





Abstract
This study uses mitochondrial DNA (mtDNA) to examine the phylogeography of the White-headed Woodpecker (Picoides albolarvatus), one of the least-studied woodpeckers in North America. A mismatch distribution and calculation of Tajima's D indicate that the overall phylogeographic history of the species is characterized by a recent range expansion that probably occurred after the start of the Pleistocene. In addition, a nested clade phylogeographic analysis indicates that additional processes such as allopatric fragmentation and restricted gene flow have influenced the evolutionary history of this species. Traditionally, the White-headed Woodpecker has been split into two subspecies whose distributions meet in the northern part of the Transverse Ranges in California. The two subspecies differ morphologically, with the southern subspecies having a larger bill in proportion to its body size than the northern subspecies. Geographical variation in mtDNA is concordant with a division at the Transverse Ranges that corresponds to the morphological variation seen between the two subspecies. An analysis of molecular variance indicates that 27% of the genetic variation results from differences between the northern and southern subspecies. Furthermore, birds in the northern part of the range differ from those in the southern part of the range by at least one base substitution. These results agree with the hypothesis that the larger bill of the southern subspecies is the result of recent local adaptation to feeding on the large cones of Coulter pines (Pinus coulteri).
Resumen
Filogeografía Intraespecífica Y Divergencia Adaptativa En Picoides Albolarvatus
Este estudio utiliza ADN mitocondrial (ADNmt) para examinar la filogeografía de Picoides albolarvatus, una de las especies de carpinteros menos estudiada de Norte América. La distribución “mismatch” y el valor calculado de la D de Tajima, indican que en general la historia filogeográfica de la especie está caracterizada por una expansión reciente en el rango de distribución, que probablemente ocurrió después del inicio del Pleistoceno. Adicionalmente, un análisis filogeográfico de clados anidados indica que otros procesos tales como una fragmentación alopátrica y flujo de genes restringido, han influenciado la historia evolutiva de esta especie. Tradicionalmente, P. albolarvatus ha sido dividido en dos subespecies cuyas distribuciones convergen en la parte norte de las Montañas Transversales en California. Las dos subespecies se diferencian morfológicamente. La subespecie ubicada hacia el sur tiene un pico más largo en proporción al tamaño del cuerpo, comparado con la subespecie ubicada más al norte. La variación geográfica en el ADNmt es concordante con la división en las Montañas Transversales, que a su vez corresponde a la variación morfológica observada entre las dos subespecies. Un análisis de varianza molecular indica que el 27% de la variación geneética es el resultado de las diferencias entre la subespecie del norte y la del sur. Incluso las aves en la parte norte del rango de distribución se diferencian de aquellas en la parte sur del rango en al menos una sustitución de bases. Estos resultados concuerdan con la hipótesis de que el pico más largo de la subespecie del sur es el resultado de una adaptación local reciente a la alimentación basada en los conos grandes de Pinus coulteri.
Introduction
The glacial cycles of the Quaternary period have greatly affected the distributions of fauna and flora throughout North America (Hewitt 2000). In particular, the resulting vicariant events, range restrictions, and range expansions have created drastically different evolutionary histories for the taxa of western North America. The ability to study, in detail, how taxa reacted to these climatic changes has been achieved with the advent of phylogeographical (Avise 2000) and coalescent (Barrowclough and Groth 1999, Zink et al. 2000) analyses of mitochondrial DNA sequences. In addition to the biogeographic history of species, many of these studies have provided new information relevant to previous morphological studies of geographic variation (Barrowclough et al. 1999, 2004, Zink et al. 2002). In this study, we use phylogeographical methods to examine the genetic structure and population history of the White-headed Woodpecker (Picoides albolarvatus).
White-headed Woodpeckers live in pine-dominated, montane coniferous forests from California to the Pacific Northwest (Fig. 1). Habitat requirements vary geographically; however, in general, the species is associated with ponderosa pine (Pinus ponderosa), including old-growth and mixed coniferous forests, where it requires broken-topped snags and stumps for nest trees (Grinnell and Miller 1944, Verner 1980, Raphael and White 1984, Milne and Hejl 1989, Garrett et al. 1996). The species is only found above 850 m throughout most of its range (Garrett et al. 1996). As a consequence of these habitat and elevation requirements the distribution of the White-headed Woodpecker is discontinuous, and in some areas the species occurs in small, isolated habitat fragments (Fig. 1). White-headed Woodpeckers are sedentary and territorial (Garrett et al. 1996). For example, in central Oregon they have been observed occupying the same territory throughout the year and using the same breeding site in multiple consecutive years (Garrett et al. 1996). Robinson (1957) found that breeding pairs may be monogamous and remain together throughout the winter. These life-history characteristics suggest the accumulation of local genetic variation, which makes it possible to assess population structure and make inferences regarding historical population patterns.
