P. dulcis, and P. armeniaca, while 137 gene families were specific to P. mongolica (Fig. 3A). We also identified 330 expanded gene families and 1,094 contracted gene families (Fig. 3B and C, Supplementary Figs. S6 and S7), suggesting that the majority of gene families in P. mongolica contracted, rather than expanded, during adaptive evolution.
GO enrichment analysis revealed that the P. mongolica-specific gene families were primarily enriched in the biological functions of metabolic process, cellular process localization, membrane part, cell part, binding, and catalytic activity, among others (Table 2). KEGG enrichment analysis revealed that these species-specific gene families were primarily enriched in the metabolic pathways of pyrimidine metabolism, protein processing in endoplasmic reticulum, butanoate metabolism, propanoate metabolism, terpenoid backbone biosynthesis, lysine degradation, fatty acid degradation, valine, leucine and isoleucine degradation, fatty acid metabolism, and pyruvate metabolism, among others (Table 3). KEGG enrichment analysis of expanded gene families found that these were enriched in the cyanoamino acid, tryptophan, arginine, proline, phenylalanine, stilbenoid, and starch and sucrose metabolism pathways (Supplementary Fig. S6E). The majority of the contracted gene families were enriched in sesquiterpene and triterpene biosynthesis, homologous recombinant, endocytosis, and starch and sucrose metabolism (Supplementary Fig. S7E). We also identified 350 positively selected genes in P. mongolica (Supplementary Table S14) and subjected these to GO and KEGG enrichment analyses (Supplementary Fig. S8). KEGG enrichment analysis indicated that the majority of positively selected genes were enriched in starch and sucrose metabolism, RNA degradation, sphingolipid metabolism, glycerolipid metabolism, and pyrimidine metabolism. These metabolic processes may be related to the characteristic cold and drought tolerance exhibited by P. mongolica.
GO enrichment analysis of Prunus mongolica-specific gene
| GO_classify 1 | GO_classify 2 | All gene | Specific |
|---|---|---|---|
| Cellular component | Extracellular region | 501 | 0 |
| Cellular component | Nucleoid | 18 | 0 |
| Cellular component | Cell junction | 175 | 0 |
| Cellular component | Membrane-enclosed lumen | 450 | 0 |
| Cellular component | Protein-containing complex | 2,064 | 0 |
| Cellular component | Organelle | 6,376 | 0 |
| Cellular component | Other organism | 141 | 0 |
| Cellular component | Other organism part | 141 | 0 |
| Cellular component | Extracellular region part | 69 | 0 |
| Cellular component | Organelle part | 2,418 | 0 |
| Cellular component | Synapse part | 2 | 0 |
| Cellular component | Synapse | 3 | 0 |
| Cellular component | Symplast | 42 | 0 |
| Cellular component | Supramolecular complex | 125 | 0 |
| Molecular function | Structural molecule activity | 375 | 0 |
| Molecular function | Transporter activity | 1,039 | 0 |
| Molecular function | Antioxidant activity | 127 | 0 |
| Molecular function | Protein tag | 13 | 0 |
| Molecular function | Cargo receptor activity | 2 | 0 |
| Molecular function | Translation regulator activity | 6 | 0 |
| Molecular function | Nutrient reservoir activity | 61 | 0 |
| Molecular function | Molecular transducer activity | 179 | 0 |
| Molecular function | Toxin activity | 3 | 0 |
| Molecular function | Molecular function regulator | 333 | 0 |
| Molecular function | Molecular carrier activity | 24 | 0 |
| Molecular function | Transcription regulator activity | 724 | 0 |
| Biological process | Reproduction | 441 | 0 |
| Biological process | Immune system process | 69 | 0 |
| Biological process | Behaviour | 5 | 0 |
| Biological process | Cell proliferation | 9 | 0 |
| Biological process | Carbon utilization | 7 | 0 |
| Biological process | Nitrogen utilization | 8 | 0 |
| Biological process | Reproductive process | 438 | 0 |
| Biological process | Biological adhesion | 2 | 0 |
| Biological process | Signalling | 535 | 0 |
| Biological process | Multicellular organismal process | 488 | 0 |
| Biological process | Developmental process | 680 | 0 |
| Biological process | Growth | 79 | 0 |
| Biological process | Locomotion | 14 | 0 |
| Biological process | Pigmentation | 1 | 0 |
| Biological process | Rhythmic process | 22 | 0 |
| Biological process | Response to stimulus | 2,092 | 0 |
| Biological process | Multi-organism process | 279 | 0 |
| Biological process | Biological regulation | 3,177 | 0 |
| Biological process | Cellular component organization or biogenesis | 1,586 | 0 |
| Biological process | Detoxification | 150 | 0 |
| Biological process | Localization | 1,841 | 2 |
| Cellular component | Cell | 8,596 | 4 |
| Cellular component | Cell part | 8,596 | 4 |
| Cellular component | Membrane | 6,095 | 7 |
| Cellular component | Membrane part | 5,331 | 7 |
| Biological process | Cellular process | 8,531 | 19 |
| Molecular function | Catalytic activity | 8,741 | 29 |
| Biological process | Metabolic process | 8,538 | 29 |
| Molecular function | Binding | 10,468 | 30 |
| #Total_gene | 19,944 | 131 |
| GO_classify 1 | GO_classify 2 | All gene | Specific |
|---|---|---|---|
| Cellular component | Extracellular region | 501 | 0 |
| Cellular component | Nucleoid | 18 | 0 |
| Cellular component | Cell junction | 175 | 0 |
| Cellular component | Membrane-enclosed lumen | 450 | 0 |
