





Abstract.
The Cerulean Warbler (Setophaga cerulea) is one of the most rapidly declining warblers in North America. Our previous genetic work suggested that this migratory songbird has no significant population genetic structure in the northern and western parts of its breeding range, and for conservation purposes we assigned all sampled populations to a single genetic management unit. Here, we expand this work to include the entire breeding range of the Cerulean Warbler, given recent evidence for morphological and ecological differentiation between the northern and southern sections of that range. We assayed variation in four microsatellite loci and an 841-base-pair fragment of the mitochondrial control region, finding support for low but significant population-genetic structure in the mitochondrial marker. Estimates of population-genetic structure for pairs of sampling locations did not detect a clear geographic pattern of differentiation, and a hierarchical AMOVA did not reveal significant structure between northern and southern locations. These findings, together with previously published evidence for high rates of contemporary dispersal among breeding locations, may support the hypothesis of a long history of gene flow among ecologically distinct Cerulean Warbler populations or may suggest that more recent anthropogenic dispersal has homogenized the genetic signature of differentiation that may have existed in pre-settlement populations.
Resumen.
Setophaga cerulea es una de las especies de arañero que está declinando más rápidamente en América del Norte. Nuestros trabajos genéticos previos sugirieron que esta ave canora migratoria no tiene una estructura genética poblacional significativa en las partes norte y oeste de su rango reproductivo, y para propósitos de conservación asignamos todas las poblaciones muestreadas a una única unidad genética. Aquí, siguiendo evidencia reciente de diferenciación morfológica y ecológica entre las secciones del norte y de sur de ese rango, expandimos este trabajo para incluir todo el rango reproductivo de S. cerulea. Analizamos la variación en cuatro loci de microsatélites y en un fragmento de 841 pares de bases de la región de control mitocondrial, encontrando apoyo para una estructura genética poblacional baja pero significativa en el marcador mitocondrial. Las estimaciones de estructura genética poblacional de pares de localidades de muestreo no revelaron un claro patrón geográfico de diferenciación, y un análisis de varianza molecular (AMOVA) jerárquico no reveló una estructura significativa entre las localidades norte y sur. Estos resultados, junto con evidencia publicada anteriormente de altas tasas de dispersión contemporánea a lo largo del rango reproductivo, puede apoyar la hipótesis de una larga historia de flujo génico entre poblaciones ecológicamente distintivas de S. cerulea, o pueden sugerir que la dispersión antropogénica más reciente ha homogeneizado la firma genética de diferenciación que puede haber existido antes del establecimiento europeo.
Introduction
The Cerulean Warbler (Setophaga cerulea) is a species of particular conservation concern. Formerly abundant in the central and eastern United States and southern Canada, the species has experienced major reductions and range fragmentation (Hamel 2000). Dependence on extensive, intact, mature forest on its breeding grounds (Rosenberg et al. 2000, Jones and Robertson 2001, Barg et al. 2006) makes the species particularly sensitive to deforestation and fragmentation of mature and old-growth ecosystems (Hamel 2000). In Canada, the Committee on the Status of Endangered Wildlife in Canada has identified the Cerulean Warbler as a species of special concern (COSEWIC 2003). It is also listed as a species of special concern in the U.S.A. and is classified as “special concern,” “special interest,” or “rare” in most states within its breeding range (Hamel 2000). Link and Sauer (2002) estimated the Cerulean Warbler to be declining at a rate of 3.04% each year, making it one of the most rapidly declining warblers in North America, and habitat loss is likely the major contributing factor (Hamel 2000).