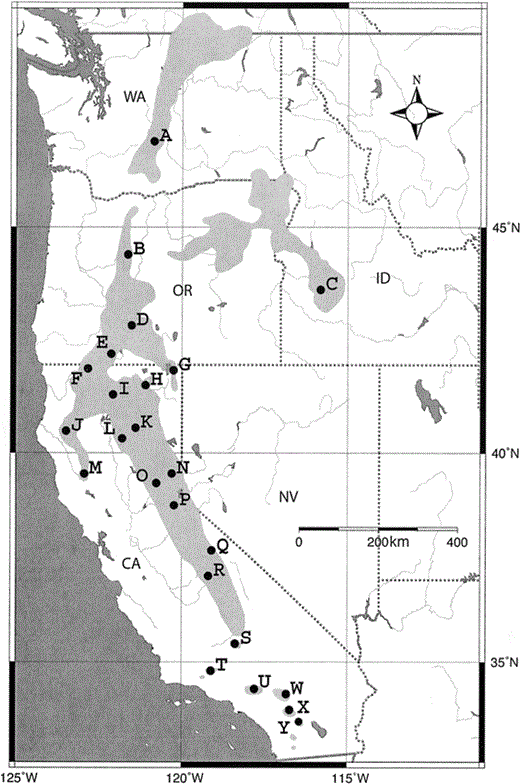
Figure 1. Geographic distribution of the White-headed Woodpecker including sampling localities lettered to correspond to the Appendix. Lightly shaded area indicates the distribution of the White-headed Woodpecker, and dark shaded area is water. State abbreviations: CA = California, ID = Idaho, NV = Nevada, OR = Oregon, and WA = Washington
White-headed Woodpeckers display measurable variation in bill morphology corresponding to two allopatrically distributed subspecies. The nominate race, P. a. albolarvatus, is found throughout most of the range excluding the southern tip (Fig. 1, populations A–T; Grinnell and Miller 1944, Winkler et al. 1995, Garrett et al. 1996). The southern subspecies, P. a. gravirostris, occupies mountaintops or “sky-islands” in southern California from the San Gabriel Mountains to the southern edge of the range in San Diego County (Fig. 1, populations U–Y; Grinnell and Miller 1944, Winkler et al. 1995, Garrett et al. 1996). Individuals from Mount Pinos (population T) have been considered to belong to both subspecies, but are possibly intermediate (Grinnell 1902, Garrett et al. 1996).
Although the two subspecies are similar in body size, P. a. gravirostris generally possesses a longer bill than P. a. albolarvatus (Grinnell 1902, Garrett et al. 1996). In addition, P. a. gravirostris shows a greater degree of sexual dimorphism in bill size than P. a. albolarvatus. This corresponds to significant sexual differences in foraging preferences observed in the southern subspecies, with males showing a stronger bias toward foraging on Coulter pines (Pinus coulteri, Koch et al. 1970, Hilkevitch 1974). Koch et al. (1970) hypothesized that P. a. gravirostris possesses a longer bill as a result of feeding on the large, spiked cones of Coulter pines throughout much of its range. In contrast, P. a. albolarvatus feeds on the smaller cones of ponderosa pine and sugar pine (Pinus lambertiana, Garrett et al. 1996) found throughout the range of this subspecies. Whether or not these differences in morphology and behavior correlate to molecular variation that is congruent with the current subspecies designation is unknown.
In this study, we use mitochondrial DNA (mtDNA) markers to study the phylogeography of White-headed Woodpeckers using several methodological approaches (AMOVA, mismatch distributions, minimum spanning haplotype network, and nested clade phylogeographical analysis) to establish possible historical genetic patterns of geographical variation. We then compare these results to current subspecies designations and discuss the conservation implications of our results. Throughout much of its range, the White-headed Woodpecker is relatively rare. Although locally common in California, government agencies in British Columbia, Washington, Idaho, and Oregon have classified the species into various categories of conservation concern. Thus, understanding genetic patterns within the species is essential to making informed management decisions.
Methods
Taxon Sampling
To examine mtDNA sequence variation of White-headed Woodpeckers, we sampled 78 individuals from 24 populations (Fig. 1, Appendix). Individuals were considered to be members of the same population if they were sampled within 40 km of each other. Sampling was more intensive in California than in other areas because permits to collect White-headed Woodpeckers were not granted by authorities for all regions. Thus, we had to rely on existing museum collections and feathers from ongoing cavity-nesting studies to obtain material from outside California. For the birds we collected, breast muscle, heart, and liver tissues were immediately preserved in 100% ethanol or frozen at −80°C, and voucher specimens were deposited in the San Diego State University Vertebrate Collections.
Molecular Laboratory Methods
A QIAmp DNA MiniKit (Qiagen, Valencia, California) tissue protocol was used to extract DNA from breast muscle, while DNA from feather samples was extracted using Chelex® beads (Walsh et al. 1991). Avian-specific primers for fragments of cytochrome b (cyt b, 1140 base pairs [bp]) and ATP synthase 6 (ATPase6, 637 bp) were used to amplify template DNA. Three overlapping segments of cyt b were amplified using primer pairs L14851 with H15297, L15206 with H15710, and L15656 with H16058 (Groth 1998). Primers A8PWL (Hunt et al. 2001) and H9906 (Sgariglia and Burns 2003) were used for ATPase6. Cyt b has been used extensively in avian phylogenetic studies and, along with ATPase6, has been shown to be a useful marker for examining population-level relationships within species (Sgariglia and Burns 2003, Burns and Barhoum 2006).