| Cellular component | Protein-containing complex | 2,064 | 0 |
| Cellular component | Organelle | 6,376 | 0 |
| Cellular component | Other organism | 141 | 0 |
| Cellular component | Other organism part | 141 | 0 |
| Cellular component | Extracellular region part | 69 | 0 |
| Cellular component | Organelle part | 2,418 | 0 |
| Cellular component | Synapse part | 2 | 0 |
| Cellular component | Synapse | 3 | 0 |
| Cellular component | Symplast | 42 | 0 |
| Cellular component | Supramolecular complex | 125 | 0 |
| Molecular function | Structural molecule activity | 375 | 0 |
| Molecular function | Transporter activity | 1,039 | 0 |
| Molecular function | Antioxidant activity | 127 | 0 |
| Molecular function | Protein tag | 13 | 0 |
| Molecular function | Cargo receptor activity | 2 | 0 |
| Molecular function | Translation regulator activity | 6 | 0 |
| Molecular function | Nutrient reservoir activity | 61 | 0 |
| Molecular function | Molecular transducer activity | 179 | 0 |
| Molecular function | Toxin activity | 3 | 0 |
| Molecular function | Molecular function regulator | 333 | 0 |
| Molecular function | Molecular carrier activity | 24 | 0 |
| Molecular function | Transcription regulator activity | 724 | 0 |
| Biological process | Reproduction | 441 | 0 |
| Biological process | Immune system process | 69 | 0 |
| Biological process | Behaviour | 5 | 0 |
| Biological process | Cell proliferation | 9 | 0 |
| Biological process | Carbon utilization | 7 | 0 |
| Biological process | Nitrogen utilization | 8 | 0 |
| Biological process | Reproductive process | 438 | 0 |
| Biological process | Biological adhesion | 2 | 0 |
| Biological process | Signalling | 535 | 0 |
| Biological process | Multicellular organismal process | 488 | 0 |
| Biological process | Developmental process | 680 | 0 |
| Biological process | Growth | 79 | 0 |
| Biological process | Locomotion | 14 | 0 |
| Biological process | Pigmentation | 1 | 0 |
| Biological process | Rhythmic process | 22 | 0 |
| Biological process | Response to stimulus | 2,092 | 0 |
| Biological process | Multi-organism process | 279 | 0 |
| Biological process | Biological regulation | 3,177 | 0 |
| Biological process | Cellular component organization or biogenesis | 1,586 | 0 |
| Biological process | Detoxification | 150 | 0 |
| Biological process | Localization | 1,841 | 2 |
| Cellular component | Cell | 8,596 | 4 |
| Cellular component | Cell part | 8,596 | 4 |
| Cellular component | Membrane | 6,095 | 7 |
| Cellular component | Membrane part | 5,331 | 7 |
| Biological process | Cellular process | 8,531 | 19 |
| Molecular function | Catalytic activity | 8,741 | 29 |
| Biological process | Metabolic process | 8,538 | 29 |
| Molecular function | Binding | 10,468 | 30 |
| #Total_gene | 19,944 | 131 |
GO enrichment analysis of Prunus mongolica-specific gene
| GO_classify 1 | GO_classify 2 | All gene | Specific |
|---|---|---|---|
| Cellular component | Extracellular region | 501 | 0 |
| Cellular component | Nucleoid | 18 | 0 |
| Cellular component | Cell junction | 175 | 0 |
| Cellular component | Membrane-enclosed lumen | 450 | 0 |
| Cellular component | Protein-containing complex | 2,064 | 0 |
| Cellular component | Organelle | 6,376 | 0 |
| Cellular component | Other organism | 141 | 0 |
| Cellular component | Other organism part | 141 | 0 |
| Cellular component | Extracellular region part | 69 | 0 |
| Cellular component | Organelle part | 2,418 | 0 |
| Cellular component | Synapse part | 2 | 0 |
| Cellular component | Synapse | 3 | 0 |
| Cellular component | Symplast | 42 | 0 |
| Cellular component | Supramolecular complex | 125 | 0 |
| Molecular function | Structural molecule activity | 375 | 0 |
| Molecular function | Transporter activity | 1,039 | 0 |
| Molecular function | Antioxidant activity | 127 | 0 |
| Molecular function | Protein tag | 13 | 0 |
| Molecular function | Cargo receptor activity | 2 | 0 |
| Molecular function | Translation regulator activity | 6 | 0 |
| Molecular function | Nutrient reservoir activity | 61 | 0 |
| Molecular function | Molecular transducer activity | 179 | 0 |
| Molecular function | Toxin activity | 3 | 0 |
| Molecular function | Molecular function regulator | 333 | 0 |
| Molecular function | Molecular carrier activity | 24 | 0 |
| Molecular function | Transcription regulator activity | 724 | 0 |
| Biological process | Reproduction | 441 | 0 |
| Biological process | Immune system process | 69 | 0 |
| Biological process | Behaviour | 5 | 0 |
| Biological process | Cell proliferation | 9 | 0 |
| Biological process | Carbon utilization | 7 | 0 |
| Biological process | Nitrogen utilization | 8 | 0 |
| Biological process | Reproductive process | 438 | 0 |
| Biological process | Biological adhesion | 2 | 0 |
| Biological process | Signalling | 535 | 0 |
| Biological process | Multicellular organismal process | 488 | 0 |
| Biological process | Developmental process | 680 | 0 |
| Biological process | Growth | 79 | 0 |
| Biological process | Locomotion | 14 | 0 |
| Biological process | Pigmentation | 1 | 0 |
| Biological process | Rhythmic process | 22 | 0 |
| Biological process | Response to stimulus | 2,092 | 0 |
| Biological process | Multi-organism process | 279 | 0 |
| Biological process | Biological regulation | 3,177 | 0 |
| Biological process | Cellular component organization or biogenesis | 1,586 | 0 |