The Cerulean Warbler Atlas Project revealed that breeding sites are concentrated in several distinct areas across the species' range (Rosenberg et al. 2000). For conservation purposes, to help delineate appropriate management units, it is important to investigate whether birds breeding in geographically isolated areas are genetically distinct. A study of the Cerulean Warbler's isotopic signatures yielded estimates of long-distance dispersal that were large (28.2% among breeding males, 9.4% among natal-year birds), suggesting that regions far enough apart to be isotopically different readily exchange genes (Girvan et al. 2007). However, birds in the northern and southern portions of the range differ morphologically (Jones et al. 2005) and may travel to different wintering grounds (Girvan et al. 2007). Specifically, body size varies according to Bergmann's rule (Mayr 1956, Jones et al. 2005), and deuterium/hydrogen ratios (δD) suggest that Cerulean Warblers breeding north of Tennessee winter in northern South America (Venezuela, Colombia, and Ecuador), whereas birds from Tennessee winter farther south, in Peru and Bolivia (Girvan et al. 2007). These findings suggest that birds breeding in the north of the range may differ genetically from those in the south.
Although many passerines have star-shaped phytogenies due to post-Pleistocene expansion and high levels of contemporary dispersal (e.g., Milá et al. 2000), significant population genetic structure has been found in some species of Setophaga. Estimates of FST range from 0.008 across the geographically restricted range of the Golden-cheeked Warbler (S. chrysoparia) (Lindsay et al. 2008), to 0.068–0.133 among subspecies of the Yellow-rumped Warbler (S. coronata) (Milá et al. 2007) and 0.025–0.567 among subspecies of the Yellow Warbler (S. petechia) (Milot et al. 2000). A lack of genetic structure in the Cerulean Warbler may be the product of post-glacial expansion and a history of natural gene flow among ecologically distinct populations. Alternatively, contemporary anthropogenic dispersal, as refugees disperse from lost habitat, may have homogenized the genetic signature of differentiation if it have existed before deforestation (Porter 1999).
Veit et al. (2005) investigated variation in five microsatellite loci and 366 base pairs (bp) of sequence from domains II and III of the mitochondrial control region in 154 Cerulean Warblers from Ontario, Illinois, Tennessee, and Arkansas. In the present study, we expand on this research to include more comprehensive geographic sampling and a more variable molecular marker. Exclusion of highly variable domain I of the mitochondrial control region from previous genetic analyses may mean that subtle genetic signatures of population structure went undetected.
Methods
Sample Collection
In 2002, blood samples were collected from 104 adult Cerulean Warblers breeding in eight regions covering six states and one province (Table 1). Individuals were caught by mist net, according to methods in Veit et al. (2005), and were banded with numbered aluminum bands through the banding programs of the U.S.A. and Canada. Two to 50 µL of blood were collected from the brachial vein onto filter paper and allowed to dry. Samples are archived at -80 °C at Queen's University, Department of Biology. We extracted DNA by either protease K digestion and phenol—chloroform extraction (Sambrook et al. 1989) or the Animal Blood Spin-Column protocol described in the DNeasy tissue kit from Qiagen (Mississauga, ON). We combined these 104 samples with the 161 samples collected by Veit et al. (2005) between 1997 and 1999. Of the resulting 265 samples we successfully amplified 241 and used them in the following molecular analyses (Table 1, Fig. 1).
Locations and numbers of Cerulean Warblers from eight regions from which DNA was amplified.