Initially, the polymerase chain reaction (PCR) was used to isolate and amplify the mtDNA fragments. This PCR was amplified in 10 µl volumes (94°C for 3 sec, 43°C for <1 sec, 71°C for 30 sec; 40 cycles) in capillary tubes placed inside a Rapid Cycler (Idaho Technology, Inc., Salt Lake City, Utah). The resulting product was then run through 2% agarose gel, stained for a sufficient amount of time in ethidium bromide, extracted from the gel with 750 µl pasteur pipets, melted, and reamplified in 40 µl volumes (94°C for 12 sec, 52°C for 4 sec, 71°C for 26 sec; 41 cycles). A GeneClean Kit (Bio101, Vista, California) was used to purify the PCR product. The purified product was cycle sequenced (96°C for 60 sec, 96°C for 30 sec, 50°C for 15 sec, 60°C for 4 min; 28 cycles) using Big Dye (Applied Biosystems, Foster City, California) terminator reaction mix. Samples were then passed through Sephadex beads contained in spin columns (3000 rpm for 3 min) before being sequenced on an ABI 377 DNA Sequencer (Applied Biosystems). Heavy and light strands of mtDNA fragments were sequenced for all 78 samples. DNA fragments were read and aligned in SEQUENCHER v. 3.1 (Gene Codes Corporation, Ann Arbor, Michigan), and translated in SE-AL v. 1.0 (Rambaut 1995). Unambiguous alignment of overlapping sequence fragments, lack of stop codons, and absence of heterozygosity during editing provided strong support for mitochondrial authenticity of sequence data. All sequences were deposited into GenBank (Accession numbers DQ158909–DQ159064).
Statistical Analysis
We used ARLEQUIN 2.0 (Schneider et al. 2000) to compute the mismatch distribution, analysis of molecular variation (AMOVA), raggedness index, Tajima's D, and nucleotide diversity (π). An AMOVA using pairwise distances was used to estimate a measure of the proportion of nucleotide variation divided among populations (Φ st; Excoffier et al. 1992). Because of marked differences in morphology and behavior between subspecies, an additional AMOVA was performed with populations grouped into subspecies. For most populations, assignment to subspecies was made using geography. However, it was unclear which subspecies was present at Mount Pinos (population T), and we were unable to sort these individuals to subspecies using morphological measurements. Thus, for this one population we had to rely on DNA sequence data to assign individuals to a subspecies. Most individuals from this population were assigned to the southern subspecies, as originally described by Grinnell (1902). However, one individual (T6) was included in the northern subspecies; this bird was assumed to be a recent migrant based on its DNA sequence (see results).
To examine the demographic history of populations and subspecies, a mismatch distribution of pairwise differences was constructed. In this analysis, the null hypothesis of an expanding population (Rogers and Harpending 1992, Rogers 1995) produces a unimodal distribution, while a ragged distribution indicates a population in stable equilibrium. Agreement between the expected and observed distributions under a sudden-expansion model was tested following Schneider and Excoffier (1999). Additionally, we calculated the raggedness index, which yields values smaller than 0.04 for an expanding population and larger values for stable populations (Harpending 1994).
Evidence of an expanding population was explored using Tajima's D (Tajima 1989). Assuming neutrality, an excess of recent mutations relative to equilibrium expectations for the number of segregating sites leads to significantly negative values of Tajima's D and rejection of population stasis. We also calculated π, the average number of base differences per site for homologous sequences (Nei 1987), for each population from which more than one individual was sampled. Nucleotide diversity estimates have been used to infer the direction of range expansions for many species of birds (Zink et al. 2001), with the assumption that smaller nucleotide diversity values will be observed in populations that recently colonized new areas.
Relationships among individuals were investigated through haplotype network construction and a nested clade phylogeographical analysis (NCPA; Templeton 1998, 2004). A haplotype network was constructed using the parsimony-based algorithm developed by Templeton et al. (1992), implemented in the program TCS v. 1.13 (Clement et al. 2000). A haplotype network approach was preferred over tree-building methods because mitochondrial data tend to form nonbifurcating relationships at the population genetic level (Crandall and Templeton 1996). Inferred haplotype relationships were nested according to published rules (Templeton et al. 1987, Templeton and Sing 1993, Crandall 1996). The nested haplotype network, in conjunction with locality and genetic data, was incorporated into a random permutational analysis using GEODIS v. 2.0 (Posada et al. 2000). For each nested clade possessing genetic and geographic variation, both clade distance (Dc) and nested clade distance (Dn) were calculated. Clade distance (Dc) is the average geographic distance of each individual in a clade to the geographic center of all individuals in that particular clade. Thus, clade distance is a measure of the geographic spread of a particular clade. Nested clade distance (Dn) measures how far the geographic center of a particular clade is to the geographic center of all clades nested at the same level. Thus, nested clade distance indicates how geographically dispersed a particular clade is in relation to closely related clades. These distance values can be used to infer the population-level evolutionary processes and events that may have influenced the current distribution of haplotypes. To make these evolutionary inferences, clades with statistically significant values (0.05 level) for either of the two measures of geographical distance (Dc and Dn) were interpreted using an inference key (Templeton 2004), to separate current population structure (e.g., ongoing gene flow) from historical processes (e.g., range expansion and fragmentation events).
The root of the intraspecific network was inferred through neutral coalescent theory by calculating root probabilities using TCS v. 1.13. This method uses the topology of the unrooted tree (tip versus interior distinction and connectedness) and haplotype frequency (common versus rare) to assign outgroup weights or root probabilities to haplotypes (Castelloe and Templeton 1994, Crandall et al. 1994). This method identifies the likely ancestral haplotype as the haplotype with the most connections and greatest frequency.
