





MNAzymes (nucleic acid enzymes formed from multiple partial enzymes) can be linked to PCR to provide a highly specific method for target detection and quantification. We investigated the feasibility of multiplexing MNAzyme quantitative PCR (qPCR) methods.
We combined MNAzyme components with PCR primers and standard qPCR reagents to perform MNAzyme qPCR and reverse-transcription qPCR (RT-qPCR) assays with a set of universal reporter probes. Assays were performed on single targets and in multiplex formats that combined up to 5 different targets in a single reaction.
A comparison of 3 targets amplified in single and triplex formats showed no significant differences with respect to detection limit or amplification efficiency. Likewise, we successfully converted single-target assays for 11 transcripts of interest to triplex assays containing 2 reference transcripts without having to optimize or modify the conditions. A quintuplex RT-qPCR that simultaneously quantified 5 transcripts with 5 universal probes produced high amplification efficiencies and r2 values for all transcripts. Despite the large numbers of oligonucleotides in the reactions, we observed no false-positive signals, owing to the requirement of 4 target-specific binding events to produce a signal. A quadruplex assay that combined MNAzymes with methylation-specific PCR to measure epigenetic biomarkers of prostate cancer was capable of detecting a single methylated DNA allele in a BACKGROUND of 1000–10 000 unmethylated alleles. The MNAzyme qPCR was compatible with a rapid-cycling protocol.
MNAzymes offer a flexible and unique approach to qPCR that is specific, sensitive, and easily multiplexed. The universal nature of MNAzyme reporter probes removes the need for target-specific probes, thereby making the development of new assays easier and cheaper.
MNAzymes2 are nucleic acid enzymes formed from multiple partial enzymes (1). These complexes were engineered from DNAzymes to further increase the functionality of this highly robust, nonprotein class of enzymes. Although DNAzymes had been evolved to detect and cleave a target (2), MNAzymes were reengineered to detect a target and cleave a separate substrate while leaving the target intact. This uncoupling of detection from catalytic readout led to the development of enzymes with greater flexibility and utility, thereby providing a powerful new tool for genetic analysis. The catalytic potential of an MNAzyme exists in 2 short “partzymes,” each of which contains (i) a target-binding sensor arm, (ii) a partial catalytic core, and (iii) a probe-binding domain (Fig. 1). These partzymes can facilitate catalysis only when their partial catalytic cores form a complete core, which occurs when the complementarity between the partzymes' sensor arms and the target facilitates the partzymes' precise, adjacent alignment on a target (RNA, DNA, or amplicons). Once assembled, MNAzymes can bind universal reporter probes and cleave them between fluorophore and quencher moieties to generate a florescence signal that can be monitored in real time (Fig. 1).
Schematic representation of MNAzyme qPCR.
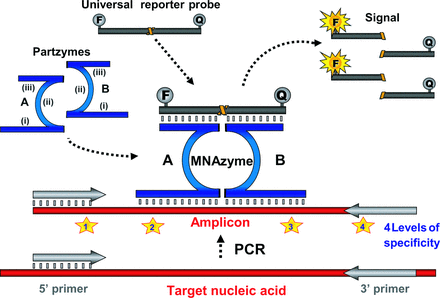
MNAzyme qPCR reactions contain the template (i.e,. RNA or DNA), PCR primers, partzyme A, partzyme B, and the universal reporter probe. The partzymes are composed of (i) a target-binding domain, (ii) a partial catalytic core, and (iii) a probe-binding domain. Upon generation of amplicons, the 2 partzymes can bind to the sequence complementary to their target-sensing arms. The fluorescently labeled reporter probe also binds to the partzymes, and once the active catalytic core is formed, the probe is cleaved, producing a signal that is indicative of successful amplification of the target gene or transcript. F, fluorophore; Q, quencher.
The unrivaled sensitivity and convenience of real-time PCR, or quantitative PCR (qPCR), has revolutionized nucleic acid research. Various qPCR methods monitor the accumulation of amplicons during the PCR by measuring the change in fluorescence associated with the binding of intercalating dyes or the changes associated with fluorescently labeled target-binding probes [reviewed in (3)]. Although approaches that use intercalating dyes are an inexpensive alternative to probe-based methods, they are not well suited to multiplexing and may lack the specificity necessary for clinical applications. Assays that use target-specific probes have higher specificity, but a new probe needs to be synthesized and optimized for every new target; consequently, multiplexing generally requires considerable optimization. The MNAzyme qPCR approach combines the ability to use one or more universal probes from a series of such probes, along with MNAzymes, each of which requires 2 distinct target-binding events (Fig. 2). This design constitutes an improved and simplified approach to multiplex PCR that has exquisite specificity and requires less optimization. MNAzyme qPCR is the only real-time PCR technology that allows target-specific interrogation with a generic readout (1).