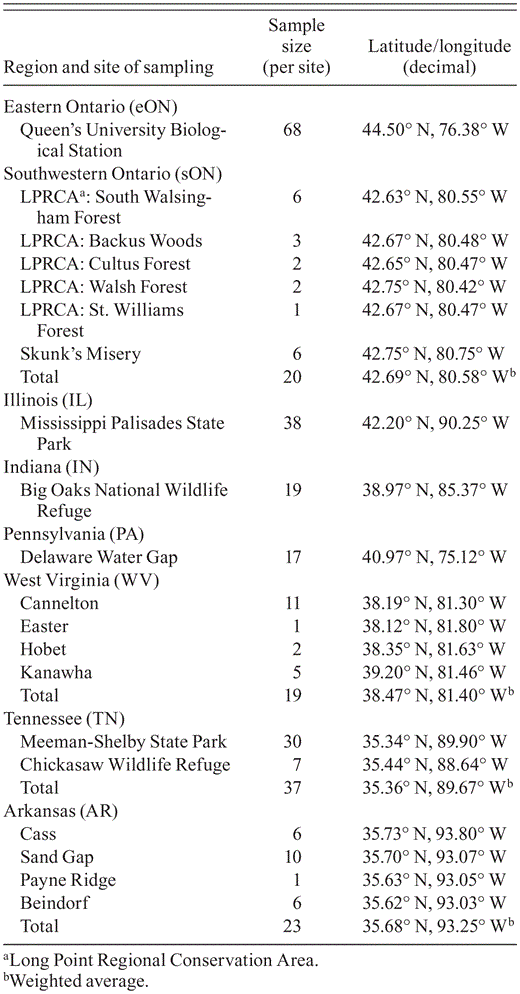
Locations and numbers of Cerulean Warblers from eight regions from which DNA was amplified.

Breeding range of the Cerulean Warbler in gray (adapted from Rosenberg et al. 2000). Sampling sites indicated by black diamonds. Pie charts depict the relative proportions of private haplotypes (white), and haplotypes shared at two or more locations (colored). The circles' size indicates number of individuals sampled per population.
Mitochondrial Sequence
We screened DNA preparations from the samples taken in 2002 with primers specific to the Cerulean Warbler, MCRL554 and MCRH922 (Veit et al. 2005), which amplify a fragment of domains II and III of the mitochondrial control region. The fragment was amplified in 15-µL volumes containing 1.5 mM MgCl2, 50 mM KCl, 5 mM Tris (pH 8.0), 10 mM Tris (pH 8.5), 0.1% gelatin, 0.18 mg mL-1 bovine serum albumen, 0.26 µM of each primer, 200 µM dNTPs, 1.5 µL DNA, and 0.5 U Taq polymerase. This was then heated to 94 °C for 3 min for denaturing, followed by 32 cycles of 30 sec at 94 °C for denaturing, 30 sec at 50 °C for annealing, and 30 sec at 72 °C for extension. Finally, the cocktail was brought to 72 °C for 5 min for extension. PCR products were sequenced with both primers on a 3730xl automated sequencer from Applied Biosystems at Genome Quebec (Montreal). All sequences were confirmed by visual comparison with the chromatogram output, and any ambiguous sites were designated as “N.” The sequences generated by the light- and heavy-strand primers were identical where they overlapped by approximately 300 bp, and we compiled them to give a 366-bp fragment.
We extracted DNA from the liver tissue of a single female (CERW-1, archived in the laboratory of V.L.F. at Queen's University), and manually sequenced domain I from this preparation with the external primer tRNAgluL16749 (5′-CTCCGAGATTTACGGCCTGAAAAG-3′) and the control-region primer MCRH258 (5′-GGGTAACGCTTGCTGCTTGGCTG-3′) designed by Veit (1999). We compared the sequence from this reaction with that for domain I generated with the same primers from blood samples. We observed two different sequence copies at the 5′ end of domain I and presume the extra copy present in DNA extracted from blood to represent a nuclear homolog (Ibarguchi et al. 2006). We generated primers specific to mtDNA by comparison of sequence from blood and tissue samples: MCRL78 (5′-TTGCTTTAAGGGTATGTATAG-3′) and MCRH623 (5′-GTGAACATAGTCACTTCTCG-3′). We used these primers to obtain sequence for approximately 495 bp from 241 samples by following the protocol described above. For most individuals, this sequence overlapped by about 20 bp with the previously acquired domain II and III sequence. We collated sequence from the two fragments to obtain a total of 841 bp of sequence for 241 individuals and used the program TCS version 1.21 (Clement et al. 2000) to ascribe individual sequence data to haplotypes and to construct an unrooted haplotype network.