| Biological process | Detoxification | 150 | 0 |
| Biological process | Localization | 1,841 | 2 |
| Cellular component | Cell | 8,596 | 4 |
| Cellular component | Cell part | 8,596 | 4 |
| Cellular component | Membrane | 6,095 | 7 |
| Cellular component | Membrane part | 5,331 | 7 |
| Biological process | Cellular process | 8,531 | 19 |
| Molecular function | Catalytic activity | 8,741 | 29 |
| Biological process | Metabolic process | 8,538 | 29 |
| Molecular function | Binding | 10,468 | 30 |
| #Total_gene | 19,944 | 131 |
| GO_classify 1 | GO_classify 2 | All gene | Specific |
|---|---|---|---|
| Cellular component | Extracellular region | 501 | 0 |
| Cellular component | Nucleoid | 18 | 0 |
| Cellular component | Cell junction | 175 | 0 |
| Cellular component | Membrane-enclosed lumen | 450 | 0 |
| Cellular component | Protein-containing complex | 2,064 | 0 |
| Cellular component | Organelle | 6,376 | 0 |
| Cellular component | Other organism | 141 | 0 |
| Cellular component | Other organism part | 141 | 0 |
| Cellular component | Extracellular region part | 69 | 0 |
| Cellular component | Organelle part | 2,418 | 0 |
| Cellular component | Synapse part | 2 | 0 |
| Cellular component | Synapse | 3 | 0 |
| Cellular component | Symplast | 42 | 0 |
| Cellular component | Supramolecular complex | 125 | 0 |
| Molecular function | Structural molecule activity | 375 | 0 |
| Molecular function | Transporter activity | 1,039 | 0 |
| Molecular function | Antioxidant activity | 127 | 0 |
| Molecular function | Protein tag | 13 | 0 |
| Molecular function | Cargo receptor activity | 2 | 0 |
| Molecular function | Translation regulator activity | 6 | 0 |
| Molecular function | Nutrient reservoir activity | 61 | 0 |
| Molecular function | Molecular transducer activity | 179 | 0 |
| Molecular function | Toxin activity | 3 | 0 |
| Molecular function | Molecular function regulator | 333 | 0 |
| Molecular function | Molecular carrier activity | 24 | 0 |
| Molecular function | Transcription regulator activity | 724 | 0 |
| Biological process | Reproduction | 441 | 0 |
| Biological process | Immune system process | 69 | 0 |
| Biological process | Behaviour | 5 | 0 |
| Biological process | Cell proliferation | 9 | 0 |
| Biological process | Carbon utilization | 7 | 0 |
| Biological process | Nitrogen utilization | 8 | 0 |
| Biological process | Reproductive process | 438 | 0 |
| Biological process | Biological adhesion | 2 | 0 |
| Biological process | Signalling | 535 | 0 |
| Biological process | Multicellular organismal process | 488 | 0 |
| Biological process | Developmental process | 680 | 0 |
| Biological process | Growth | 79 | 0 |
| Biological process | Locomotion | 14 | 0 |
| Biological process | Pigmentation | 1 | 0 |
| Biological process | Rhythmic process | 22 | 0 |
| Biological process | Response to stimulus | 2,092 | 0 |
| Biological process | Multi-organism process | 279 | 0 |
| Biological process | Biological regulation | 3,177 | 0 |
| Biological process | Cellular component organization or biogenesis | 1,586 | 0 |
| Biological process | Detoxification | 150 | 0 |
| Biological process | Localization | 1,841 | 2 |
| Cellular component | Cell | 8,596 | 4 |
| Cellular component | Cell part | 8,596 | 4 |
| Cellular component | Membrane | 6,095 | 7 |
| Cellular component | Membrane part | 5,331 | 7 |
| Biological process | Cellular process | 8,531 | 19 |
| Molecular function | Catalytic activity | 8,741 | 29 |
| Biological process | Metabolic process | 8,538 | 29 |
| Molecular function | Binding | 10,468 | 30 |
| #Total_gene | 19,944 | 131 |
KEGG enrichment analysis of Prunus mongolica-specific gene
| ID | Description | GeneRatio | BgRatio | enrich_factor | pvalue | qvalue | geneID |
|---|---|---|---|---|---|---|---|
| ko00240 | Pyrimidine metabolism | 4/51 | 114/9379 | 6.45 | 0.0033 | 0.0456 | Pmo02G005780 Pmo02G008730 Pmo04G020390 Pmo07G001640 |
| ko00650 | Butanoate metabolism | 2/51 | 33/9379 | 11.15 | 0.0137 | 0.0941 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00640 | Propanoate metabolism | 2/51 | 49/9379 | 7.51 | 0.0289 | 0.1014 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00900 | Terpenoid backbone biosynthesis | 2/51 | 53/9379 | 6.94 | 0.0335 | 0.1014 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00310 | Lysine degradation | 2/51 | 56/9379 | 6.57 | 0.0370 | 0.1014 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00071 | Fatty acid degradation | 2/51 | 68/9379 | 5.41 | 0.0526 | 0.1120 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00280 | Valine, leucine and isoleucine degradation | 2/51 | 75/9379 | 4.9 | 0.0626 | 0.1223 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko01212 | Fatty acid metabolism | 2/51 | 86/9379 | 4.28 | 0.0794 | 0.1358 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00620 | Pyruvate metabolism | 2/51 | 108/9379 | 3.41 | 0.1166 | 0.1772 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko04141 | Protein processing in endoplasmic reticulum | 4/51 | 381/9379 | 1.93 | 0.1519 | 0.1814 | Pmo03G005840 Pmo03G005850 Pmo03G005860 Pmo07G002920 |
| ko00380 | Tryptophan metabolism | 2/51 | 128/9379 | 2.87 | 0.1534 | 0.1814 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00630 | Glyoxylate and dicarboxylate metabolism | 2/51 | 131/9379 | 2.81 | 0.1591 | 0.1814 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00460 | Cyanoamino acid metabolism | 2/51 | 179/9379 | 2.05 | 0.2543 | 0.2677 | Pmo06G015160 Pmo08G002680 |