Strategy for multiplexing MNAzymes.
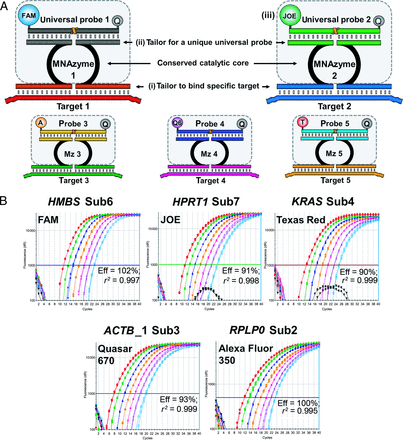
(A), Tailor sensor arms to bind to each specific target (i). The catalytic core and probe-binding domains are universal (hatched boxes). However, each probe domain in a multiplex must be tailored to a unique sequence (ii), and each probe in a multiplex must be labeled with a different fluorophore (iii). (B), Amplification plots generated from the quintuplex MNAzyme RT-qPCR reaction (500 ng to 120 pg). Eff, efficiency; r2, coefficient of determination (i.e., the correlation coefficient squared).
The unique MNAzyme qPCR approach has advantages for multiplex analysis, because new targets can be added to a reaction without requiring synthesis of a new probe and the approach eliminates any concerns about cross-reactivity between probes. Fig. 2 outlines the strategy for multiplexing MNAzymes, which requires only that the target-sensing arms of partzymes be tailored to the sequence of interest. The partial catalytic cores remain constant, and the probe arms are matched to one of a set of well-characterized universal probes that have been optimized for reliable, consistent performance in multiplex assays.
The present study has demonstrated the utility of a selection of the versatile applications of MNAzyme qPCR, including quantification of DNA and RNA, methylation analysis, and rapid-cycling qPCR. In particular, these studies showcase the advantages of MNAzyme qPCR for multiplex analysis.
Materials and Methods
REPORTER PROBES
MNAzyme activity was evaluated by monitoring the cleavage of a dual-labeled nucleic acid reporter probe composed of DNA and RNA nucleotides. The probes were labeled with a fluorophore at the 5′ end and with a quencher at the 3′ end (Table 1).
Probe sequences and fluorescent labeling.
| Probea | Sequence (5′ → 3′)b |
|---|---|
| Sub2-A3B Sub2-FB Sub2-Q7B2 | AAGGTTTCCTCguCCCTGGGCA |
| Sub3-JB Sub3-Q6B2 | CAGCACAACCguCACCAACCG |
| Sub4-TRB2 | CATGGCGCACguTGGGAGAAGTA |
| Sub6-FB Sub6-Q5B2 Sub6-TRB2 | ATCACGCCTCguTCCTCCCAG |
| Sub7-JB Sub7-TRB2 | TTAACATGGCACguTGGCTGTGATA |
| Probea | Sequence (5′ → 3′)b |
|---|---|
| Sub2-A3B Sub2-FB Sub2-Q7B2 | AAGGTTTCCTCguCCCTGGGCA |
| Sub3-JB Sub3-Q6B2 | CAGCACAACCguCACCAACCG |
| Sub4-TRB2 | CATGGCGCACguTGGGAGAAGTA |
| Sub6-FB Sub6-Q5B2 Sub6-TRB2 | ATCACGCCTCguTCCTCCCAG |
| Sub7-JB Sub7-TRB2 | TTAACATGGCACguTGGCTGTGATA |
Fluorophore–quencher combinations used with each probe in this study: A3, Alexa Fluor™ 350; F, 6-FAM (6-carboxyfluorescein); J, JOE (2,7-dimethoxy-4,5-dichloro-6-carboxyfluorescein); Q5, Quasar® 570; Q6, Quasar 670; Q7, Quasar 705; TR, Texas Red®-X; B, Black Hole Quencher® 1; B2, Black Hole Quencher 2.