Microsatellite Markers
Following Veit et al. (2005) and Dawson et al. (1997), we screened samples for allelic diversity at four microsatellite loci, excluding locus Dpµ15, which was found to have a significant excess of homozygotes, indicating the presence of a null allele (Veit et al. 2005). Forward primers for Dpµ01, Dpµ03, Dpµ16 and Cµµ28 were end-labeled with 33P-γ-dATP. We amplified 10-µL PCR reaction volumes containing 1.5 mM MgCl2, 50 mM KCl, 5 mM Tris (pH 8.0), 10 mM Tris (pH 8.5), 0.1% gelatin, 0.18 mg mL-1 bovine serum albumen, 0.05 µM unlabeled forward primer, 0.09 µM reverse primer, 200 µM dNTPs, 1 µL DNA, and 0.5 U Taq polymerase by the same temperature profile described above, at 50 °C annealing. After amplification, PCR products were visualized and scored from 4% denaturing Polyacrylamide gels. Alternatively, we labeled 104 samples (20 from eastern Ontario, 15 from Illinois, 19 from Indiana, 18 from Pennsylvania, 22 from West Virginia, and 10 from Tennessee) with a fluorescent dye and sized them with a 4200 DNA analyzer (Li-Cor; Lincoln, NE). We amplified and scored most samples (209 individuals) by both techniques to confirm that allele size could be confidently and consistently inferred by either method and to validate the pooling of data sets.
Grouping Samples
Many samples used in this study were from sites clustered within a larger region (e.g., sites Easter, Hobet, and Kanawha within West Virginia), or from the same location from different years (e.g., 1997–2002 within eastern Ontario). We did several tests to determine the validity of pooling samples from nearby sites and over multiple years. Veit et al. (2005) tested for microgeographic structure among sites sampled in southwestern Ontario, Tennessee and Arkansas and found no statistically significant evidence for genetic structure. Tests for microgeographic structure among sites in West Virginia, samples unique to this study, found no significant evidence for structure at this spatial scale (ΦST = -0.050, P = 0.768; FST = 0.006, P = 0.323; see below for details of methods), supporting the grouping of samples from nearby sites to represent larger, geographically defined sampling locations. We also found no evidence for genetic differentiation between eastern Ontario samples collected from 1997 to 1999 (originally included in Veit et al. 2005) and 2002 (this study) (ΦST = -0.015, P = 0.880; FST = 0.006, P = 0.106). These samples were pooled for all subsequent analyses.
Statistical Analyses
Analyses of mitochondrial sequence. We tested the assumption that variation in mitochondrial sequences is neutral in Arlequin version 3.11 (Excoffier et al. 2005), using the Ewens—Watterson test (Slatkin 1996, Watterson 1978) and Chakraborty's test of population amalgamation (Chakraborty 1990). The tests could not be performed for samples from Indiana, Pennsylvania, or West Virginia because all individuals from these locations had different haplotypes.
Using Arlequin, we performed an analysis of molecular variance (AMOVA) with K2P-corrected distances (Kimura 1980) to compare the correlation of control-region haplotypes within a location to haplotypes chosen at random from any location (ΦST) (Excoffier et al. 1992). In addition to estimating global ΦST and ΦST for pairs of sampling locations, we performed a hierarchical AMOVA to test explicitly for genetic differentiation between northern locations (eastern Ontario, southwestern Ontario, Illinois, Indiana, Pennsylvania, and West Virginia) and southern locations (Tennessee and Arkansas). We corrected pairwise estimates of ΦST for multiple tests by the Benjamini—Yekutieli method (new á = 0.01; Benjamini and Yekutieli 2001).
We used an online database (http://www.lat-long.com/) to obtain the latitude and longitude of our sampling sites according to the place's name, then calculated the geographic distance between locations by averaging the latitude and longitude across all sites, weighted by the number of individuals sampled at each site (Table 1) and entering pairwise comparisons into an online distance calculator (Michels 1997; http://jan.ucc.nau.edu/~cvm/latlongdist.html). We used Mantel's test to assess whether Slatkin's linearized FST (Mantel 1967) and log-transformed geographic distance were correlated.
