





Abstract
I genotyped lek-mating Long-tailed Manakins (Chiroxiphia linearis) at Monteverde and Santa Rosa, Costa Rica, 115 km apart. Cavalli-Sforza distance was 0.04, DLR was 0.18, and RST and θ were both 0.02. Bayesian clustering analysis indicated that both populations were part of a single cluster rather than from distinct clusters. I present a binomial test for probability of allelic absence as a function of sample size. Genotypic likelihood tests assigned 50% of Monteverde birds to Santa Rosa, versus 26% of Santa Rosa birds to Monteverde. Two lines of evidence supported the idea of asymmetric gene flow up the elevational gradient from Santa Rosa to Monteverde. Low differentiation at this spatial scale, despite intense sexual selection, suggests that sexual selection alone is unlikely to promote rapid divergence leading to speciation. Reduced gene flow, produced by geographic barriers or behavioral factors, may also be required.
Evidencia de Flujo Génico Mediante ADN Microsatelital en Chiroxiphia linearis, un Ave Neotropical con Estrategia Reproductiva Tipo “Lek”
Resumen. Determiné el genotipo de individuos de Chiroxiphia linearis en dos poblaciones separadas por 115 km, Monteverde y Santa Rosa en Costa Rica. La distancia de Cavalli-Sforza fue 0.04, DLR fue 0.18, y tanto el valor de RST como el de θ fue 0.02. Un análisis de agrupamiento bayesiano indicó que ambas poblaciones pertenecen a un mismo grupo y no a dos grupos diferentes. Presento una prueba binomial para determinar la probabilidad de ausencia alélica como una función del tamaño muestral. La prueba de probabilidad genotípica asignó al 50% de los individuos de Monteverde a la población de Santa Rosa, mientras que un 26% de los individuos de Santa Rosa fue asignado a Monteverde. Dos líneas de evidencia apoyan la idea de flujo génico asimétrico hacia arriba del gradiente altitudinal entre Santa Rosa y Monteverde. A pesar de la intensa presión selectiva sexual, la baja diferenciación a esta escala espacial sugiere que probablemente la selección sexual por sí sola no promueve la rápida divergencia que conduce a la especiación. También se requeriría reducción del flujo génico a través de barreras geográficas y factores conductuales.
The Neotropics are a major repository of avian biodiversity and the home of the manakins (Pipridae), most of whose species have lek mating systems characterized by intense sexual selection. A number of major theories related to speciation have empirical bases in Neotropical ornithology. Among these are the hypothesis that riverine barriers promote speciation (Wallace 1853, Capparella 1991), the role of vicariance events along the Andes (Cracraft and Prum 1988, Brumfield and Capparella 1996), and the role of forest refugia in speciation (Endler 1982, Mayr and O'Hara 1986). Despite the abundance of species and their importance to evolutionary theory, relatively few studies have addressed genetic differentiation in Neotropical birds, particularly at fine spatial scales. Numerous authors have suggested that sexual selection may promote speciation (e.g., Lande 1981, West Eberhard 1983, Uy and Borgia 2000). Long-tailed Manakins (Chiroxiphia linearis) are lek-mating birds of tropical dry forests from southern Mexico to Costa Rica. Because Long-tailed Manakins have one of the highest variances in male mating success yet demonstrated in vertebrates (McDonald 1989, 1993), one might expect that rapid interpopulation divergence would occur. In this paper, in the light of possible factors promoting divergence and speciation, I assess genetic variation in the Long-tailed Manakin between a mid-elevation (1300 m) and sea-level site separated by 115 km.