Uppercase bases represent DNA; lowercase bases represent RNA.
| Probea | Sequence (5′ → 3′)b |
|---|---|
| Sub2-A3B Sub2-FB Sub2-Q7B2 | AAGGTTTCCTCguCCCTGGGCA |
| Sub3-JB Sub3-Q6B2 | CAGCACAACCguCACCAACCG |
| Sub4-TRB2 | CATGGCGCACguTGGGAGAAGTA |
| Sub6-FB Sub6-Q5B2 Sub6-TRB2 | ATCACGCCTCguTCCTCCCAG |
| Sub7-JB Sub7-TRB2 | TTAACATGGCACguTGGCTGTGATA |
| Probea | Sequence (5′ → 3′)b |
|---|---|
| Sub2-A3B Sub2-FB Sub2-Q7B2 | AAGGTTTCCTCguCCCTGGGCA |
| Sub3-JB Sub3-Q6B2 | CAGCACAACCguCACCAACCG |
| Sub4-TRB2 | CATGGCGCACguTGGGAGAAGTA |
| Sub6-FB Sub6-Q5B2 Sub6-TRB2 | ATCACGCCTCguTCCTCCCAG |
| Sub7-JB Sub7-TRB2 | TTAACATGGCACguTGGCTGTGATA |
Fluorophore–quencher combinations used with each probe in this study: A3, Alexa Fluor™ 350; F, 6-FAM (6-carboxyfluorescein); J, JOE (2,7-dimethoxy-4,5-dichloro-6-carboxyfluorescein); Q5, Quasar® 570; Q6, Quasar 670; Q7, Quasar 705; TR, Texas Red®-X; B, Black Hole Quencher® 1; B2, Black Hole Quencher 2.
Uppercase bases represent DNA; lowercase bases represent RNA.
TEMPLATE AND TARGETS
Human genomic DNA (gDNA) and total RNA were extracted from cell lines LNCap, IM9, and K562 with the QIAamp DNA and Blood Mini Kit (Qiagen) and the QIAamp RNA and Blood Mini Kit (Qiagen), respectively, and in accordance with the manufacturer's instructions. Where applicable, LNCap and K562 gDNA were bisulfite modified with the EZ-DNA Methylation Kit (Zymo Research Corporation) according to the manufacturer's instructions. The DNA or RNA was then used as template in subsequent MNAzyme qPCR or reverse-transcription qPCR (RT-qPCR) reactions to amplify several genes (for full sequence information, see Supplemental Data in the Data Supplement that accompanies the online version of this article at http://www.clinchem.org/content/vol59/issue2).
PRIMERS AND PARTZYMES
Tables 1 and 2 in the online Data Supplement list the sequences of PCR primers and partzymes, along with the concentration used in each of the following experimental examples.
STANDARD MULTIPLEX MNAzyme qPCR CONDITIONS
Unless specifically indicated, MNAzyme qPCR reactions contained 40 nmol/L forward primer and 200 nmol/L each of reverse primer, partzyme A, partzyme B, and reporter probe in a 25-μL final reaction volume containing 10 mmol/L MgCl2, 200 μmol/L of each deoxynucleoside triphosphate, 10 U RNasin® (Promega), 1× ImmoBuffer (Bioline), and DNA polymerase (as specified below). Thermocycling conditions were as follows: 95 °C for 2 min; 5 cycles of 95 °C for 15 s and 55 °C for 60 s; and 40 cycles of 95 °C for 15 s and 52 °C for 60 s. All experiments were performed on the Mx3005P QPCR System (Agilent Technologies), with the exception of the methylation-specific (MSP) MNAzyme qPCR, which was performed on the SmartCycler® instrument (Cepheid). Nuclease-free water (NF-H2O) was used for all dilutions and for all no-template control reactions.
SINGLEPLEX VS TRIPLEX MNAzyme qPCR
We amplified the TFRC3 [transferrin receptor (p90, CD71)], B2M (beta-2-microglobulin), and KRAS (v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog) genes (see Table 3 in the online Data Supplement) with 2 U MyTaqHS™ DNA Polymerase (Bioline) with a final MgCl2 concentration of 8 mmol/L and either IM9 gDNA (125, 31, 8, 2, or 0.5 ng) or NF-H2O.