Results
Sequence Variation
Twenty-five unique haplotypes were identified among the 78 White-headed Woodpeckers sequenced. Of the 1777 total base pairs, 1742 (98%) characters were constant, 18 (1%) characters were variable but uninformative, and 17 (1%) were phylogenetically informative. Variation within the sequence included 31 transitions and four transversions. The uncorrected p-distance averaged 0.25% (range: 0%–0.68%) among all individuals.
Descriptive Statistics
Average nucleotide diversity (π) for this species was 0.002. The overall AMOVA yielded a Φ st of 0.33 (Table 1), with 67% of the molecular variation explained by within-population differences and 33% by among-population differences. When populations were separated into subspecific groups (Table 1), 27% of the variation was explained by differences between the northern and southern subspecies. However, most of the variation was still due to differences within populations (58%).
Analysis of molecular variance of White-headed Woodpeckers. Results are shown first for an analysis in which populations were not divided into groups and then for an analysis in which populations were divided into northern and southern subspecies. p-values are the probability of getting a more extreme Φ-statistic by chance. The Φ-statistic indicates the proportion of nucleotide variation divided among populations and ranges from 0 to 1.0, with completely subdivided populations having a Φ-statistic of 1.0
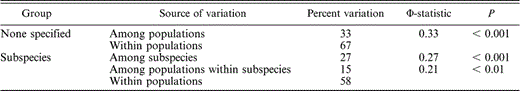
Analysis of molecular variance of White-headed Woodpeckers. Results are shown first for an analysis in which populations were not divided into groups and then for an analysis in which populations were divided into northern and southern subspecies. p-values are the probability of getting a more extreme Φ-statistic by chance. The Φ-statistic indicates the proportion of nucleotide variation divided among populations and ranges from 0 to 1.0, with completely subdivided populations having a Φ-statistic of 1.0
Tajima's D test for neutrality of all individuals (D = −2.0, p = 0.01) was consistent with the model of an expanding population. In addition, the mismatch distribution for all individuals showed a unimodal distribution (Fig. 2), with a raggedness index of 0.02 and a mean of 2.69, and was not significantly different (p = 0.86) from a recently expanding population. Smaller π values were not always found in a particular region of the distribution (Table 2). Therefore, the direction of a potential range expansion could not be inferred.
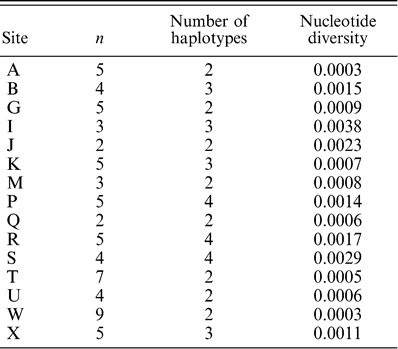
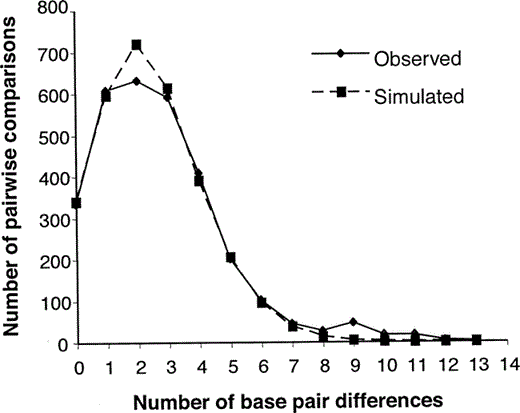
Figure 2. Mismatch distribution of White-headed Woodpecker haplotypes. Solid line indicates observed distribution and dashed line indicates expected distribution under a model of sudden expansion
Haplotype Network and Nested Clade Phylogeographical Analysis
Haplotype network construction resulted in a single network (Fig. 3), with all connections between haplotypes falling within a 95% plausible set of relationships. A minor ambiguity in the form of a loop was resolved using rules of parsimony, neutral coalescent theory, and estimates of tip and interior probabilities (Crandall and Templeton 1993, Crandall et al. 1994). These rules assume that associations between haplotypes that share localities are favored, and rare haplotypes (tips) are derived from more common haplotypes (interiors).
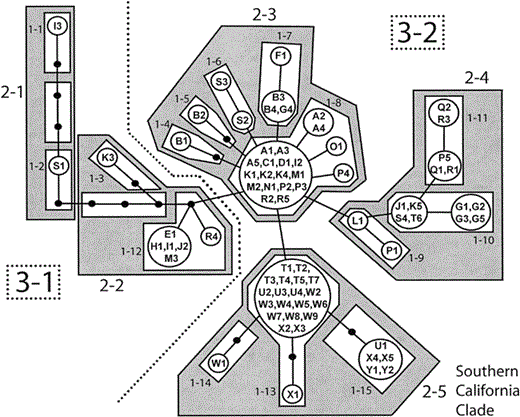
Figure 3. Nested haplotype network for White-headed Woodpeckers. Each branch represents a single base mutation. Black circular nodes represent unsampled haplotypes; lettered nodes represent sampled haplotypes. Each letter with subscript number represents a single individual from its corresponding locality (Fig. 1, Appendix). One-step clades are contained within white boxes, two-step clades within shaded boxes, three-step clades with a dashed line, and the total cladogram is one four-step clade
A total of 39 haplotypes, 25 of which were sampled, were found in the final network. The network was nested into one-step clades, two-step clades, three-step clades, and a four-step total cladogram (Fig. 3). GEODIS identified significant Dc or Dn values for one one-step clade, two two-step clades, one three-step clade, and the four-step total cladogram (Table 3). The inference key identified allopatric fragmentation, restricted gene flow, and range expansion for these clades (Table 3, Fig. 4).