| ko01200 | Carbon metabolism | 2/51 | 310/9379 | 1.19 | 0.5065 | 0.4950 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00940 | Phenylpropanoid biosynthesis | 2/51 | 362/9379 | 1.02 | 0.5913 | 0.5395 | Pmo06G015160 Pmo08G002680 |
| ko00500 | Starch and sucrose metabolism | 2/51 | 453/9379 | 0.81 | 0.7135 | 0.6102 | Pmo06G015160 Pmo08G002680 |
| ko04144 | Endocytosis | 1/51 | 276/9379 | 0.67 | 0.7829 | 0.6302 | Pmo07G002920 |
| ko03040 | Spliceosome | 1/51 | 350/9379 | 0.53 | 0.8570 | 0.6515 | Pmo07G002920 |
| ID | Description | GeneRatio | BgRatio | enrich_factor | pvalue | qvalue | geneID |
|---|---|---|---|---|---|---|---|
| ko00240 | Pyrimidine metabolism | 4/51 | 114/9379 | 6.45 | 0.0033 | 0.0456 | Pmo02G005780 Pmo02G008730 Pmo04G020390 Pmo07G001640 |
| ko00650 | Butanoate metabolism | 2/51 | 33/9379 | 11.15 | 0.0137 | 0.0941 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00640 | Propanoate metabolism | 2/51 | 49/9379 | 7.51 | 0.0289 | 0.1014 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00900 | Terpenoid backbone biosynthesis | 2/51 | 53/9379 | 6.94 | 0.0335 | 0.1014 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00310 | Lysine degradation | 2/51 | 56/9379 | 6.57 | 0.0370 | 0.1014 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00071 | Fatty acid degradation | 2/51 | 68/9379 | 5.41 | 0.0526 | 0.1120 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00280 | Valine, leucine and isoleucine degradation | 2/51 | 75/9379 | 4.9 | 0.0626 | 0.1223 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko01212 | Fatty acid metabolism | 2/51 | 86/9379 | 4.28 | 0.0794 | 0.1358 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00620 | Pyruvate metabolism | 2/51 | 108/9379 | 3.41 | 0.1166 | 0.1772 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko04141 | Protein processing in endoplasmic reticulum | 4/51 | 381/9379 | 1.93 | 0.1519 | 0.1814 | Pmo03G005840 Pmo03G005850 Pmo03G005860 Pmo07G002920 |
| ko00380 | Tryptophan metabolism | 2/51 | 128/9379 | 2.87 | 0.1534 | 0.1814 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00630 | Glyoxylate and dicarboxylate metabolism | 2/51 | 131/9379 | 2.81 | 0.1591 | 0.1814 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00460 | Cyanoamino acid metabolism | 2/51 | 179/9379 | 2.05 | 0.2543 | 0.2677 | Pmo06G015160 Pmo08G002680 |
| ko01200 | Carbon metabolism | 2/51 | 310/9379 | 1.19 | 0.5065 | 0.4950 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00940 | Phenylpropanoid biosynthesis | 2/51 | 362/9379 | 1.02 | 0.5913 | 0.5395 | Pmo06G015160 Pmo08G002680 |
| ko00500 | Starch and sucrose metabolism | 2/51 | 453/9379 | 0.81 | 0.7135 | 0.6102 | Pmo06G015160 Pmo08G002680 |
| ko04144 | Endocytosis | 1/51 | 276/9379 | 0.67 | 0.7829 | 0.6302 | Pmo07G002920 |
| ko03040 | Spliceosome | 1/51 | 350/9379 | 0.53 | 0.8570 | 0.6515 | Pmo07G002920 |
KEGG enrichment analysis of Prunus mongolica-specific gene
| ID | Description | GeneRatio | BgRatio | enrich_factor | pvalue | qvalue | geneID |
|---|---|---|---|---|---|---|---|
| ko00240 | Pyrimidine metabolism | 4/51 | 114/9379 | 6.45 | 0.0033 | 0.0456 | Pmo02G005780 Pmo02G008730 Pmo04G020390 Pmo07G001640 |
| ko00650 | Butanoate metabolism | 2/51 | 33/9379 | 11.15 | 0.0137 | 0.0941 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00640 | Propanoate metabolism | 2/51 | 49/9379 | 7.51 | 0.0289 | 0.1014 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00900 | Terpenoid backbone biosynthesis | 2/51 | 53/9379 | 6.94 | 0.0335 | 0.1014 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00310 | Lysine degradation | 2/51 | 56/9379 | 6.57 | 0.0370 | 0.1014 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00071 | Fatty acid degradation | 2/51 | 68/9379 | 5.41 | 0.0526 | 0.1120 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00280 | Valine, leucine and isoleucine degradation | 2/51 | 75/9379 | 4.9 | 0.0626 | 0.1223 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko01212 | Fatty acid metabolism | 2/51 | 86/9379 | 4.28 | 0.0794 | 0.1358 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00620 | Pyruvate metabolism | 2/51 | 108/9379 | 3.41 | 0.1166 | 0.1772 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko04141 | Protein processing in endoplasmic reticulum | 4/51 | 381/9379 | 1.93 | 0.1519 | 0.1814 | Pmo03G005840 Pmo03G005850 Pmo03G005860 Pmo07G002920 |
| ko00380 | Tryptophan metabolism | 2/51 | 128/9379 | 2.87 | 0.1534 | 0.1814 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00630 | Glyoxylate and dicarboxylate metabolism | 2/51 | 131/9379 | 2.81 | 0.1591 | 0.1814 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00460 | Cyanoamino acid metabolism | 2/51 | 179/9379 | 2.05 | 0.2543 | 0.2677 | Pmo06G015160 Pmo08G002680 |
| ko01200 | Carbon metabolism | 2/51 | 310/9379 | 1.19 | 0.5065 | 0.4950 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00940 | Phenylpropanoid biosynthesis | 2/51 | 362/9379 | 1.02 | 0.5913 | 0.5395 | Pmo06G015160 Pmo08G002680 |
| ko00500 | Starch and sucrose metabolism | 2/51 | 453/9379 | 0.81 | 0.7135 | 0.6102 | Pmo06G015160 Pmo08G002680 |
| ko04144 | Endocytosis | 1/51 | 276/9379 | 0.67 | 0.7829 | 0.6302 | Pmo07G002920 |
| ko03040 | Spliceosome | 1/51 | 350/9379 | 0.53 | 0.8570 | 0.6515 | Pmo07G002920 |