MNAzyme RT-qPCR QUANTIFICATION OF A GENE OF INTEREST AND 2 REFERENCE GENES
MNAzyme RT-qPCR was performed for transcripts of 11 genes of interest (GOIs): ACTB (actin, beta), BCR (breakpoint cluster region), HPRT1 (hypoxanthine phosphoribosyltransferase 1), KRAS, TP53 (tumor protein p53), HMBS (hydroxymethylbilane synthase), PGK1 (phosphoglycerate kinase 1), PMM1 (phosphomannomutase 1), PSMB6 [proteasome (prosome, macropain) subunit, beta type, 6], RPLP0 (ribosomal protein, large, P0), and TFRC. We also carried out MNAzyme RT-qPCR reactions with transcripts for 2 reference genes (RGs): B2M and PPIA [peptidylprolyl isomerase A (cyclophilin A)] (see Table 4 in the online Data Supplement). Thermocycling conditions were as follows: 50 °C for 30 min; 95 °C for 10 min; and 40 cycles of 95 °C for 15 s and 52 °C for 60 s. Each target was amplified individually and in combination with the 2 RGs. Reactions contained 2 U Immolase™ (Bioline), 20 U reverse transcriptase (M-MLV Reverse Transcriptase, RNase H Minus, Promega), and either 50 ng K562 total RNA or NF-H2O.
QUINTUPLEX MNAzyme RT-qPCR WITH TOTAL RNA
KRAS, RPLP0, HMBS, HPRT1, and ACTB_1 transcripts (see Table 5 in the online Data Supplement) were amplified with the following thermocycling conditions: 50 °C for 30 min; 95 °C for 10 s; 10 cycles of 95 °C for 15 s and 64 °C for 30 s (with a 1 °C decrease per cycle); and 40 cycles of 95 °C for 15 s and 51 °C for 60 s. Reactions contained 2 U Immolase, 20 U of the Promega M-MLV reverse transcriptase, and either K562 total RNA (500, 125, 31, 8, 2, 0.5, or 0.12 ng) or NF-H2O. A control reaction that lacked reverse transcriptase was also performed.
MSP QUADRUPLEX MNAzyme qPCR ASSAYS OF METHYLATED AND UNMETHYLATED gDNA
MNAzyme qPCR assays of GSTP1 (glutathione S-transferase pi 1), RARB (retinoic acid receptor, beta), APC (adenomatous polyposis coli), and ACTB (see Table 6 in the online Data Supplement) were performed with following thermocycling conditions: 95 °C for 1 min; 18 cycles of 95 °C for 15 s and 64 °C for 30 s; and 40 cycles of 95 °C for 15 s and 52 °C for 60 s. Reactions contained 6 mmol/L MgCl2, 1× additive (final concentrations: 0.2 g/L BSA, 150 mmol/L trehalose, 2 mL/L Tween 20), 1× buffer (final concentration: 67 mmol/L Tris-HCl, pH 8.8; Ambion), 5 U Taq polymerase (Applied Biosystems), and bisulfite-modified gDNA [100 ng LNCap bisulfite-modified DNA was mixed with 100 ng K562 bisulfite-modified DNA in proportions of 1 part LNCap DNA in 9.99 parts K562 DNA, 1 part in 99, 1 part in 999, or 1 part in 9 999; 100 ng LNCap (positive control); 100 ng K562 (negative control); or NF-H2O].
RAPID TRIPLEX MNAzyme qPCR
The HMBS, HFE (hemochromatosis), and HPRT1 genes (see Table 7 in the online Data Supplement) were amplified. Results obtained with standard thermocycling conditions (95 °C for 2 min; 40 cycles of 95 °C for 15 s and 53 °C for 60 s) were compared with those obtained with rapid-cycling conditions (95 °C for 2 min; 40 cycles of 95 °C for 5 s and 53 °C for 15 s). Reactions contained 2 U of MyTaqHS and either IM9 gDNA (500, 125, 31, 8, 2, or 0.5 ng) or NF-H2O.