Chains of inference of clades with significant Dc or Dn values. Dc is the average geographic distance of each individual in a clade to the geographic center of all individuals in that particular clade. Dn measures how far the geographic center of a particular clade is to the geographic center of all clades nested at the same level. Letters and numbers under “Inference chain” indicate responses to questions in the inference key of Templeton (2004). For example, for the total cladogram, the answer to question one is no, to question two is yes, to question three is no, to question four is no. As a result, for this clade the inferred pattern from the inference key is “restricted gene flow with isolation by distance.”
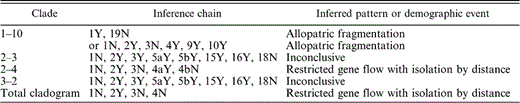
Chains of inference of clades with significant Dc or Dn values. Dc is the average geographic distance of each individual in a clade to the geographic center of all individuals in that particular clade. Dn measures how far the geographic center of a particular clade is to the geographic center of all clades nested at the same level. Letters and numbers under “Inference chain” indicate responses to questions in the inference key of Templeton (2004). For example, for the total cladogram, the answer to question one is no, to question two is yes, to question three is no, to question four is no. As a result, for this clade the inferred pattern from the inference key is “restricted gene flow with isolation by distance.”
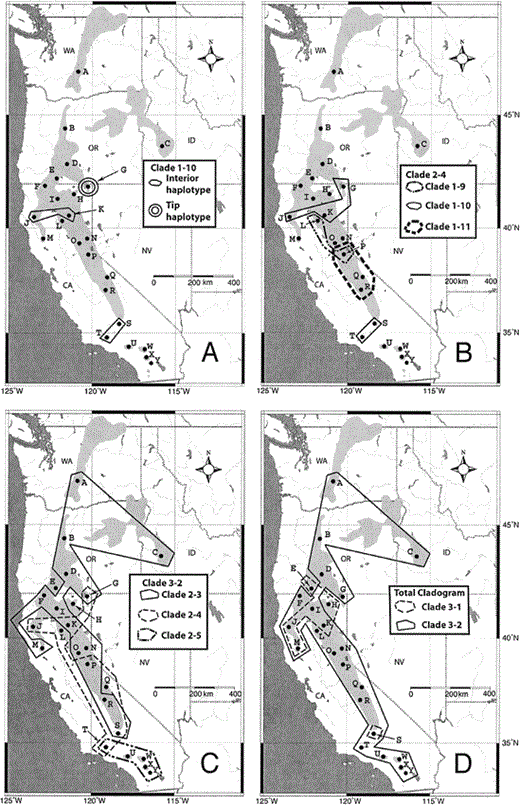
Figure 4. Diagrammatical representation of biogeographic conclusions inferred by the nested clade phylogeographical analysis (Table 3). (A) Clade 1-10, past fragmentation; (B) clade 2-4, restricted gene flow; (C) clade 3-2, possible range expansion; and (D) total cladogram, restricted gene flow. Lightly shaded area indicates the distribution of the White-headed Woodpecker, and dark shaded area is water. For clade 1-10, tip haplotype refers to the haplotype (G1, G2, G3, and G5) derived from the interior haplotype (J1, K5, S4, and T6). State abbreviations: CA = California, ID = Idaho, NV = Nevada, OR = Oregon, and WA = Washington
All individuals of the southern subspecies (populations U–Y) differed by at least one substitution from all individuals of populations A–S (most of the northern subspecies). Most individuals from the intermediate population of Mount Pinos (population T) were found in a monophyletic group with all individuals of populations U–Y. However, one individual from Mount Pinos (T6) was identical in sequence to three individuals from the northern populations (J1, K5, and S4). Other than the division between northern and southern birds, no strong geographic grouping was apparent in the haplotype network. Thus, haplotypes are commonly shared across populations in the north and across populations in the south. The northern group contained 21 sampled haplotypes spread across 19 populations. The most common haplotype in the north was found in nine populations and shared by 15 individuals (A1, A3, A5, C1, D1, I2, K1, K2, K4, M1, N1, P2, P3, R2, and R5). The southern group consisted of four sampled haplotypes in five populations. The most common southern haplotype was detected at four of the five populations and shared by 19 individuals (T1, T2, T3, T4, T5, T7, U2, U3, U4, W2, W3, W4, W5, W6, W7, W8, W9, X2, and X3). The most common northern haplotype was never found in southern birds, and the most common southern haplotype was never found in northern populations.
Identifying the Root of the Haplotype Network
Under coalescent theory, the most common northern haplotype is most likely the oldest haplotype, because it possesses the most connections, is the most geographically widespread (found in nine populations: A, C, D, I, K, M, N, P, and R), and has the highest outgroup weight (0.20, calculated in TCS v. 1.13). The most common southern haplotype may be the oldest in the network because it is shared by the most individuals and has an outgroup weight of 0.18, similar to that of the most common northern haplotype. However, the high frequency of this southern haplotype is more likely due to the fact that some of the southern populations were more heavily sampled.