Methods
Sampled Populations
As part of a long-term study of a color-banded population of Long-tailed Manakins in Monteverde, Costa Rica (10°18′N, 84°48′W, 1300 m elevation) I collected blood samples for microsatellite DNA analyses (McDonald and Potts 1994). Between 1987 and 1992, I took blood samples from 128 individuals in the Monteverde population and 39 individuals in the lowland Santa Rosa population sampled from the Bosque Humedo near the headquarters of Parque Nacional Santa Rosa, 115 km northwest of Monteverde in the drought-deciduous forests of Guanacaste province. All individuals were mist netted, individually color banded and approximately 100 μL of blood was sampled by ulnar venipuncture. Extracted DNA was assessed at four microsatellite loci (SJ133, LTMR8, LTR6, LTR15) by polyacrylamide gel electrophoresis visualized by ethidium bromide staining. The primer sequences and details of the laboratory methods are given in McDonald and Potts (1994).
Binomial Test for Allelic Presence/Absence
For microsatellite loci, the sometimes small sample sizes obtainable from natural populations may affect allelic presence/absence and potentially influence conclusions concerning population structure. I used a binomial probability approach to calculate the likelihood that private alleles (alleles unique to a population; Neel 1973, Slatkin 1985) were truly missing in the population from which I took the smaller sample. If Fi denotes the frequency of a particular allele at a given locus over both populations, the probability that any given individual has no copy of the allele is M = (1−Fi)2. The probability that the allele will be totally missing in a sample of size n is Mn.
Statistical Analyses
I calculated unbiased expected heterozygosity (Hexp) and observed heterozygosity (Hobs) and their variances, as well as allelic richness (El Mousadik and Petit 1996). To assess genetic differentiation among populations, I used two measures of genetic distance and two variants of variance-based F-statistics. These measures are very differently derived, and have been shown to perform well with microsatellite data (Takezaki and Nei 1996, Paetkau et al. 1997, Kalinowski 2002). The relatively low heterozygosities of the loci I assayed mean that problems with bounds on FST discussed by Hedrick (1999) do not apply. The distance measures were Cavalli-Sforza chord distance (Cavalli-Sforza and Edwards 1967), a purely geometric assessment of genetic structure that makes no biological assumptions, and DLR, a distance measure based on the allelic probability approach of the assignment test (Paetkau et al. 1995). F-statistic variants were RST (Slatkin 1995, Goodman 1997), and θ (Weir and Cockerham 1984). Except for RST, I did not use measures based on a stepwise mutation model, for two reasons. First, several such measures have been shown to have high variances and to perform poorly in simulations (Paetkau et al. 1997, Takezaki and Nei 1996). Second, they assume a predominance of mutation over forces such as drift. The allelic distributions (available on request from the author) were a poor fit to a stepwise model, and almost certainly reflect a strong influence of factors such as drift and migration. The Bayesian approach of Pritchard et al. (2000) assesses whether the sampled genotypes are substructured into multiple (K > 1) clusters or constitute a single Hardy-Weinberg population (K = 1). Log-likelihood ratios from Monte Carlo Markov chain sampling provide the basis for deciding what number of clusters best fits the data. To compute the descriptive statistics, fit to Hardy-Weinberg equilibrium, distance measures, F-statistic variants and Bayesian clustering, I used the programs GENETIX (Belkhir et al. 2002), FSTAT (Goudet 2002), PHYLIP (Felsenstein 1993), Doh (Brzustowski 2002), GeneClass (Cornuet 1999), and Structure (Pritchard 2000).
Assignment tests (Paetkau et al. 1995) calculate the likelihood that the genotypes of individuals match the allelic profiles of the population in which they were sampled relative to the profiles of a comparison population. Bivariate plots of the log-likelihood of a genotype in its sampled population versus its log-likelihood in a comparison population allow one to distinguish “assignment matches” (individuals assigned to the population in which they were sampled) versus assignment mismatches (individuals assigned to a population other than the one in which they were sampled). A large number of assignment mismatches indicates extensive gene flow.
Two potential sources of bias exist for assignment tests. The first is the overweighting of an individual's genotype in its sampled population, and the second is that alleles absent in other populations lead to zero assignment probabilities in any but the sampled population (Paetkau et al. 1995). To avoid these sources of bias, I used an option for allele frequency adjustment derived from the work of Titterington et al. (1981). I also used the Bayesian assignment method of GeneClass, with the leave-one-out option for dealing with missing alleles. Assumptions and rationale for this variant assignment method can be found on the GeneClass website (Cornuet 1999).