Results
MULTIPLEX MNAzyme qPCR
The KRAS, TFRC, and B2M genes were quantified in singleplex and triplex reactions in duplicate MNAzyme qPCR assays that were performed over a range of gDNA concentrations (Fig. 3A). We followed the amplification of these 3 genes by monitoring the cleavage of 3 universal MNAzyme probes (Sub2-FB, Sub3-JB, and Sub6-TRB2, respectively). The mean difference in the quantification cycle (ΔCq) between the single and the triplex results for individual genes was calculated for each of the 6 target concentrations. In all cases, the mean shift in Cq was minimal, approximately 0.12 (SD, 0.1) (Fig. 3A). Alignment of the amplification plots for single and triplex results showed neither a loss in signal intensity nor a significant change in the amplification curve/efficiency (Fig. 3A). Both the single and triplex reactions for these genes produced amplification efficiencies of 99.3% to 103.8% and r2 values ≥0.998 (see Table 8 in the online Data Supplement). All assays were highly specific: None of the no-template controls produced a signal, and there was no evidence of cross-signaling between the universal probes.
Results of multiplex MNAzyme qPCR or RT-qPCR reactions.
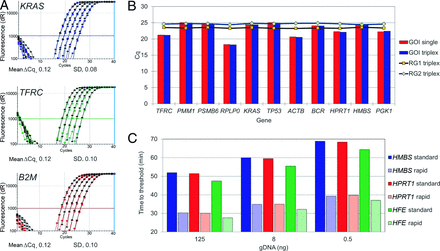
(A), Overlay of amplification plots for single and triplex MNAzyme qPCR reactions (125 ng to 500 pg). Indicated are triplex assays (black) and singleplex assays (blue, green, red). (B), Comparison of Cq values for 11 GOI and 2 RGs in single and triplex MNAzyme reactions. (C), Total time to reach the threshold value for 3 genes (HMBS, HPRT1, and HFE) with standard thermocycling protocols (solid colors) and with rapid-cycling protocols (striped colors).
To further demonstrate the ease of combining multiple MNAzyme assays, we individually amplified transcripts for the 11 GOIs along with the 2 RGs in triplex reactions. Comparison of the single and triplex reactions for each GOI showed that the Cq value did not change significantly after adding the 2 RGs (Fig. 3B; see Table 9 in the online Data Supplement). Furthermore, the Cq values for the 2 RGs showed very little variation in the context of the 11 different triplex reactions: The mean (SD) Cq values obtained for PPIA and B2M were 19.5 (0.14) and 20.6 (0.15), respectively (n = 22 for both) (see Table 9 in the online Data Supplement).
The advantages of multiplex MNAzyme qPCR analysis were further tested by assessing the ability of the technology to produce a robust quintuplex MNAzyme RT-qPCR assay. Five target assays were combined into a single reaction that contained 10 partzymes, 10 primers, and 5 probes. The quintuplex reaction produced highly specific results, with no signal detected from any of the negative controls lacking template or reverse transcriptase. The reactions for all 5 target transcripts produced calibration curves with amplification efficiencies between 93% and 103%, and r2 values ≥0.997 (Fig. 2B).
QUADRUPLEX MNAzyme MSP ASSAY
A multiplexed MSP assay consisting of 3 methylation markers (RARB, GSTP1, APC) and an endogenous control (ACTB) has previously been reported (4). MNAzymes were designed to detect sequences that were amplified by the preexisting primer sets published for that study (see Table 2 in the online Data Supplement). MNAzyme assays were performed with bisulfite-modified gDNA prepared from cell lines LNCap and K562, which are known to be methylated and unmethylated, respectively, for these 3 marker genes. We isolated gDNA samples from the LNCap and K562 lines and mixed them to produce a range of concentrations and ratios that contained methylated genes in a BACKGROUND of their unmethylated counterparts. We ran this series of dilutions in duplicate for each of the 3 methylated genes; the MNAzyme MSP qPCR assay allowed the detection of 1 methylated allele in a BACKGROUND of 1000 unmethylated alleles for both replicates of the 3 genes in a quadruplex reaction. Furthermore, the assay detected 1 methylated allele in a BACKGROUND of 10 000 unmethylated alleles in the duplicate reactions that amplified GSTP1 and in 1 of 2 replicates of the APC and RARB amplifications (see Table 10 in the online Data Supplement). No signal was detected for any of these 3 genes when unmethylated K562 DNA alone was analyzed. To confirm the presence of equal amounts of gDNA in all dilutions of LNCap DNA in K562 DNA, and in K562 DNA alone, we used an endogenous control reaction that coamplified a region of the ACTB gene devoid of any variant methylated bases as the fourth gene in the quadruplex reaction. The Cq value for ACTB was consistent for all reactions and ranged from 12.6–13.1. We detected no signal for any of the 4 genes in any of the reactions when no template DNA was added.