| ID | Description | GeneRatio | BgRatio | enrich_factor | pvalue | qvalue | geneID |
|---|---|---|---|---|---|---|---|
| ko00240 | Pyrimidine metabolism | 4/51 | 114/9379 | 6.45 | 0.0033 | 0.0456 | Pmo02G005780 Pmo02G008730 Pmo04G020390 Pmo07G001640 |
| ko00650 | Butanoate metabolism | 2/51 | 33/9379 | 11.15 | 0.0137 | 0.0941 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00640 | Propanoate metabolism | 2/51 | 49/9379 | 7.51 | 0.0289 | 0.1014 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00900 | Terpenoid backbone biosynthesis | 2/51 | 53/9379 | 6.94 | 0.0335 | 0.1014 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00310 | Lysine degradation | 2/51 | 56/9379 | 6.57 | 0.0370 | 0.1014 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00071 | Fatty acid degradation | 2/51 | 68/9379 | 5.41 | 0.0526 | 0.1120 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00280 | Valine, leucine and isoleucine degradation | 2/51 | 75/9379 | 4.9 | 0.0626 | 0.1223 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko01212 | Fatty acid metabolism | 2/51 | 86/9379 | 4.28 | 0.0794 | 0.1358 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00620 | Pyruvate metabolism | 2/51 | 108/9379 | 3.41 | 0.1166 | 0.1772 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko04141 | Protein processing in endoplasmic reticulum | 4/51 | 381/9379 | 1.93 | 0.1519 | 0.1814 | Pmo03G005840 Pmo03G005850 Pmo03G005860 Pmo07G002920 |
| ko00380 | Tryptophan metabolism | 2/51 | 128/9379 | 2.87 | 0.1534 | 0.1814 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00630 | Glyoxylate and dicarboxylate metabolism | 2/51 | 131/9379 | 2.81 | 0.1591 | 0.1814 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00460 | Cyanoamino acid metabolism | 2/51 | 179/9379 | 2.05 | 0.2543 | 0.2677 | Pmo06G015160 Pmo08G002680 |
| ko01200 | Carbon metabolism | 2/51 | 310/9379 | 1.19 | 0.5065 | 0.4950 | Pmoptg000086l_1G000010 Pmoptg000095l_1G000010 |
| ko00940 | Phenylpropanoid biosynthesis | 2/51 | 362/9379 | 1.02 | 0.5913 | 0.5395 | Pmo06G015160 Pmo08G002680 |
| ko00500 | Starch and sucrose metabolism | 2/51 | 453/9379 | 0.81 | 0.7135 | 0.6102 | Pmo06G015160 Pmo08G002680 |
| ko04144 | Endocytosis | 1/51 | 276/9379 | 0.67 | 0.7829 | 0.6302 | Pmo07G002920 |
| ko03040 | Spliceosome | 1/51 | 350/9379 | 0.53 | 0.8570 | 0.6515 | Pmo07G002920 |
P. mongolica grows in harsh, arid environments characterized by drought, cold temperatures, high winds, and sandblasting, among other stressors. Such harsh environmental conditions can negatively impact normal physiological functions, metabolic dynamics, and cell membrane structure and permeability, resulting in damage to protoplasts and deranged signalling pathways.67,68 Over long-term evolution, the gene families specific to P. mongolica likely evolved to enhance stress resistance by regulating metabolic pathways such as pyrimidine metabolism, protein processing in endoplasmic reticulum, and butanoate metabolism, among others. Additionally, in response to drought and cold stress, P. mongolica experienced expansion in gene families related to osmoregulation, scavenging radicals, and protecting cell structures through proline and sucrose metabolism. According to GO analysis, the positively selected genes in P. mongolica were found to be enriched in the biological functions of biological process, cellular component, and molecular function. Similarly to the species-specific and expanded gene families, KEGG pathway analysis of positively selected genes revealed that these genes were enriched in the same metabolic pathways, including starch and sucrose metabolism, pyrimidine metabolism, RNA degradation, sphingolipid metabolism, and glycerolipid metabolism, among others. Both positive transcriptional regulation and membrane stabilization are critical responses to environmental stress.69–71 It appears that P. mongolica maintains normal physiological functioning under harsh climatic conditions through the expression and regulation of a variety of species-specific, expanded, and positively selected genes. Furthermore, the expansion and contraction of gene families and positively selected genes may have contributed to the phenotypic diversification and speciation of P. mongolica.
WGD events (polyploidization) have played a major role in the evolutionary history of angiosperms.72,73 In order to study the evolutionary relationships between P. mongolica and other plant species, we measured the synonymous substitution rates (Ks) of orthologous gene pairs. On the basis of the distribution of Ks values of ~0.05 and ~1.37 between orthologs, two WGD events were identified in the P. mongolica genome, corresponding to divergences at ~3.57 and ~97.86 Mya, respectively. Furthermore, the analysis revealed peaks of ~0.025 for P. mongolica-P. dulcis and ~0.032 for P. mongolica-P. armeniaca, corresponding to divergences at ~1.79 and ~2.28 Mya, respectively (Fig. 4A). Analysis of the distribution of sequence divergence values for syntenic duplicate genes revealed two significant peaks for the P. mongolica genome (4DTv ~0.018 and ~0.488; Fig. 4B), further confirming that P. mongolica had experienced two WGDs. A divergence peak value (4DTv ~0.01 and ~0.002) was observed for P. dulcis and P. armeniaca (Fig. 4B), suggesting that P. mongolica diverged from P. armeniaca later than from P. dulcis.
