






Abstract
Camostat inhibits severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection in vitro. We studied the safety and efficacy of camostat in ACTIV-2/A5401, a phase 2/3 platform trial of therapeutics for COVID-19 in nonhospitalized adults.
We conducted a phase 2 study in adults with mild-to-moderate COVID-19 randomized to oral camostat for 7 days or a pooled placebo arm. Primary outcomes were time to improvement in COVID-19 symptoms through day 28, proportion of participants with SARS-CoV-2 RNA below the lower limit of quantification (LLoQ) from nasopharyngeal swabs through day 14, and grade ≥3 treatment-emergent adverse events (TEAEs) through day 28.
Of 216 participants (109 randomized to camostat, 107 to placebo) who initiated study intervention, 45% reported ≤5 days of symptoms at study entry and 26% met the protocol definition of higher risk of progression to severe COVID-19. Median age was 37 years. Median time to symptom improvement was 9 days in both arms (P = .99). There were no significant differences in the proportion of participants with SARS-CoV-2 RNA <LLoQ on days 3, 7, and 14. Through day 28, 6 (5.6%) participants in the camostat arm and 5 (4.7%) in the placebo arm were hospitalized; 1 participant in the camostat arm subsequently died. Grade ≥3 TEAEs occurred in 10.1% of camostat versus 6.5% of placebo participants (P = .35).
In a phase 2 study of nonhospitalized adults with mild-to-moderate COVID-19, oral camostat did not accelerate viral clearance or time to symptom improvement, or reduce hospitalizations or deaths.
Clinical Trials Registration. ClinicalTrials.gov identifier: NCT 04518410.
Coronavirus disease 2019 (COVID-19), caused by infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), continues to exert a heavy toll on individuals and societies worldwide [1, 2].
Antiviral strategies, including monoclonal antibodies, remdesivir, and the oral antivirals molnupiravir and ritonavir-boosted nirmatrelvir, have demonstrated efficacy for reducing COVID-19–related hospitalizations and deaths in patients with mild-to-moderate COVID-19 and high risk of clinical progression [3–5]. However, widespread implementation of the available treatment options has been hampered by (1) the need for parenteral administration for some agents, (2) emergence of resistance to monoclonal antibodies [6, 7], (3) drug–drug interactions for nirmatrelvir/ritonavir, (4) modest efficacy for molnupiravir [4], and (5) limited global availability. Medications with proven efficacy against COVID-19 are currently only recommended for persons at high risk of progression to severe disease and are not accessible for most of the world's population. Hence, there is an ongoing need for easily administered therapies that are safe, affordable, and globally available.
Camostat, an oral serine protease inhibitor, blocks transmembrane serine protease 2 (TMPRSS2), a host factor for SARS-CoV-2 entry [8]. Camostat's inhibition of infection with SARS-CoV-2 and related coronaviruses was described in silico [9, 10], and found to be effective in vitro [8, 11, 12]. In a mouse model of SARS using a higher dose of camostat, 60% of mice fed oral camostat (30 mg/kg twice daily) survived a challenge with SARS-CoV-1 that was 100% lethal in the untreated controls [13]. It has been safe in clinical trials and post-marketing surveillance in Japan and South Korea, where it is available as a relatively inexpensive medication for chronic pancreatitis and postoperative reflux esophagitis [14]. Similar to other antiviral strategies, including monoclonal antibody–based treatments and others [15–17], camostat did not show clinical efficacy in the treatment of COVID-19 in hospitalized individuals in randomized double-blind controlled trials [18, 19]. Nevertheless, several of these therapies have subsequently demonstrated clinical benefit in nonhospitalized patients, which could be due to treatment earlier in the course of disease compared with hospitalized persons. Several trials of camostat for COVID-19 in nonhospitalized individuals, however, were challenged by insufficient enrollment. An interim analysis of a planned phase 2 trial in outpatients performed after recruitment had stalled did not show an effect of camostat on viral clearance or clinical improvement [20]. Another single-center trial was terminated early for futility by the Data Safety and Monitoring Board, yet a preprint reports potential beneficial treatment effects based on a small number of participants [21]. One recently published phase 2 trial did not find clinical benefit for camostat, but, according to the authors, was “insufficiently powered for the primary endpoint” [22]. Hence, there is a need for informative results on the efficacy of camostat in outpatients from an adequately powered, randomized controlled trial.