RAPID MNAzyme qPCR
The thermocycling parameters of the MNAzyme qPCR reactions were modified to produce results more rapidly. We performed a triplex assay with both standard and reduced annealing and denaturation times. Compared with the standard protocol, rapid cycling reduced the time to detect 0.5 ng gDNA from approximately 70 min to approximately 40 min for each of the 3 genes (Fig. 3C; see Table 11 in the online Data Supplement). All genes had calibration curves with amplification efficiencies between 93% and 104% (r2 values ≥0.997) for both standard and rapid-cycling protocols (see Table 12 in the online Data Supplement).
DISCUSSION
MNAzymes are a versatile class of nucleic acid enzymes that constitute a powerful tool for the detection and analysis of genetic targets. The structure of MNAzymes allows target detection to be uncoupled from signal generation in a format that has advantages for both functionalities. First, target detection with MNAzymes requires 2 distinct target-binding events, because the 2 partzymes must hybridize adjacently on a target to form an MNAzyme. Consequently, MNAzyme-mediated target detection is exquisitely specific. Partzymes have no enzymatic activity in the absence of an appropriate target to direct their assembly and thus cannot generate false-positive signals. Second, signal generation is facilitated by cleavage of an MNAzyme substrate that can serve as a universal probe for any target. A series of universal probes has been developed, and these probes generally can be used to report on any target. Any single probe can be used with any target, and any series of probes can be used in a multiplex format to detect any series of targets. MNAzymes are particularly useful tools for detecting and monitoring qPCR reactions, particularly multiplex reactions. The specificity for each target is always high, regardless of the number of oligonucleotides present, because signal generation during the PCR requires 4 target-specific binding events, namely hybridization of 2 PCR primers as well as 2 partzymes. Thus, MNAzyme qPCR has an additional level of specificity (Fig. 1) compared with such protocols as hydrolysis probes and molecular beacons, which have 3 levels of specificity (derived from 2 primers and 1 target-specific probe). Given that specificity is of paramount importance in clinical settings, MNAzyme qPCR is ideal for in vitro diagnostic applications. This study used several examples to showcase the ease of multiplex analysis and the other benefits afforded by MNAzyme qPCR.
In one of these experiments, we amplified 3 genes (KRAS, TFRC, and B2M), both separately and together. When amplification curves from the single-gene reactions were laid over those for the triplex reaction, there was no significant change in the Cq values or in the shapes of the amplification curves. Multiplexing MNAzyme qPCR reactions therefore does not compromise the detection limit or the amplification efficiency. All reactions produced high efficiency and r2 values, and they all produced no signal in the absence of the relevant target, demonstrating that high specificity is maintained when targets are multiplexed in a single reaction.
To further demonstrate the ease of multiplexing, we amplified 11 GOIs, both individually and together with 2 RGs in a series of triplex reactions. Again, our comparisons of singleplex reactions with triplex reactions revealed no significant changes in Cq values. Furthermore, Cq values were invariant for each of the 2 RGs, regardless of which GOI they were coamplified with and despite the considerable variation in GOI expression levels. All single-gene reactions contained the same standard concentrations of MNAzyme qPCR reagents, and results for triplex reactions were obtained without further optimization or modification of reaction conditions. The internal controls for clinical diagnostic qPCR assays have strict requirements for confirming the efficiencies of extraction and amplification, and MNAzyme qPCR allows rapid development of multiplex assays to achieve this standard.
Bioline recently developed kits that incorporate MNAzymes for multiplex qPCR. These kits (DNA Extraction Control 670 and RNA Extraction Control 670) provide reagents that can be used in combination with qPCR protocols that use different chemistries, such as hydrolysis probes or molecular beacons. Sequences of control DNA or RNA are added to samples and coextracted with gDNA or total RNA. During the qPCR, the target of interest can be monitored with the investigator's preferred real-time chemistry, and the control sequences are coamplified and monitored with MNAzymes also present in the multiplex qPCR mixture. The lack of a need for optimization when combining an MNAzyme with alternative targets, as well as with alternative real-time chemistries, is a testament to the ease of multiplexing with this technology.