To estimate relative effective sizes for the populations and mutation rates for the loci I used MISAT (Nielsen 1997a, 1997b). MISAT provides a separate estimate of 4Neμ (where μ is the mutation rate) for each locus–population combination. To reconcile these estimates across all loci or all populations, I first log transformed the values and then used linear, multivariate regression to calculate coefficients that could be used to estimate overall relative mutation rates or effective sizes across both populations or all four loci. I calculated confidence intervals for the estimates by using the multivariate regression standard deviations times the t-statistic (0.975, with 5 degrees of freedom from the six total variables: two populations and four loci). The protocol for the log regression is available on request. I also estimated 4Neμ and 4 Nem (where m is the migration rate) using the infinite alleles option of the program Migrate (Beerli and Felsenstein 2001; version 1.6.9, Beerli and Felsenstein 2002). Both Migrate and MISAT make several assumptions that are likely violated in most natural populations. Nevertheless, the results may be robust even to fairly major violations of assumptions (Neigel 2002), and the assumption of overriding importance, which one trusts is met, is that no other competing forces are likely to produce the observed patterns of variation. For the outputs from MISAT and Migrate I present the estimates accompanied by a 95% confidence interval (CI).
Results
I scored four polymorphic microsatellite loci for 128 individuals from Monteverde and 39 individuals from Parque Nacional Santa Rosa. All locus-population combinations conformed to Hardy-Weinberg expectations. Table 1 presents several measures of genetic diversity for the two populations. Two loci each had an allele found only in the Monteverde sample: the 142 base-pair allele at locus SJ133 (27 of 334 total alleles in the two populations), and an allele at locus LTMR8 that was also 142 base pairs long (2 alleles in the total sample). The binomial probability-of-occurrence test indicated that the more common locus SJ133 allele was very unlikely to be missing from the Santa Rosa sample by chance or because of inadequate sampling (M39 = 0.001). For the very rare LTMR8 allele, however, the likelihood of missing the allele in the smaller Santa Rosa sample was high (M39 = 0.63).
Measures of genetic variation for two populations of Long-tailed Manakins in Costa Rica separated by 115 km. Heterozygosity values are means ± SD

Measures of genetic variation for two populations of Long-tailed Manakins in Costa Rica separated by 115 km. Heterozygosity values are means ± SD
Genetic distance and F-statistic-like measures yielded concordant, relatively low levels of genetic differentiation at this spatial scale. The Cavalli-Sforza chord distance between the two populations was 0.04. The distance measure DLR, based on the assignment test (Paetkau et al. 1995) was 0.18. Weir and Cockerham's (1984) θ and Slatkin's (1985),RST as modified by Goodman (1997) were both 0.02. Bayesian clustering analysis (Pritchard et al. 2000) indicated a single cluster (no substructuring) in the absence of prior information on the sampled population. When I included prior information for sampled population and a migration probability of 0.01, the strongest support was for a two-cluster model, with all individuals assigned to their sampled population with P > 0.9.
The assignment test (Fig. 1) showed rather little differentiation (many mismatches) between the manakin populations, but a very asymmetric pattern of deviations from the diagonal line of equal assignment likelihood. Of the 128 individuals genotyped in Monteverde, 64 (50%) were mismatches, assigned to Santa Rosa. Of the 39 individuals sampled in Santa Rosa, 10 (26%) were mismatches, assigned to Monteverde. Deviations from the line of equality were decidedly asymmetric. A large cluster of 26 of the 74 individuals assigned to Monteverde had residuals from the line of equality of greater than 2.0. In contrast, none of the 93 individuals assigned to Santa Rosa had residuals from the line of equality of more than 0.6 (Fig. 1). As a result, the values of the residuals were significantly higher on the Monteverde side (below the diagonal in Fig. 1) than on the Santa Rosa side (two-tailed t76 = 5.9, P < 0.001). All of the individuals in the strongly deviating cluster bore one or more copies of the two alleles found only in Monteverde (individuals with these Monteverde-only alleles are marked by unfilled circles in Fig. 1). No evidence existed for a sex bias in tendency to deviate from the line of equality. Of the Monteverde samples, 15 of 66 known males (23%) and eight of 30 known females (27%) were among the 26 individuals in the strongly deviating cluster.