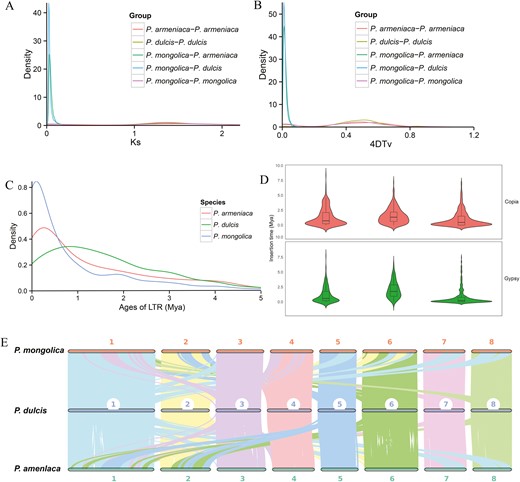
(A) Distribution of the synonymous substitution rate (Ks) between P. mongolica and P. dulcis, P. armeniaca. (B) Genome duplication in P. mongolica and genomes of related species as revealed by 4DTv analyses. (C) Distribution of insertion ages of LTR retrotransposons in the genomes of P. mongolica and genomes of related species. LTR, long terminal repeat; Mya, million years ago. (D) Copia and Gypsy elements insertion time. (E) MCscanX identified synteny blocks between P. mongolica, P. dulcis, and P. armeniaca.
LTR retrotransposons are the most abundant group of TEs in plants, and are considered the ‘engine’ of plant evolution.74 We found that the majority of LTR-RT insertion events in the P. mongolica genome occurred <1 Mya. Furthermore, the genomes of P. armeniaca, P. dulcis, and P. mongolica carried younger LTR-RTs, and the greatest proportion of LTR-RTs exhibited insertion times of ~0.25, 0.83, and 0.1 Mya, respectively (Fig. 4C), with Copia and Gypsy element insertion times of 0.98 and 0.75 Mya (Fig. 4D). This phenomenon may have resulted from rapid environmental change, such as desertification and drought, with the recent burst of LTR-RT insertions and gene duplications coinciding with the aridification of inland Asia during the late Cenozoic.75 These results suggest that the substantial increase LTR-RT insertions and tandem gene duplications within the P. mongolica genome may have contributed to the expansion of its genome and its adaptation to arid environments.
Synteny block analysis is often conducted to assess assembly quality and to investigate the evolutionary history of related species.76 To study colinear relationships within the P. mongolica, P. armeniaca, and P. dulcis genomes, we identified similar gene pairs using Diamond and syntenic blocks using MCScanX (Fig. 4E). On the basis of the order of orthologous genes, a total of 380 and 256 syntenic blocks were identified between P. mongolica-P. armeniaca and P. mongolica-P. dulcis, corresponding to 33,908 and 34,680 gene pairs in P. mongolica-P. armeniaca and P. mongolica-P. dulcis, respectively. The frequency of large-scale fragment rearrangements was determined in the P. mongolica, P. armeniaca, and P. dulcis genomes, including inversions and translocations, indicating that P. mongolica-P. dulcis had higher collinearity than P. mongolica-P. armeniaca, consistent with their close phylogenetic relationship as members of the Prunus clade (Supplementary Fig. S9).
3.4. Drought resistance and fatty acid biosynthesis genes
With an ever-increasing amount of publicly available data and published research, the model plant Arabidopsis (A. thaliana) continues to offer a convenient mechanism to identify and characterize functional genes and molecular mechanisms in other eukaryotic organisms.77 For example, in Rubus corchorifolius, key genes involved in anthocyanin and lignin biosynthesis were identified through comparison with Arabidopsis genes.78 In Euphorbia lathyris, genes related to lipid metabolism were identified by performing homology searches against the Arabidopsis genes involved in the biosynthesis of fatty acids and triacylglycerols.79
Here, we performed a homology search against the Arabidopsis genome and identified a total of 19 and 29 genes likely related to drought resistance and fatty acid/triglyceride biosynthesis, respectively, across the P. mongolica genome (Table 4 and Supplementary Table SWGD15). Among these drought resistance genes, AVP1 (Arabidopsis vacuolar H+-pyrophosphatase gene), CDKC2 (Cyclin dependent kinase group C2) and Sucrose synthase 3 (SUS3) had two copies; Alcohol dehydrogenase 1 (ADH1) had three copies; and Early-responsive to dehydration (ERD9) had six copies. All other genes had only one copy. AVP1, vacuolar proton-pumping pyrophosphatase (H+ -PPase) gene, has been shown to increase plant growth under both stressed and unstressed conditions in Arabidopsis.80CDKC2, a cyclin-dependent protein kinase, enhances plant stress tolerance by regulating the phosphorylation of SR (Serine/arginine-rich)-splicing factors.81 In Arabidopsis, this phosphorylation triggers the alternative splicing of pre-mRNAs and of stress-related genes, resulting to the induction of the stress response.81SUS3, one of the key enzymes in sucrose metabolism, is highly responsive to both internal and external environmental signals and can dramatically alter development and stress acclimation.82ADH1, of which there are three copies in P. mongolica, has been found to improve resistance to both biotic and abiotic stressors when overexpressed in Arabidopsis.83ERD9, of which there are six copies in P. mongolica, is the core environmental stress response gene in plants.84 Among these fatty acid biosynthesis genes, only Acetyl CO-enzyme A carboxylase subunit (CAC2) had two copies and the other genes had only one copy. These candidate genes are likely to prove useful for the future study of drought tolerance and fatty acid biosynthesis in P. mongolica, and will provide important genetic resources for molecular breeding experiments.