Although many reports in the literature have described duplex and triplex qPCR reactions, protocols that involve multiplexing ≥4 targets are rare, because considerable optimization of primers and probe combinations is generally required to achieve success with alternative chemistries (5). In contrast, this level of multiplexing is readily attainable with MNAzymes; we have presented examples of both quadruplex and quintuplex MNAzyme qPCR. The maintenance of sensitivity with higher-order multiplexing is shown for an MNAzyme qPCR reaction with gDNA that allowed the detection of ≤10 copies for each of all 5 genes in a quintuplex reaction (data not shown). In one RT-qPCR reaction, 5 transcripts were reverse transcribed, amplified, and monitored simultaneously with MNAzymes designed to cleave 5 unique universal probes, each labeled with a different fluorophore. The 5 probes are well characterized and perform consistently, regardless of the targets being multiplexed. Some optimization was required to achieve equivalent amplification efficiencies for all 5 targets, and this effort involved minor adjustments to the relative concentrations of primers and probes. An aim of ongoing work in our laboratory is to increase the number of universal probes available and to identify probes with improved multiple-turnover kinetics. A newly characterized sixth universal probe has recently been used in multiplex reactions that quantify 6 targets simultaneously (data not shown).
MNAzyme qPCR protocols generally use asymmetric primer ratios. Such asymmetry provides an advantage in multiplex reactions when the targets have different abundances. In asymmetric PCR, amplification is initially exponential but later becomes linear as the depletion of one of the primers leads to amplification of only 1 strand. The Cqs of all targets are achieved during the exponential phase, and the targets reach the later linear phase in the order of their relative abundance. This dynamic produces conditions that are more favorable for less abundant targets in later cycles because of less competition for reagents; hence, they continue to amplify exponentially while the more abundant targets are in their linear phase.
MNAzymes can be combined with specialized PCR protocols such as ARMS (amplification-refractory mutation system) (6) and REMS (restriction endonuclease–mediated selective) (7) PCR for the detection of single-nucleotide polymorphisms or mutations (data not shown). Alternatively, MSP PCR (8) is used to discriminate between methylated and unmethylated sequences. A quadruplex MSP MNAzyme qPCR was performed with a 2-stage thermocycling protocol in which the initial cycles favored outer primers with a higher melting temperature (Tm) and later cycles allowed amplification with inner primers having lower Tms. The protocol allowed discrimination between targets that differed by as few as 3 differentially methylated nucleotides. The sensitivity of the assay allowed detection of a single methylated allele in a BACKGROUND of 1000–10 000 unmethylated alleles. Given that the total amount of gDNA in the reaction was 100 ng, this sensitivity corresponds to the detection of 3–30 copies of the methylated targets. The quadruplex reaction contained 26 oligonucleotides, which consisted of 2 sets of primers, 1 set of partzymes for each of the 4 targets, and 4 MNAzyme universal probes. Again, no BACKGROUND signal was observed in the absence of bisulfite-modified DNA, a result consistent with the very high specificity that characterizes MNAzyme qPCR. This sensitivity and specificity make the assay suitable for detecting circulating tumor DNA in patients with prostate cancer.
Another desirable characteristic of clinical assays is speed. We used a triplex assay to test the ability to use MNAzyme qPCR under rapid-cycling conditions. A signal above the threshold of detection was evident for all 3 targets within 40 min in reactions containing 500 pg gDNA.
In general, MNAzyme qPCR is very user friendly. MNAzymes can be used with preexisting primer pairs with minimal or no optimization of primer sequences and reaction conditions. The flexible nature of MNAzymes allows the development of universal reaction conditions such that only highly specialized assays require optimization of reaction conditions (see Section 4 in the Supplemental Data of the online Data Supplement). Typical amplicon sizes in MNAzyme qPCR are approximately 80 bp, which accommodates the binding of 2 primers and 2 partzymes. Shorter amplicons of approximately 60 bp are also compatible with MNAzyme qPCR reactions that use a strategy of overlapping primers and partzymes. Finally, the technology has been tested on a wide variety of PCR platforms, including the Mx3005P (Stratagene), Eco™ (Illumina), CFX96™ (Bio-Rad Laboratories), the LightCycler® 480 System (Roche Applied Science), SmartCycler® (Cepheid), Rotor-Gene® Q (Qiagen), and the ABI 7500, ABI 7900, ABI 7700, ABI 7000 systems (Life Technologies).