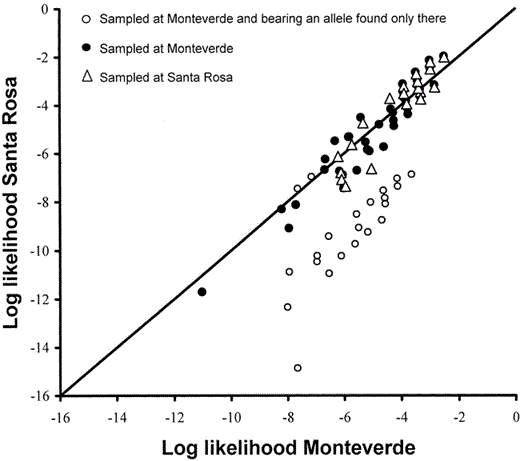
Assignment likelihoods for two populations of Long-tailed Manakins in Costa Rica separated by 115 km. Genotypes above the diagonal are more likely in the Santa Rosa population; those below the diagonal are more likely in the Monteverde population. Most individuals cluster near the diagonal line of equal probability, indicating relatively low population distinctiveness. Half the individuals sampled at Monteverde (64 of 128; multiple points are superimposed on both sides of the line) were “misassigned” to Santa Rosa, while 26% of those sampled at Santa Rosa were “misassigned” to Monteverde (many superimposed points on the Santa Rosa side of the line). A cluster of 26 individuals deviating strongly from the line of equality is evident on the Monteverde side (below the diagonal; minimum residual 2.0; the points for six individuals coincide with other points). The cluster is largely responsible for a significant difference in mean deviation between the Monteverde-assigned genotypes and those assigned to Santa Rosa. All the individuals in the cluster bore one or both of the alleles found only at Monteverde. Symbols: circles = individuals sampled at Monteverde; unfilled circles = individuals bearing an allele found only at Monteverde; filled circles = sampled at Monteverde but not bearing an allele unique to Monteverde; triangles = individuals sampled at Santa Rosa
The point estimate for the effective size of the Santa Rosa population as estimated by MISAT (Nielsen 1997b) was smaller than that of Monteverde (0.83 relative to an arbitrarily set value of 1.0 for Monteverde, CI = 0.42 to 1.67). Because the confidence interval overlaps equality (1.0), the MISAT results do not allow one to conclude that the population sizes differ. Migrate (Beerli and Felsenstein 2002) estimated 4Neμ as 0.73 for Monteverde (CI = 0.68 to 0.77) and 0.12 for Santa Rosa (CI = 0.11 to 0.14). Assuming equivalent mutation rates for the four loci in the two populations, Migrate therefore estimated a considerably larger Ne for the Monteverde population. Migrate estimated an asymmetric migration parameter (4Nem) of 0.73 from Monteverde to Santa Rosa (CI = 0.42 to 1.28) and 1.99 from Santa Rosa to Monteverde (CI = 1.43 to 2.72).
Discussion
Long-tailed Manakins showed rather low levels of differentiation across the 115-km scale addressed in this study. Bayesian clustering analysis provided no support for multiple clusters (substructure) in the absence of prior information on population origin. Given prior information on population origin, the program Structure assigned all individuals to the location in which they were sampled. The result suggests that little structure is evident in the data in the absence of pre-existing information on the sampling location, further supporting the hypothesis of gene flow extensive enough to prevent major divergence of the populations. Examples from published studies with FST-like measures (FST, RST, or θ, as described in Methods) suggest that gene flow is comparable to or higher than that found in other species (Table 2).