Due to its mechanism of action and in vitro activity against SARS-CoV-2, camostat is an attractive candidate for the treatment of COVID-19. Hence, we studied the safety and antiviral and clinical efficacy of orally administered camostat in nonhospitalized adults with mild-to-moderate COVID-19 within the Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV)-2/A5401 platform trial.
METHODS
Trial Design
ACTIV-2/A5401 is a randomized, controlled, multicenter platform trial for efficient, concurrent testing of multiple agents for the treatment of mild-to-moderate COVID-19 within a common trial infrastructure. It entails an initial placebo-controlled phase 2 study and, if predefined safety and efficacy criteria are met, a subsequent phase 3 evaluation. The protocol was approved by a central institutional review board (IRB), Advarra (Pro00045266), with additional local IRB review and approval as required by participating sites. All participants provided written informed consent.
Study Population
The study population consisted of nonhospitalized adults (≥18 y of age) with a positive SARS-CoV-2 antigen or nucleic acid test on an upper respiratory sample collected within 10 days prior to study entry. Participants were required to have ongoing COVID-19 symptoms within 24 hours prior to study entry and began study intervention within 10 days after self-reported onset of COVID-19 symptoms. Pregnancy, breastfeeding, and severe chronic liver or kidney disease were exclusionary. Full eligibility criteria are provided in the ACTIV-2 protocol (available at: https://www.nih.gov/research-training/medical-research-initiatives/activ/covid-19-therapeutics-prioritized-testing-clinical-trials; accessed 24 January 2023).
Randomization and Study Intervention
All participants underwent a 2-step randomization: the first occurred with equal probability to one of however many agent groups were open to enrollment at the site and for which a participant was eligible. Subsequently, within each agent group, there was an immediate second randomization to active treatment or placebo with a randomization ratio of r:1, where r was the number of agents in the same phase of evaluation (phase 2 or phase 3) that a participant was eligible to receive. Randomization was stratified on time from symptom onset at study entry (≤5 d vs >5 d) and risk of COVID-19 progression (higher vs lower) (criteria described in the (Supplementary Methods).
A pooled placebo control group was constructed including all participants who were eligible for randomization to the camostat group and were randomized to placebo for camostat or placebo for another agent in phase 2 evaluation. Thereby, all placebo participants who contributed to this trial were enrolled during the same period as participants receiving camostat.
Participants randomized to the camostat group received 200 mg oral camostat (2 tablets, 100 mg each) or matching placebo every 6 hours for 7 days.
Study Procedures
Clinical assessments included safety evaluations and site collection of nasopharyngeal (NP) swabs on study days 0 (start of treatment), 3, 7, 14, and 28. Participants completed a daily symptom diary (Supplementary Figure 1) from day 0 through day 28, including a daily global assessment question of whether they had returned to their usual (pre–COVID-19) health and grading of 13 targeted COVID-19 symptoms as absent, mild, moderate, and severe by self-assessment. A total symptom score was calculated for each day by assigning “absent” a score of 0, “mild” 1, “moderate” 2, and “severe” 3, and summing scores for all symptoms (range of 0–39). Quantitative NP SARS-CoV-2 RNA levels were determined as previously described [23]. The assay limit of detection (LoD) was 1.4 log10 copies/mL, the lower limit of quantification (LLoQ) was 2 log10 copies/mL, and the upper limit of quantification (ULoQ) was initially 7, then 8 log10 copies/mL. Assays were rerun with dilution for samples with RNA greater than the ULoQ.
Outcome Measures
Primary outcome measures were as follows: (1) NP SARS-CoV-2 RNA level below the LLoQ at days 3, 7, and 14; (2) time to improvement of all 13 targeted COVID-19 symptoms through day 28 from the start of investigational treatment (day 0) to the first of 2 consecutive days when all symptoms scored as moderate or severe at day 0 were scored as mild or absent, and all targeted symptoms scored as mild or absent at day 0 were scored as absent; and (3) development of a grade 3 or higher treatment-emergent adverse event (TEAE) through 28 days.