Discussion
Phylogeography of the White-headed Woodpecker
Overall, the late Quaternary history of the White-headed Woodpecker is characterized by a range expansion and the isolation of northern and southern populations. In addition, the nested clade phylogeographical analysis (NCPA) revealed that additional processes such as allopatric fragmentation and restricted gene flow influenced the phylogeographic history of the White-headed Woodpecker.
For example, within Clade 1-10, there is a single haplotype shared by four (G1, G2, G3, and G5) of the five individuals from the Warner Mountains (Fig. 4A). NCPA inferred that differentiation of this haplotype from its closest relative was the result of allopatric fragmentation, which agrees with the geological data. The Warner Mountains are separated from the continuous northern California range of White-headed Woodpeckers in the Sierra Nevada by lava fields known as the Modoc Plateau. Coniferous forests covered the Modoc Plateau at the end of the Pliocene; however, widespread desertification occurred during the Holocene (Woolfenden 1996), resulting in the present-day mixture of pinyon woodland and shrubsteppe habitats. The Modoc Plateau area is also a contact zone between the Oak Titmouse (Baeolophus inornatus) and its sister species, the Juniper Titmouse (B. ridgwayi, Cicero 2004), and a hybrid zone between the Northern and California Spotted Owl lineages (Strix occidentalis, Barrowclough et al. 2005).
Clade 2-4 consists of three one-step clades that contain individuals ranging from the Warner Mountains in the north to the southern Sierra Nevada in the south (Fig. 4B). NCPA inferred restricted gene flow with isolation by distance for this clade. Glaciation of the Sierra Nevada and other mountains of northern California during the Pleistocene could have limited movements of White-headed Woodpeckers during this time, resulting in restricted gene flow. Many other vertebrate taxa also show genetic discontinuity along the Sierra Nevada (Matocq 2002 and references therein), indicating that glacial cycles likely affected the genetic patterns of a variety of organisms in the region.
Clade 3-2 contains the majority of individuals and genetic diversity for the overall cladogram (Fig. 4C). The NCPA inference key could not discriminate between range expansion (including colonization) and restricted dispersal with gene flow for this clade. However, an inference of range expansion would agree with the results of the mismatch distribution, raggedness index, and Tajima's D calculation for the White-headed Woodpecker. Different histories for the two tip clades (Clade 2-4 and Clade 2-5) in relation to the interior clade (Clade 2-3) could confound the ability to make inferences for Clade 3-2. Thus, we examined each tip clade separately. Clade 2-5 has a distribution that does not overlap the interior Clade 2-3 and is completely restricted to the “sky-islands” of southern California. This distribution would be expected if colonization was recent, especially given that Clade 3-2 is only separated by one substitution from the rest of the cladogram. In contrast, the distribution of tip Clade 2-4 mostly overlaps that of interior Clade 2-3. This type of distribution is commonly the result of restricted gene flow with isolation by distance, the other possibility identified by NCPA. Thus, each tip clade in Clade 3-2 could have experienced a separate history, making an overall inference for this clade difficult.
Although range expansion may be an important process influencing overall differentiation within the White-headed Woodpecker, the NCPA indicated that the relationship between the two oldest clades (3-1 and 3-2) was likely influenced by restricted gene flow with isolation by distance. Clade 3-2 is the more geographically widespread of these two subclades, covering the entire length of the species range (Fig. 4D). The more restricted Clade 3-1 is found across northern California, with only one member of this clade found in the southern Sierra Nevada. The extensive overlap of Clade 3-1 with the widespread Clade 3-2 drives the inference of restricted gene flow. An inference of restricted gene flow agrees with the sedentary and territorial nature of this species.
Comparative Phylogeography
Concordant patterns in the phylogeography of diverse taxa can indicate how common geological and historical events have shaped lineage diversification across species (Bermingham and Avise 1986, Zink 2002). Alternatively, incongruent histories indicate how unique ecological characteristics of a species influenced its ability to respond to particular biogeographic events. The phylogeographic histories of a variety of organisms that have distributions at least partially overlapping that of the White-headed Woodpecker have now been studied. In particular, a number of studies have focused on species found within the California Floristic Province (Calsbeek et al. 2003, LaPointe and Rissler 2005). Calsbeek et al. (2003) analyzed data from many of these studies and found that most animal taxa displayed a north-south division in the vicinity of the Transverse Ranges. For example, the rubber boa (Charina bottae) shows a genetic break between northern and southern forms in the vicinity of Mount Pinos (Rodríguez-Robles et al. 2001), similar to what we found for the White-headed Woodpecker.
Unfortunately, comparing phylogeographic histories is difficult because differences in sampling design, markers used, or methods of analyses among studies may affect interpretations. However, two recent studies used the same markers and analyses as our study. Burns and Barhoum (2006) studied the phylogeography of the Wrentit (Chamaea fasciata) and Sgariglia and Burns (2003) studied the phylogeography of the California Thrasher (Toxostoma redivivum). Although the latter species use different habitats than the White-headed Woodpecker, nearby populations were sampled in most cases. While a southern ancestry was inferred for the Wrentit and the California Thrasher, the White-headed Woodpecker more likely has a northern origin. All three species have an overall history characterized by a recent range expansion, and all three species show a north-south genetic break in the vicinity of the Transverse Ranges. Apart from these similarities, each of the three species experienced unique events in its phylogeographic history.