Analyses of microsatellite variation. To determine whether microsatellite loci deviate from Hardy—Weinberg equilibrium, we ran exact tests using a Markov chain, with 100 000 randomizations, in Arlequin (Guo and Thompson 1992). We also tested microsatellite loci for deviations from linkage disequilibrium in Arlequin, with 10 000 randomizations.
To calculate FST as an index of population genetic structure based on the frequencies of alleles sampled from each location (Wright 1951) across all four microsatellite loci, we performed an AMOVA in Arlequin. We did not calculate RST, an analog of FSTthat assumes a stepwise model of mutation, for this data set because two loci Cµµ28 and Dpµ03) contained complex repeats so were unlikely to have evolved in a simple stepwise pattern. As with the mitochondrial data set we estimated FSTfor the global sample, for pairs of sampling locations, and for northern vs. southern locations in a hierarchical AMOVA. Pairwise estimates of FST were corrected for multiple tests as above, by the method of Benjamini and Yekutieli (2001). We used the program FSTAT (Goudet 1995) to calculate FISas an index of inbreeding.
Results
Mitochondrial Sequence
Individual sequences from the control region of 241 Cerulean Warblers constituted 169 unique haplotypes (Fig. 1, Appendix 1, available at http://dx.doi.org/10.1525/cond.2012.120031; GenBank KC481710-KC481878. Most haplotypes differed by only one or two nucleotide substitutions. Tests of neutrality were not significant for most locations and so provide no evidence against neutral evolution. Eastern Ontario was the only exception to this rule: the result of Chakraborty's test of population amalgamation was significant (for “k” or more haplotypes P = 0.02).
Number of control-region haplotypes (NH), number of private haplotypes (NpNPH), gene diversity (He), and nucleotide diversity (π) with pairwise estimates of Φ by location. For abbreviations for locations, see Table 1.
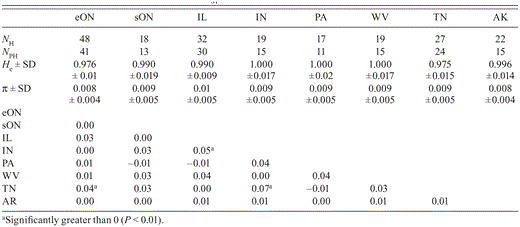
Number of control-region haplotypes (NH), number of private haplotypes (NpNPH), gene diversity (He), and nucleotide diversity (π) with pairwise estimates of Φ by location. For abbreviations for locations, see Table 1.
Haplotype diversity was similar and very high across locations. In Indiana, Pennsylvania, and West Virginia, each individual sampled had a distinct haplotype. Nucleotide diversity was also similar among locations (Table 2).
Most haplotypes (97%) were private, although they occurred at very low frequency in each location and were often represented by only a single individual (Appendix 2, available at http://dx.doi.org/10.1525/cond.2012.120031). By location, Illinois had the greatest proportion of haplotypes that were private (93.8%), and Pennsylvania had the smallest (64.7%) (Fig. 1, Table 2). Few haplotypes were common to multiple sampling locations (Fig. 1, Appendix 2).
Across the species, we found population genetic structure to be weak but statistically significant: ΦST = 0.017 (P < 0.01). Three pairwise estimates of ΦST between sampling locations were significant after correction for multiple tests: Tennessee and eastern Ontario, Tennessee and Indiana, and Indiana and Illinois (Table 2). A hierarchical AMOVA did not detect significant differentiation between northern and southern locations (FCT = 0.0046, P = 0.32); variation within northern and southern locations was low but significant (FSC = 0.014, P = 0.01). Mantel's test did not detect a relationship between geographic distance and genetic differentiation in the populations sampled (r = -0.013, P > 0.71).