Secondary outcome measures included all-cause hospitalization and death through day 28, quantitative levels of SARS-CoV-2 RNA from NP swabs, time to the first of 4 consecutive days when all symptoms were absent, time to return to usual (pre–COVID-19) health for 2 consecutive days, time to return to usual health for 4 consecutive days, time-averaged total daily symptom score, and progression of 1 or more targeted COVID-19 symptoms to a worse status than at day 0.
Study medication adherence over the 7-day treatment period was evaluated by the percentage of the 28 doses of camostat or matching placebo that were missed by self-report and tablet count.
Statistical Analysis
The primary analysis population included all participants randomized to camostat and the pooled placebo group for camostat who started treatment (modified intent-to-treat [mITT] population). The safety population only included participants who received at least 1 dose of the study drug.
The planned sample size of 220 participants (∼110 each receiving camostat or pooled placebo) was chosen to provide at least 82% power to detect a 20% absolute difference in the percentage less than the LLoQ for SARS-CoV-2 at any time point with a 2-sided α = 0.05.
Proportions of participants with SARS-CoV-2 RNA less than the LLoQ were compared between arms across measurement times using Poisson regression adjusted for day 0 log10-transformed SARS-CoV-2 RNA level (the fitting of the planned log-binomial model failed because of numerical convergence issues) and summarized with a risk ratio (RR) and 95% confidence interval (CI) at each time. Missing data were assumed to be missing completely at random and were ignored. A joint test across multiple time points was assessed using a 2-sided Wald test. SARS-CoV-2 RNA levels were compared using Wilcoxon rank-sum tests for each post-entry study visit.
Time to symptom improvement and return to usual pre–COVID-19 health were compared between arms using Wilcoxon's test adapted for handling censored data (Gehan-Wilcoxon test). The time-averaged total daily symptom score was compared using a 2-sided Wilcoxon test. The proportion of participants with symptom progression was compared using log-binomial regression and summarized with an RR and 95% CI.
Proportions of participants experiencing a grade 3 or higher TEAE through study day 28 were compared between arms using log-binomial regression and summarized with an RR, 95% CI, and P value based on a Wald test. The cumulative proportion of participants hospitalized or who died was estimated using Kaplan-Meier methods and compared using a ratio of proportions.
All comparisons used a 2-sided 5% type I error rate without adjustment for multiple comparisons. Statistical analyses were conducted in SAS version 9.4 (SAS Institute). See the Supplementary Methods for additional details regarding statistical methods.
RESULTS
Characteristics of the Participants
Between February 10 and 26 April 2021, a total of 224 participants were randomized at 54 US sites, 113 to camostat and 111 to placebo (Figure 1). After exclusion of 8 participants who did not initiate the study intervention, the mITT population included 216 participants (109 randomized to camostat, 107 to placebo). Among the 107 participants in the pooled placebo arm, 51 (48%) were randomized to the placebo for camostat and 56 (52%) to the placebo for a different agent (Table 1). Eighteen participants discontinued the study before day 28, and 198 (92%) were still in follow-up at day 28 (Figure 1). Fifty-nine percent in both arms reported no missed doses (Supplementary Table 1). The proportion with at least 4 missed doses was 19.3% for the active camostat arm and 23.5% for those receiving placebo for camostat (RR: .82: 95% CI: .44, 1.53).
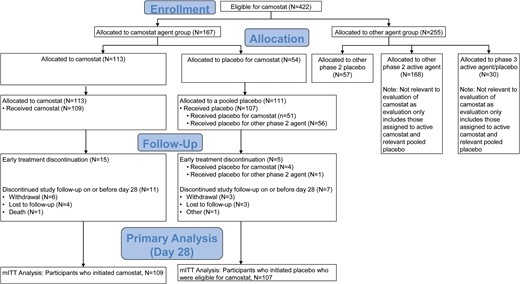
Enrollment and randomization. Participants were recruited between 10 February and 26 April 2021. Of 422 participants who were eligible to receive camostat in the study, 167 were allocated to the camostat agent group; 113 of those were randomized to camostat and 54 to oral placebo matching camostat. Of 255 allocated to the other agent groups, 57 participants who had been randomized to placebo from another phase 2 agent group, together with the 54 participants who received placebo matching camostat, formed the pooled placebo group for the primary analysis. The participants who received at least 1 dose of study treatment (109 in the camostat and 107 in the placebo arms) formed the mITT population. Abbreviation: mITT, modified intent-to-treat.