This report has outlined the technical advantages of MNAzyme qPCR for clinical diagnostics; however, parallel work in our laboratories also has demonstrated the suitability of MNAzymes for use with a wide variety of clinical specimens, including DNA or RNA extracted from blood (peripheral blood, plasma, serum), fresh frozen tissue, paraffin-embedded tissue, sputum, and urine (data not shown). Furthermore, MNAzyme qPCR has proven performance in a clinical setting. The technology was used to monitor the expression of an anti-HIV ribozyme targeted to HIV in RNA extracted from peripheral blood mononuclear cells that were obtained from patients enrolled in a phase II clinical trial (9). In this setting, a duplex MNAzyme RT-qPCR assay produced robust and reliable results for hundreds of patient samples and controls over a 2-year period; not a single false positive was detected among negative-control reactions carried out over the course of the trial.
In summary, the development of MNAzyme single-tube, 1-step qPCR and RT-qPCR assays introduces a competitive technology into the field of qPCR. This report has described our development and evaluation of this technology has highlighted its multiple advantages over other qPCR detection methods, which include its exquisite specificity and simplified multiplex protocols. Given that the assays use universal probes, QC procedures may be simplified because the same probes can be used for all targets and all applications. MNAzyme qPCR provides a viable alternative to other real-time chemistries for clinical diagnostic testing, for which sensitivity, speed, specificity, robustness, and ability to incorporate multiple internal controls are highly desirable.
2 Nonstandard abbreviations:
- MNAzyme
nucleic acid enzymes formed from multiple partial enzymes
- qPCR
quantitative PCR
- gDNA
genomic DNA
- RT-PCR
reverse-transcription PCR
- MSP
methylation-specific
- NF-H2O
nuclease-free water
- GOI
gene of interest
- RG
reference gene
- ΔCq
difference in quantification cycle
- ARMS
amplification-refractory mutation system
- REMS
restriction endonuclease–mediated selective (PCR)
- Tm
melting temperature (at which 50% of double-stranded DNA or DNA/RNA hybrids are denatured).
3 Human genes:
- TFRC
transferrin receptor (p90, CD71) [GenBank nos. NT_029928.13 and NM_003234]
- B2M
beta-2-microglobulin [NT_010194.8 and NM_004048]
- KRAS
v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog [NT_009714.16 and NM_033360]
- ACTB
actin, beta [NT_007819.17 and NM_001101]
- BCR
breakpoint cluster region [NM_004327.3]
- HPRT1
hypoxanthine phosphoribosyltransferase 1 [NT_011786.16]
- TP53
tumor protein p53 [NM_000546.5]
- HMBS
- PGK1
phosphoglycerate kinase 1 [NM_000291]
- PMM1
phosphomannomutase 1 [NM_002676]
- PSMB6
proteasome (prosome, macropain) subunit, beta type, 6 [NM_002798.1]
- RPLP0
ribosomal protein, large, P0 [NM_001002]
- PPIA
peptidylprolyl isomerase A (cyclophilin A) [NM_021130]
- GSTP1
glutathione S-transferase pi 1 [NT_167190.1]
- RARB
retinoic acid receptor, beta [NT_022517.18]
- APC
adenomatous polyposis coli [NT_034772.6]
- HFE
hemochromatosis [NT_007592.15].
Author Contributions:All authors confirmed they have contributed to the intellectual content of this paper and have met the following 3 requirements: (a) significant contributions to the conception and design, acquisition of data, or analysis and interpretation of data; (b) drafting or revising the article for intellectual content; and (c) final approval of the published article.
Authors' Disclosures or Potential Conflicts of Interest:Upon manuscript submission, all authors completed the author disclosure form. Disclosures and/or potential conflicts of interest:
Employment or Leadership: None declared.
Consultant or Advisory Role: None declared.
Stock Ownership: E. Mokany, SpeeDx Pty Ltd.; C.J. Fuery, SpeeDx Pty Ltd.; A.V. Todd, SpeeDx Pty Ltd.
Honoraria: None declared.
Research Funding: None declared.
Expert Testimony: None declared.
Patents: E. Mokany, EP1948822, AU2006302729, and NZ567403; A.V. Todd, EP1948822, AU2006302729, and NZ567403.
Role of Sponsor: No sponsor was declared.
Acknowledgments
We thank K. Nejedla and K. Bird for technical assistance.
References

{kind=link}
{kind=link}
{kind=link}