High rates of molecular evolution in domain I of the control region have the potential to generate homoplasy. As a result, the unrooted haplotype tree constructed with TCS was characterized by many ambiguous connections. Since no clear phylogeographic pattern was evident, we do not present the tree here.
Microsatellite Markers
In microsatellites, genotype frequencies did not differ from Hardy—Weinberg expectations (all P > 0.002) after corrections for multiple comparisons (n = 56) following the Benjamini—Yekutieli method (Benjamini and Yekutieli 2001) (Appendices 3 and 4, available at http://dx.doi.org/10.1525/cond.2012.120031). Similarly, no pairwise estimates of linkage disequilibrium were significant after correction (all P > 0.001). By locus, average allelic richness ranged from 5 (Dpµ03) to 15.9 (Dpµ01 Table 3). No significant population genetic structure was detected in nuclear data: the global estimate of FST was 0.002 (P > 0.20), and no estimates for pairwise comparisons of sampling locations were significant (Table 3). All estimates of FISwere significantly different from zero. Mantel's test did not detect a relationship between geographic distance and genetic differentiation among the locations sampled (r = -0.00, P > 0.82).
Discussion
Neutral Genetic Diversity
Although the Cerulean Warbler is rapidly declining, diversity at neutral loci remains high. The number of controlregion haplotypes in this species is large in comparison to that published in other studies. For example, Bull et al. (2010) sequenced a 486-bp fragment of the mitochondrial control region of 206 Orange-crowned Warblers (Oreothlypis celata) of several subspecies and reported only 92 haplotypes. Estimates of allelic richness and FIS across microsatellite loci also support a fairly high level of diversity at neutral loci. However, diversity at neutral loci does not mean that the species has access to a large pool of genetic variability at adaptively important loci, or that it is protected from the harmful effects of genetic drift and inbreeding depression (Reed and Frankham 2001). These factors should continue to be considered of potential importance in a species experiencing a steep population decline.
Number of microsatellite alleles (NA), gene diversity (He), and FIS for each locus, average gene diversity ( He± SD) and FIS over all loci, and pairwise estimates of FSTby location (see Table 1 for abbreviations).a
He± SD) and FIS over all loci, and pairwise estimates of FSTby location (see Table 1 for abbreviations).a
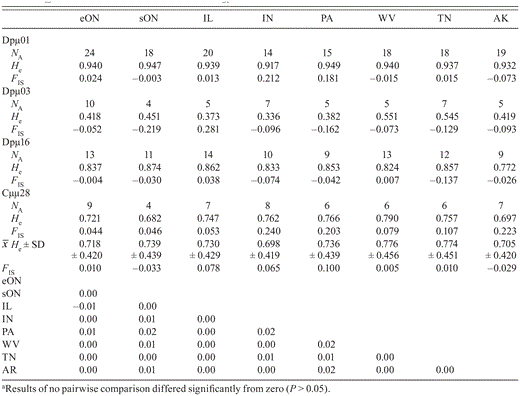
Number of microsatellite alleles (NA), gene diversity (He), and FIS for each locus, average gene diversity ( He± SD) and FIS over all loci, and pairwise estimates of FSTby location (see Table 1 for abbreviations).a
Tests of Neutrality
Chakraborty's test of population amalgamation was significant for eastern Ontario. This result is unlikely to be due to insufficient or nonrandom sampling since the sample from that location was the largest. Haplotype frequencies in 19971999 and 2002 did not differ (see Results, Grouping Samples). Rather, departures from neutrality in eastern Ontario may indicate that this population is not in migration-drift equilibrium because of recent population expansion (Tajima 1989). After farms in this area were abandoned in the early 1900s, succession led to the re-establishment of mature forests, which may have facilitated the Cerulean Warbler's population expansion (Robbins et al. 1989).