Demographic and Clinical Characteristics
| Characteristics | Camostat (n = 109) | Placebo (n = 107) | Total (N = 216) |
|---|---|---|---|
| Age | |||
| Median age (Q1, Q3), y | 37 (29, 49) | 39 (29, 48) | 37 (29, 48) |
| ≥ 60 y, n (%) | 3 (3) | 8 (7) | 11 (5) |
| Female sex, n (%) | 63 (58) | 55 (51) | 118 (55) |
| Transgender spectrum, n (%) | 1 (1) | 1 (1) | 2 (1) |
| Race,a n (%) | |||
| Asian | 3 (3) | 2 (2) | 5 (2) |
| Black | 4 (4) | 15 (14) | 19 (9) |
| Multiple | 1 (1) | 1 (1) | 2 (1) |
| Other | 3 (3) | 4 (4) | 7 (3) |
| White | 98 (90) | 85 (79) | 183 (85) |
| Hispanic or Latinx,a n (%) | 61 (56) | 49 (46) | 110 (51) |
| Time from symptom onset | |||
| Median (Q1, Q3), d | 6 (4, 7) | 6 (3, 7) | 6 (4, 7) |
| ≤ 5 days, n (%) | 51 (47) | 46 (43) | 97 (45) |
| Days from positive COVID-19 test to randomization, median (Q1, Q3) | 3 (2, 6) | 3 (2, 6) | 3 (2, 6) |
| Higher risk for progression to severe COVID-19, n (%) | 30 (28) | 26 (24) | 56 (26) |
| BMI, median (Q1, Q3), kg/m2 | 28.2 (24.0, 31.0) | 27.7 (24.4, 31.8) | 28.2 (24.2, 31.5) |
| Vaccinated against COVID-19, n (%) | 6 (6) | 5 (5) | 11 (5) |
| Agent group,b n (%) | |||
| Camostat 200 mg | 109 (100) | 51 (48) | 160 (74) |
| AZD7442 300 mg intravenous | 0 (0) | 8 (7) | 8 (4) |
| AZD7442 600 mg intramuscular | 0 (0) | 36 (34) | 36 (17) |
| SNG001 (inhaled IFN-β1a) | 0 (0) | 12 (11) | 12 (6) |
| SARS-CoV-2 variant data,c n | 95 | 89 | 184 |
| Alpha, n (%) | 48 (51) | 53 (60) | 101 (55) |
| Beta, n (%) | 1 (1) | 3 (3) | 4 (2) |
| Gamma, n (%) | 11 (12) | 5 (6) | 16 (9) |
| Delta, n (%) | 1 (1) | 0 (0) | 1 (1) |
| Epsilon, n (%) | 8 (8) | 4 (4) | 12 (7) |
| Iota, n (%) | 13 (14) | 11 (12) | 24 (13) |
| Lambda, n (%) | 0 (0) | 1 (1) | 1 (1) |
| Mu, n (%) | 1 (1) | 0 (0) | 1 (1) |
| Other, n (%) | 12 (13) | 12 (13) | 24 (13) |
| Characteristics | Camostat (n = 109) | Placebo (n = 107) | Total (N = 216) |
|---|---|---|---|
| Age | |||
| Median age (Q1, Q3), y | 37 (29, 49) | 39 (29, 48) | 37 (29, 48) |
| ≥ 60 y, n (%) | 3 (3) | 8 (7) | 11 (5) |
| Female sex, n (%) | 63 (58) | 55 (51) | 118 (55) |
| Transgender spectrum, n (%) | 1 (1) | 1 (1) | 2 (1) |
| Race,a n (%) | |||