Variants of Wright's F-statistics, used as measures of differentiation among populations, for avian taxa analyzed with microsatellite markers. Taxonomic order follows Monroe and Sibley (1993)
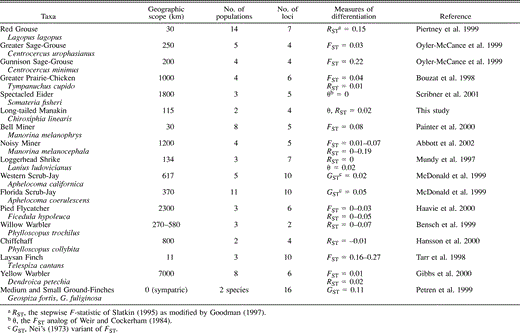
Variants of Wright's F-statistics, used as measures of differentiation among populations, for avian taxa analyzed with microsatellite markers. Taxonomic order follows Monroe and Sibley (1993)
The asymmetry of the assignment test suggests the possibility that gene flow is largely one-way, from Santa Rosa to Monteverde, a conclusion supported by the asymmetric migration estimate from the program Migrate. A caution in assessing the low resolution of the assignment testing (high frequency of mismatches) is that more loci might improve the resolving power. The binomial test developed here indicated that locus SJ133's 142 base-pair allele, which was found only in Monteverde, was sufficiently common there that it should have shown up in Santa Rosa if gene flow were occurring in that direction. Monteverde is at the altitudinal limits of the range (1300 m), raising the possibility that lowland populations act as sources and higher-elevation populations act as sinks. The alternative possibility of a bottleneck in the lowlands but not at Monteverde seem unlikely, but further evidence on paired high and low elevation populations would be instructive.
The relatively low level of differentiation between the two Long-tailed Manakin populations has implications for questions of genetic structure and speciation in birds. It suggests that sexual selection alone may not suffice to generate genetic divergence at small spatial scales. The habitat of Long-tailed Manakins is nearly continuous across their range from southern Mexico to northwestern Costa Rica. No large rivers occur, and their distribution is entirely west of the continental divide, so that no mountain ranges separate populations. Both sexes appear to disperse widely. With negligible geographic barriers and no behavioral checks on dispersal, little differentiation has developed despite intense sexual selection. In some species of birds, behaviorally induced reduction in the dispersal of one or both sexes might interact with sexual selection to promote rapid divergence. For other species, geographic barriers might interact with sexual selection to produce accelerated divergence. The occurrence of localized forms of Chiroxiphia in the Andean foothills of South America, including the Yungas Manakin (C. boliviana) and a distinctive subspecies of the Blue-backed Manakin (C. pareola regina) whose crown is yellow rather than red (Parker and Remsen 1987), lend support to the latter possibility even within the genus Chiroxiphia.
The traditional chasm separating population genetics from systematics has blurred in recent years, largely through the pioneering efforts of John Avise (2000) in creating the field of phylogeography, based on intraspecific analyses of mitochondrial DNA phylogenies. Microsatellite analyses at a range of scales also contribute to this synthesis (Gibbs et al. 2000, McDonald and Potts 1997, McDonald et al. 1999, Petren et al. 1999) and will likely continue to play a major role in illuminating the fine structure of incipient speciation.
Acknowledgments
Many Earthwatch Institute volunteers helped with data collection and provided financial and logistical support. D. Lawson helped with the fieldwork in Santa Rosa. J. Gilardi, K. Buchanan, and R. Clay were particularly helpful in gathering data in Monteverde. K. Gerow helped with the binomial test for likelihood of allelic absence given differing sample sizes. Two anonymous reviewers and the editor made comments that substantially improved the manuscript. The study was funded in part by an award from the National Geographic Society's Committee for Research and Exploration, as well as by funding from the Harry Frank Guggenheim Foundation.
Literature Cited

{kind=link}