Population Genetic Structure
Mitochondrial sequences indicate weak but statistically significant population genetic structure among female Cerulean Warblers. The lack of statistically significant structure in biparentally inherited microsatellite loci may suggest sex-biased dispersal since male-biased dispersal may cause stronger differentiation among females than within the species as a whole. However, although debate about the patterns and processes underlying sex-biased dispersal in birds continues, dispersal of females tends to be greater (Clobert et al. 2001). Alternatively, disagreement between mitochondrial and nuclear markers may be a product of markers differing in mutation rates and the smaller effective population size of the mitochondrial genome. Our use of only four loci may also have limited our ability to detect subtle patterns of differentiation.
Although estimates of ΦST between some pairs of sampling locations were significant, the differences show no clear geographic pattern. Tennessee was genetically distinct from both eastern Ontario and Indiana. Results from Mantel's test did not support geographic distance as a potential mechanism for genetic differentiation. In fact, pairwise comparisons revealed significant differences between Indiana and Illinois, two sampling locations relatively near each other.
Factors other than geographic distance could influence population genetic structure. A difference in winter range between birds breeding in northern and southern locations (Girvan et al. 2007) might influence gene flow and genetic structure, since dispersal may be more likely within than between northern and southern locations (Baker et al. 1990, Arnaud 2003, but see Russell et al. 2005). Research by Jones et al. (2005) into latitudinal variation in body size suggested that gene flow between northern and southern populations may be low enough to allow some degree of local adaptation, if the morphological variation is genetic and not simply due to environmental factors. However, body-size variation with latitude is influenced by diet (Ho et al. 2010), and support for genetic differentiation between northern and southern locations is lacking in the neutral markers we used.
A history of rangewide gene flow (historical association) in the Cerulean Warbler is a likely explanation for the weak global mitochondrial structure we found and for the lack of a clear geographic pattern in significant pairwise comparisons. However, there is an alternative to explanations invoking historical association: refugees from lost habitat have the potential to change the nature of gene flow between populations dramatically (Porter 1999). Rapid clearance of old-growth forests in the last two centuries may have caused Cerulean Warblers to disperse to new breeding locations. Porter argued that the establishment of individuals in new breeding locations reduces genetic differentiation among populations and increases variation within populations. Although the extent of genetic structure in the Cerulean Warbler before deforestation is unknown, forest clearance must be considered a potential agent of genetic homogenization since high rates of contemporary migration between populations have the potential to obscure or erase historical structure. The high rates of dispersal by breeding adults in comparison to those of natal-year birds observed by Girvan et al. (2007) and the high levels of haplotypic and allelic diversity despite a significant rangewide decline are consistent with a role for habitat loss in encouraging gene flow among contemporary breeding populations.
Conservation Recommendations
We found no evidence that Cerulean Warblers constitute more than one genetically distinct management unit, despite morphological differences among populations. However, the potential for recent anthropogenic disturbance to elevate rangewide gene flow must not be overlooked. Continuing long-distance dispersal by refugees from lost habitat may have obscured historical associations in this species, and further research into this question should be undertaken before the historical presence or absence of locally adapted, evolutionarily significant populations is fully understood. Changes in the pattern and extent of gene flow may be another dimension in which human land use has affected bird populations of conservation concern.
Acknowledgments
Special thanks are due to M. Lisa Veit, who carried out the previous molecular work on the Cerulean Warbler, and Kate Girvan for collecting samples. Scott Taylor, Gabriela Ibarguchi, and Jamie Morris-Pocock provided both assistance in the lab and advice on data analysis. This work was made possible by grants from the Natural Sciences and Engineering Research Council (NSERC) to V. L. F. for the lab work and to R. J. R. for research in the field. Research contributed by K. D. M. was funded by a post-doctoral fellowship from NSERC. Financial support was provided to P. E. D. by Queen's University through the Student Work Experience Program.
Literature Cited

{kind=link}