| Asian | 3 (3) | 2 (2) | 5 (2) |
| Black | 4 (4) | 15 (14) | 19 (9) |
| Multiple | 1 (1) | 1 (1) | 2 (1) |
| Other | 3 (3) | 4 (4) | 7 (3) |
| White | 98 (90) | 85 (79) | 183 (85) |
| Hispanic or Latinx,a n (%) | 61 (56) | 49 (46) | 110 (51) |
| Time from symptom onset | |||
| Median (Q1, Q3), d | 6 (4, 7) | 6 (3, 7) | 6 (4, 7) |
| ≤ 5 days, n (%) | 51 (47) | 46 (43) | 97 (45) |
| Days from positive COVID-19 test to randomization, median (Q1, Q3) | 3 (2, 6) | 3 (2, 6) | 3 (2, 6) |
| Higher risk for progression to severe COVID-19, n (%) | 30 (28) | 26 (24) | 56 (26) |
| BMI, median (Q1, Q3), kg/m2 | 28.2 (24.0, 31.0) | 27.7 (24.4, 31.8) | 28.2 (24.2, 31.5) |
| Vaccinated against COVID-19, n (%) | 6 (6) | 5 (5) | 11 (5) |
| Agent group,b n (%) | |||
| Camostat 200 mg | 109 (100) | 51 (48) | 160 (74) |
| AZD7442 300 mg intravenous | 0 (0) | 8 (7) | 8 (4) |
| AZD7442 600 mg intramuscular | 0 (0) | 36 (34) | 36 (17) |
| SNG001 (inhaled IFN-β1a) | 0 (0) | 12 (11) | 12 (6) |
| SARS-CoV-2 variant data,c n | 95 | 89 | 184 |
| Alpha, n (%) | 48 (51) | 53 (60) | 101 (55) |
| Beta, n (%) | 1 (1) | 3 (3) | 4 (2) |
| Gamma, n (%) | 11 (12) | 5 (6) | 16 (9) |
| Delta, n (%) | 1 (1) | 0 (0) | 1 (1) |
| Epsilon, n (%) | 8 (8) | 4 (4) | 12 (7) |
| Iota, n (%) | 13 (14) | 11 (12) | 24 (13) |
| Lambda, n (%) | 0 (0) | 1 (1) | 1 (1) |
| Mu, n (%) | 1 (1) | 0 (0) | 1 (1) |
| Other, n (%) | 12 (13) | 12 (13) | 24 (13) |
Percentages may not total 100 because of rounding.
Abbreviations: BMI, body mass index; COVID-19, coronavirus disease 2019; Q, quartile; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Race and ethnicity were self-reported by the participants.
Including participants in the camostat treatment and placebo arms, and from the placebo arms for 3 other investigational agents. The pooled placebo arm included participants who received placebo from the camostat agent group and from 3 other agent groups, AZD7442 intravenous administration (AZD7442 300 mg IV), AZD 7442 intramuscular administration (AZD7442 600 mg IM), and SNG001 (inhaled IFN-β1a).
Percentages for SARS-CoV-2 variant data are given as percentages of those samples available for sequencing.