Genes involved in drought resistance in Prunus mongolica genome
| Genes | Full name | EC number | Candidate genes |
|---|---|---|---|
| ADH1 | Alcohol dehydrogenase 1 | EC:1.1.1.1 | Pmo08G001370, Pmo08G001390, Pmo08G001410 |
| AO4 | Aldehyde oxidase 4 | EC:1.2.1.28 | Pmo06G013050 |
| AVP1 | (AVP1) | EC:7.1.3.1 | Pmo03G007910, Pmo07G022760 |
| β-AM1 | β-Amylase 1 | EC:3.2.1.2 | Pmo01G004640 |
| CDKC2 | Cyclin dependent kinase group C2 | EC:2.7.11.22 | Pmo01G015660, Pmo06G016710 |
| CER1 | Eceriferum 1 | EC:4.1.99.5 | Pmo02G009550 |
| CHIA | Chitinase A | EC:3.2.1.14 | Pmo02G026230 |
| ERD9 | Early-responsive to dehydration | EC:2.5.1.18 | Pmo04G013040, Pmo04G013050, Pmo04G013060, Pmo04G013070, Pmo04G013080, Pmo04G013090 |
| GolS2 | Galactinol synthase2 | EC:2.4.1.123 | Pmo06G027030 |
| GSTU19 | Glutathione s-transferase tau 19 | EC:2.5.1.18 | Pmo04G013080 |
| HA1 | H(+)-ATPASE 1 | EC:7.1.2.1 | Pmo03G002540 |
| HPR | Hydroxypryruate reductase | EC:1.1.1.29 | Pmo07G023950 |
| NCED3 | Nine-cis-exoxycarotenoid dioxygenase 3 | EC:1.13.11.51 | Pmo04G013330 |
| PHS1 | α-Glucan phosphorylase 1 | EC:2.4.1.1 | Pmo03G022850 |
| SELO | Selenoprotein O | EC:2.7.7.108 | Pmo06G001270 |
| SUS3 | Sucrose synthase 3 | EC:2.4.1.13 | Pmo01G010740, Pmo08G022980 |
| UBC33 | Ubiquitin-conjugating enzyme 33 | EC:2.3.2.23 | Pmo03G002630 |
| UBC34 | Ubiquitin-conjugating enzyme 34 | EC:2.3.2.23 | Pmo03G002630 |
| WOL | Wooden leg | EC:2.7.13.3 | Pmo01G029440 |
| Genes | Full name | EC number | Candidate genes |
|---|---|---|---|
| ADH1 | Alcohol dehydrogenase 1 | EC:1.1.1.1 | Pmo08G001370, Pmo08G001390, Pmo08G001410 |
| AO4 | Aldehyde oxidase 4 | EC:1.2.1.28 | Pmo06G013050 |
| AVP1 | (AVP1) | EC:7.1.3.1 | Pmo03G007910, Pmo07G022760 |
| β-AM1 | β-Amylase 1 | EC:3.2.1.2 | Pmo01G004640 |
| CDKC2 | Cyclin dependent kinase group C2 | EC:2.7.11.22 | Pmo01G015660, Pmo06G016710 |
| CER1 | Eceriferum 1 | EC:4.1.99.5 | Pmo02G009550 |
| CHIA | Chitinase A | EC:3.2.1.14 | Pmo02G026230 |
| ERD9 | Early-responsive to dehydration | EC:2.5.1.18 | Pmo04G013040, Pmo04G013050, Pmo04G013060, Pmo04G013070, Pmo04G013080, Pmo04G013090 |
| GolS2 | Galactinol synthase2 | EC:2.4.1.123 | Pmo06G027030 |
| GSTU19 | Glutathione s-transferase tau 19 | EC:2.5.1.18 | Pmo04G013080 |
| HA1 | H(+)-ATPASE 1 | EC:7.1.2.1 | Pmo03G002540 |
| HPR | Hydroxypryruate reductase | EC:1.1.1.29 | Pmo07G023950 |
| NCED3 | Nine-cis-exoxycarotenoid dioxygenase 3 | EC:1.13.11.51 | Pmo04G013330 |
| PHS1 | α-Glucan phosphorylase 1 | EC:2.4.1.1 | Pmo03G022850 |
| SELO | Selenoprotein O | EC:2.7.7.108 | Pmo06G001270 |
| SUS3 | Sucrose synthase 3 | EC:2.4.1.13 | Pmo01G010740, Pmo08G022980 |
| UBC33 | Ubiquitin-conjugating enzyme 33 | EC:2.3.2.23 | Pmo03G002630 |
| UBC34 | Ubiquitin-conjugating enzyme 34 | EC:2.3.2.23 | Pmo03G002630 |
| WOL | Wooden leg | EC:2.7.13.3 | Pmo01G029440 |
Genes involved in drought resistance in Prunus mongolica genome
| Genes | Full name | EC number | Candidate genes |
|---|---|---|---|
| ADH1 | Alcohol dehydrogenase 1 | EC:1.1.1.1 | Pmo08G001370, Pmo08G001390, Pmo08G001410 |
| AO4 | Aldehyde oxidase 4 | EC:1.2.1.28 | Pmo06G013050 |
| AVP1 | (AVP1) | EC:7.1.3.1 | Pmo03G007910, Pmo07G022760 |
| β-AM1 | β-Amylase 1 | EC:3.2.1.2 | Pmo01G004640 |
| CDKC2 | Cyclin dependent kinase group C2 | EC:2.7.11.22 | Pmo01G015660, Pmo06G016710 |
| CER1 | Eceriferum 1 | EC:4.1.99.5 | Pmo02G009550 |
| CHIA | Chitinase A | EC:3.2.1.14 | Pmo02G026230 |
| ERD9 | Early-responsive to dehydration | EC:2.5.1.18 | Pmo04G013040, Pmo04G013050, Pmo04G013060, Pmo04G013070, Pmo04G013080, Pmo04G013090 |
| GolS2 | Galactinol synthase2 | EC:2.4.1.123 | Pmo06G027030 |
| GSTU19 | Glutathione s-transferase tau 19 | EC:2.5.1.18 | Pmo04G013080 |
| HA1 | H(+)-ATPASE 1 | EC:7.1.2.1 | Pmo03G002540 |
| HPR | Hydroxypryruate reductase | EC:1.1.1.29 | Pmo07G023950 |
| NCED3 | Nine-cis-exoxycarotenoid dioxygenase 3 | EC:1.13.11.51 | Pmo04G013330 |
| PHS1 | α-Glucan phosphorylase 1 | EC:2.4.1.1 | Pmo03G022850 |