Subspecific Taxonomy
The molecular variation found in this study is congruent with the division of the White-headed Woodpecker into a northern and southern subspecies based on morphological variation. With the exception of one individual, the two subspecies form monophyletic groups, and differences between northern and southern subspecies accounted for 27% of the variation in the AMOVA. Our results contrast with those typically found when comparing patterns of genetic and morphological diversity among avian subspecies. For the majority of birds examined to date, subspecific designation based on morphology typically does not agree with molecular data (Zink 2004).
The currently recognized subspecific division of the White-headed Woodpecker is based on morphological differences described by Grinnell and Miller (1944). More recently, Garrett et al. (1996) presented morphological data for the two subspecies, including measurements of bill length, wing-chord length, tarsus length, and tail length; the only significant difference (-test, p < 0.05) between the subspecies was the longer bill possessed by the southern subspecies, P. a. gravirostris. On average, bills of southern birds were 11% larger than those of northern birds (Garrett et al. 1996).
The differences in bill size between the two subspecies correspond to ecological differences in their habitats. Coulter pines are present throughout the range of the southern subspecies, including the Mount Pinos population (T), and absent in the range of the northern subspecies. Cones of this species are the heaviest in the world and possess long apical spines (2.5–3.0 cm) at the tips of the scales. These exceptionally long spines could protect the large seeds from woodpeckers with smaller bills, and could have driven the evolution of longer bills in southern populations to successfully take advantage of this food source. Foraging observations (Koch et al. 1970, Hilkevitch 1974) are consistent with the idea that a preference for Coulter pine cones has driven the evolution of larger bills in P. a. gravirostris.
The contact zone of the northern and southern subspecies is located at Mount Pinos (population T). The Mount Pinos population was assigned to P. a. albolarvatus by Grinnell and Miller (1944) and considered possibly intermediate by Garrett et al. (1996). We did not observe any southern haplotypes in the Piute Mountains (population S), or any northern haplotypes in the San Gabriel Mountains (population U). Between these two areas, we found only one (T6) of seven Mount Pinos individuals with a northern haplotype; the rest of the individuals possessed southern haplotypes. Individuals of southern ancestry may be more common and outcompete those of northern ancestry in this area due to the presence of Coulter pines. The contact zone may continue to the unsampled “sky-island” population of the Tehachapi Mountains, located between Mount Pinos and the Piute Mountains. This project was unable to collect specimens from the Tehachapi Mountains because this area is completely encompassed by private property; however, if the difference in the subspecies distribution is a result of and maintained by the presence of Coulter pines, then the population in the Tehachapi Mountains should be composed of individuals genetically similar to northern populations.
Conservation Implications
A long history of isolation may not be needed to create functionally divergent populations, because substantial adaptive divergence and reproductive isolation can occur despite high levels of gene flow (Dieckmann and Doebeli 1999). These factors can lead to distinct population groups such as Evolutionarily Significant Units (ESUs; Ryder 1986). If two groups display adaptive distinctiveness and historical and reproductive isolation, they should be managed independently of each other. This concept depends on two categories of exchangeability, genetic and ecological, which serve as null hypotheses (Crandall et al. 2000). Genetic exchangeability is rejected when there is evidence of restricted gene flow between populations. Ecological exchangeability is rejected when there is evidence for population differentiation due to natural selection or genetic drift, such as differences in morphology, habitat, and life history traits. The genetic, ecological, and behavioral differences exhibited between the northern P. a. albolarvatus and southern P. a. gravirostris confirm the existence of two independently evolving lineages. Genetic exchangeability in the White-headed Woodpecker is rejected by genetic differences between P. a. albolarvatus and P. a. gravirostris, although these differences have likely only recently evolved. Ecological exchangeability is rejected by the evidence for differences in behavior (foraging differences), morphology (bill size differences), and habitat (presence of Coulter pines in the range of P. a. gravirostris) that led to adaptive divergence. Thus, results from this study indicate P. a. albolarvatus and P. a. gravirostris should be managed as two distinct ESUs.
Both subspecies face management challenges. Available habitat for northern populations of White-headed Woodpeckers in the state of Washington has been greatly reduced through timber harvest over the last century and the degradation of suitable habitat due to fire suppression (Buchanan et al. 2003). Reduction in suitable habitat for northern White-headed Woodpeckers is also a problem throughout Oregon (Dixon 1995). The subspecies found in southern California faces habitat alteration from development and logging, magnified by the small geographic size of each “sky-island.” Additionally, recent drought, beetle infestations, and fires have compromised the size and quality of suitable habitat in southern California. Because of the small size of most habitat fragments in the south, any one of the few White-headed Woodpecker populations in southern California could easily be extirpated.