Demographic and Clinical Characteristics
| Characteristics | Camostat (n = 109) | Placebo (n = 107) | Total (N = 216) |
|---|---|---|---|
| Age | |||
| Median age (Q1, Q3), y | 37 (29, 49) | 39 (29, 48) | 37 (29, 48) |
| ≥ 60 y, n (%) | 3 (3) | 8 (7) | 11 (5) |
| Female sex, n (%) | 63 (58) | 55 (51) | 118 (55) |
| Transgender spectrum, n (%) | 1 (1) | 1 (1) | 2 (1) |
| Race,a n (%) | |||
| Asian | 3 (3) | 2 (2) | 5 (2) |
| Black | 4 (4) | 15 (14) | 19 (9) |
| Multiple | 1 (1) | 1 (1) | 2 (1) |
| Other | 3 (3) | 4 (4) | 7 (3) |
| White | 98 (90) | 85 (79) | 183 (85) |
| Hispanic or Latinx,a n (%) | 61 (56) | 49 (46) | 110 (51) |
| Time from symptom onset | |||
| Median (Q1, Q3), d | 6 (4, 7) | 6 (3, 7) | 6 (4, 7) |
| ≤ 5 days, n (%) | 51 (47) | 46 (43) | 97 (45) |
| Days from positive COVID-19 test to randomization, median (Q1, Q3) | 3 (2, 6) | 3 (2, 6) | 3 (2, 6) |
| Higher risk for progression to severe COVID-19, n (%) | 30 (28) | 26 (24) | 56 (26) |
| BMI, median (Q1, Q3), kg/m2 | 28.2 (24.0, 31.0) | 27.7 (24.4, 31.8) | 28.2 (24.2, 31.5) |
| Vaccinated against COVID-19, n (%) | 6 (6) | 5 (5) | 11 (5) |
| Agent group,b n (%) | |||
| Camostat 200 mg | 109 (100) | 51 (48) | 160 (74) |
| AZD7442 300 mg intravenous | 0 (0) | 8 (7) | 8 (4) |
| AZD7442 600 mg intramuscular | 0 (0) | 36 (34) | 36 (17) |
| SNG001 (inhaled IFN-β1a) | 0 (0) | 12 (11) | 12 (6) |
| SARS-CoV-2 variant data,c n | 95 | 89 | 184 |
| Alpha, n (%) | 48 (51) | 53 (60) | 101 (55) |
| Beta, n (%) | 1 (1) | 3 (3) | 4 (2) |
| Gamma, n (%) | 11 (12) | 5 (6) | 16 (9) |
| Delta, n (%) | 1 (1) | 0 (0) | 1 (1) |
| Epsilon, n (%) | 8 (8) | 4 (4) | 12 (7) |
| Iota, n (%) | 13 (14) | 11 (12) | 24 (13) |
| Lambda, n (%) | 0 (0) | 1 (1) | 1 (1) |
| Mu, n (%) | 1 (1) | 0 (0) | 1 (1) |
| Other, n (%) | 12 (13) | 12 (13) | 24 (13) |
| Characteristics | Camostat (n = 109) | Placebo (n = 107) | Total (N = 216) |
|---|---|---|---|
| Age | |||
| Median age (Q1, Q3), y | 37 (29, 49) | 39 (29, 48) | 37 (29, 48) |
| ≥ 60 y, n (%) | 3 (3) | 8 (7) | 11 (5) |
| Female sex, n (%) | 63 (58) | 55 (51) | 118 (55) |
| Transgender spectrum, n (%) | 1 (1) | 1 (1) | 2 (1) |
| Race,a n (%) | |||
| Asian | 3 (3) | 2 (2) | 5 (2) |
| Black | 4 (4) | 15 (14) | 19 (9) |
| Multiple | 1 (1) | 1 (1) | 2 (1) |
| Other | 3 (3) | 4 (4) | 7 (3) |
| White | 98 (90) | 85 (79) | 183 (85) |
| Hispanic or Latinx,a n (%) | 61 (56) | 49 (46) | 110 (51) |
| Time from symptom onset | |||
| Median (Q1, Q3), d | 6 (4, 7) | 6 (3, 7) | 6 (4, 7) |
| ≤ 5 days, n (%) | 51 (47) | 46 (43) | 97 (45) |
| Days from positive COVID-19 test to randomization, median (Q1, Q3) | 3 (2, 6) | 3 (2, 6) | 3 (2, 6) |
| Higher risk for progression to severe COVID-19, n (%) | 30 (28) | 26 (24) | 56 (26) |
| BMI, median (Q1, Q3), kg/m2 | 28.2 (24.0, 31.0) | 27.7 (24.4, 31.8) | 28.2 (24.2, 31.5) |
| Vaccinated against COVID-19, n (%) | 6 (6) | 5 (5) | 11 (5) |
| Agent group,b n (%) | |||
| Camostat 200 mg | 109 (100) | 51 (48) | 160 (74) |
| AZD7442 300 mg intravenous | 0 (0) | 8 (7) | 8 (4) |
| AZD7442 600 mg intramuscular | 0 (0) | 36 (34) | 36 (17) |
| SNG001 (inhaled IFN-β1a) | 0 (0) | 12 (11) | 12 (6) |
| SARS-CoV-2 variant data,c n | 95 | 89 | 184 |
| Alpha, n (%) | 48 (51) | 53 (60) | 101 (55) |
| Beta, n (%) | 1 (1) | 3 (3) | 4 (2) |
| Gamma, n (%) | 11 (12) | 5 (6) | 16 (9) |
| Delta, n (%) | 1 (1) | 0 (0) | 1 (1) |
| Epsilon, n (%) | 8 (8) | 4 (4) | 12 (7) |
| Iota, n (%) | 13 (14) | 11 (12) | 24 (13) |
| Lambda, n (%) | 0 (0) | 1 (1) | 1 (1) |
| Mu, n (%) | 1 (1) | 0 (0) | 1 (1) |
| Other, n (%) | 12 (13) | 12 (13) | 24 (13) |
Percentages may not total 100 because of rounding.