| SELO | Selenoprotein O | EC:2.7.7.108 | Pmo06G001270 |
| SUS3 | Sucrose synthase 3 | EC:2.4.1.13 | Pmo01G010740, Pmo08G022980 |
| UBC33 | Ubiquitin-conjugating enzyme 33 | EC:2.3.2.23 | Pmo03G002630 |
| UBC34 | Ubiquitin-conjugating enzyme 34 | EC:2.3.2.23 | Pmo03G002630 |
| WOL | Wooden leg | EC:2.7.13.3 | Pmo01G029440 |
| Genes | Full name | EC number | Candidate genes |
|---|---|---|---|
| ADH1 | Alcohol dehydrogenase 1 | EC:1.1.1.1 | Pmo08G001370, Pmo08G001390, Pmo08G001410 |
| AO4 | Aldehyde oxidase 4 | EC:1.2.1.28 | Pmo06G013050 |
| AVP1 | (AVP1) | EC:7.1.3.1 | Pmo03G007910, Pmo07G022760 |
| β-AM1 | β-Amylase 1 | EC:3.2.1.2 | Pmo01G004640 |
| CDKC2 | Cyclin dependent kinase group C2 | EC:2.7.11.22 | Pmo01G015660, Pmo06G016710 |
| CER1 | Eceriferum 1 | EC:4.1.99.5 | Pmo02G009550 |
| CHIA | Chitinase A | EC:3.2.1.14 | Pmo02G026230 |
| ERD9 | Early-responsive to dehydration | EC:2.5.1.18 | Pmo04G013040, Pmo04G013050, Pmo04G013060, Pmo04G013070, Pmo04G013080, Pmo04G013090 |
| GolS2 | Galactinol synthase2 | EC:2.4.1.123 | Pmo06G027030 |
| GSTU19 | Glutathione s-transferase tau 19 | EC:2.5.1.18 | Pmo04G013080 |
| HA1 | H(+)-ATPASE 1 | EC:7.1.2.1 | Pmo03G002540 |
| HPR | Hydroxypryruate reductase | EC:1.1.1.29 | Pmo07G023950 |
| NCED3 | Nine-cis-exoxycarotenoid dioxygenase 3 | EC:1.13.11.51 | Pmo04G013330 |
| PHS1 | α-Glucan phosphorylase 1 | EC:2.4.1.1 | Pmo03G022850 |
| SELO | Selenoprotein O | EC:2.7.7.108 | Pmo06G001270 |
| SUS3 | Sucrose synthase 3 | EC:2.4.1.13 | Pmo01G010740, Pmo08G022980 |
| UBC33 | Ubiquitin-conjugating enzyme 33 | EC:2.3.2.23 | Pmo03G002630 |
| UBC34 | Ubiquitin-conjugating enzyme 34 | EC:2.3.2.23 | Pmo03G002630 |
| WOL | Wooden leg | EC:2.7.13.3 | Pmo01G029440 |
4. Conclusion
Here, we present a high-quality, chromosome-level P. mongolica genome assembly encompassing eight pseudochromosomes with a total length of 233.17 Mb. Within this highly complete genomic assembly, we identified 23,798 protein-coding genes, 625 tRNAs, 832 rRNAs, 217 miRNAs, and 283 snRNAs. Phylogenetic analysis based on 1,538 single-copy orthologous genes revealed that P. mongolica and P. persica are the most closely related, diverging from P. dulcis 4.2 Mya. Ks and 4DTv analyses indicated that the P. mongolica genome experienced two WGD events. These results suggest that the substantial increase in LTR-RT insertions and tandem gene duplications within the P. mongolica genome may have contributed to the expansion of its genome and its adaptation to arid environments. We also identified a number of candidate genes involved in drought resistance and fatty acid biosynthesis. These candidate genes are likely to prove useful for the future studies of drought tolerance and fatty acid biosynthesis in P. mongolica, and will provide important genetic resources for molecular breeding and improvement experiments in Prunus species. This high-quality reference genome will also accelerate the study of the adaptation of xerophytic plants to drought.
Supplementary data
Supplemental data are available at DNARES online.
Acknowledgements
The authors would like to thank TopEdit (www.topeditsci.com) for its linguistic assistance during the preparation of this manuscript.
Funding
This work was supported by the Ningxia Science and Technology Program (2022BBF02032, 2020YFH0005).
Conflict of interest
None declared.
Data availability
The genome assembly and raw sequencing data for P. mongolica have been deposited to the National Genomics Data Center (NGDC, https://ngdc.cncb.ac.cn/),85,86 with the project number of PRJCA013466 and the genome accession number of GWHBQDV00000000.1. These data were also deposited to NCBI with the project number of PRJNA930130. The detailed information of annotation are available on FigShare at the link: https://doi.org/10.6084/m9.figshare.22214515.v1.
References
Author notes
†Qiang Zhu, Yali Wang, and Ning Yao contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}