Acknowledgments
Funds for this project were provided by the American Museum of Natural History (Frank M. Chapman Memorial Fund), Los Angeles Audubon Society (Ralph W. Schreiber Ornithology Research Award), San Diego State University Evolutionary Biology Graduate Scholarship, and the National Science Foundation (grant IBN 0217817). The U.S. Fish and Wildlife Service and California Department of Fish and Game provided collecting permits. For additional tissues we thank collectors and curators associated with the Natural History Museum of Los Angeles County, Museum of Vertebrate Zoology at U.C. Berkeley, the Burke Museum of Natural History and Culture, and the Louisiana State University Museum of Natural Science Collection of Genetic Resources. We also thank R. Frenzel (Oregon Cascades White-headed Woodpecker Research Station) for supplying feather and tissue samples from Oregon populations, and J. Dudley (USDA Forest Service, Rocky Mountain Research Station) for feather samples from Idaho. For use of specimens and specimen data we thank the San Diego Museum of Natural History (P. Unitt), the Natural History Museum of Los Angeles County (K. Garrett), the Louisiana State University Museum of Natural Science (S. Cardiff and J. V. Remsen) and the Slater Museum of Natural History (G. Shugart). For comments on and assistance with various aspects of this project, we thank A. Bohonak, P. Pryde, K. Garrett, P. Unitt, L. Klatsky, T. Shephard, R. Alexander, T. Reeder, E. Sgariglia, S. Crews, J. Bland, C. Moore, R. Dixon, G. Barrowclough, L. Frabotta, S. Cardiff, and R. Sedano. Comments from D. Dobkin, T. Sachtleben, and two anonymous reviewers improved the manuscript.
Literature Cited
Appendix
Locality and voucher information for 78 individual White-headed Woodpeckers from 25 localities (A–Y). Each individual is labeled according to its population, and population letters (A through Y) correspond to Figure 1. For example, the five individuals from population A are labeled A1, A2, A3, A4, and A5. SDSU = Vertebrate Collections, San Diego State University; MVZ = Museum of Vertebrate Zoology, University of California at Berkeley; LSUMZ = Louisiana State University Museum of Natural Science Collection of Genetic Resources, UWBM = Burke Museum of Natural History and Culture; LACM = Natural History Museum of Los Angeles County
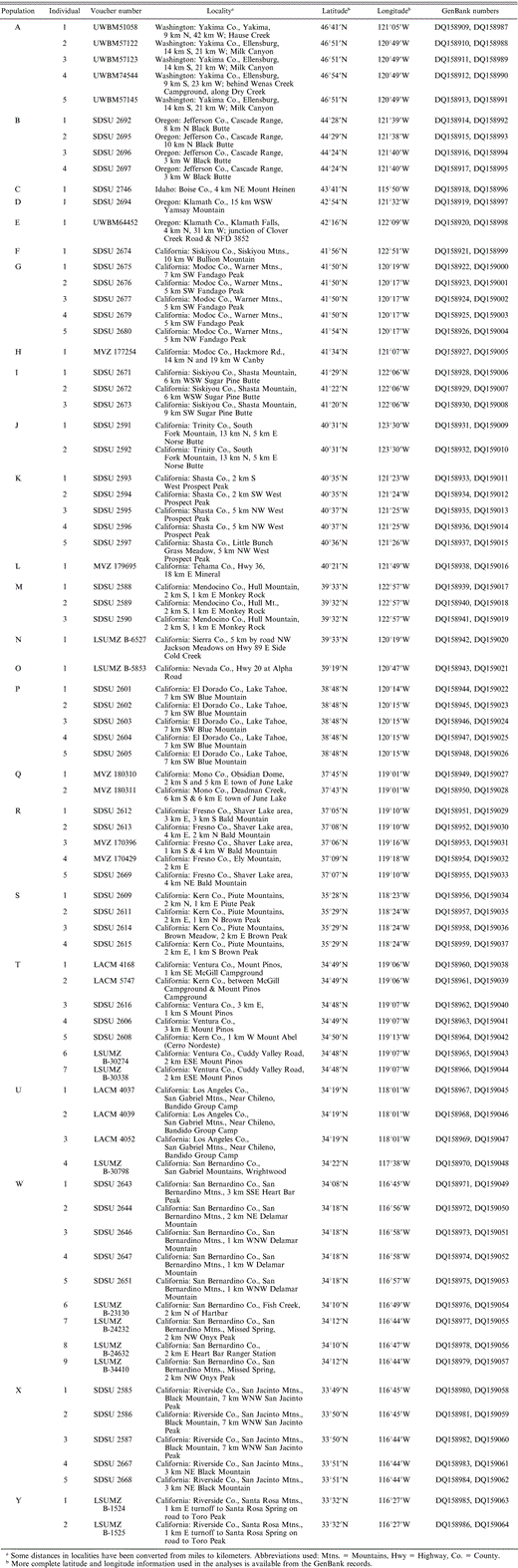
Locality and voucher information for 78 individual White-headed Woodpeckers from 25 localities (A–Y). Each individual is labeled according to its population, and population letters (A through Y) correspond to Figure 1. For example, the five individuals from population A are labeled A1, A2, A3, A4, and A5. SDSU = Vertebrate Collections, San Diego State University; MVZ = Museum of Vertebrate Zoology, University of California at Berkeley; LSUMZ = Louisiana State University Museum of Natural Science Collection of Genetic Resources, UWBM = Burke Museum of Natural History and Culture; LACM = Natural History Museum of Los Angeles County
Author notes
Corresponding author. [email protected]

{kind=link}
{kind=link}
{kind=link}
{kind=link}