Abbreviations: BMI, body mass index; COVID-19, coronavirus disease 2019; Q, quartile; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Race and ethnicity were self-reported by the participants.
Including participants in the camostat treatment and placebo arms, and from the placebo arms for 3 other investigational agents. The pooled placebo arm included participants who received placebo from the camostat agent group and from 3 other agent groups, AZD7442 intravenous administration (AZD7442 300 mg IV), AZD 7442 intramuscular administration (AZD7442 600 mg IM), and SNG001 (inhaled IFN-β1a).
Percentages for SARS-CoV-2 variant data are given as percentages of those samples available for sequencing.
Baseline characteristics were similar across arms (Table 1). The median age was 37 years, 55% were female sex at birth, 85% identified as White, 9% as Black, and 51% as Hispanic or Latinx. Forty-five percent reported 5 or fewer days of symptoms at study entry (median of 6 days since symptom onset in both arms), and 56 (26%) were at higher risk of progression to severe COVID-19. Symptoms reported by more than half of participants at enrollment were cough (85%), fatigue (85%), body/muscle pains/aches (71%), nasal obstruction/congestion (70%), headaches (70%), nasal discharge (55%), shortness of breath (51%), and chills (51%) (Supplementary Figure 2). The predominant SARS-CoV-2 variant was Alpha in 55% of the participants with available variant sequencing data.
Antiviral Efficacy
The proportion of participants with SARS-CoV-2 RNA less than the LLoQ from NP swabs increased across study visits, ranging from 18% with RNA less than the LLoQ at day 0 to 96% at day 28 (Figure 2A), and with nonsignificant differences favoring placebo at day 3 (camostat vs placebo: 32% vs 40%; RR [95% CI]: .76 [.51, 1.13]), day 7 (64% vs 68%; .97 [.79, 1.19]), and day 14 (87% vs 88%; 1.05 [.90, 1.22]) and overall (P = .56) across all visits (Supplementary Table 2). Distributions of SARS-CoV-2 RNA levels did not differ significantly between arms at any study visit (Figure 2B). At day 3, median (quartile [Q] 1, Q3) NP SARS-CoV-2 RNA levels were 3.2 (LoD to <LLoQ, 5.0) log10 copies/mL in the camostat arm and 2.7 (<LoD, 3.9) log10 copies/mL in the placebo arm (P = .10) (Supplementary Table 3). On days 7, 14, and 28, the median SARS-CoV-2 RNA levels were less than the LLoQ for both arms. In sensitivity analyses limited to participants with quantifiable RNA on day 0, camostat did not lead to higher proportions of participants with NP SARS-CoV-2 RNA less than the LLoQ or lower viral loads (Supplementary Tables 4 and 5). Among participants who initiated study intervention 5 days or fewer from symptom onset, the median (Q1, Q3) SARS-CoV-2 RNA level at day 3 was 4.4 (2.9, 5.6) log10 copies/mL in the camostat arm and 3.1 (LoD to <LLoQ, 6.1) log10 copies/mL in the placebo arm.
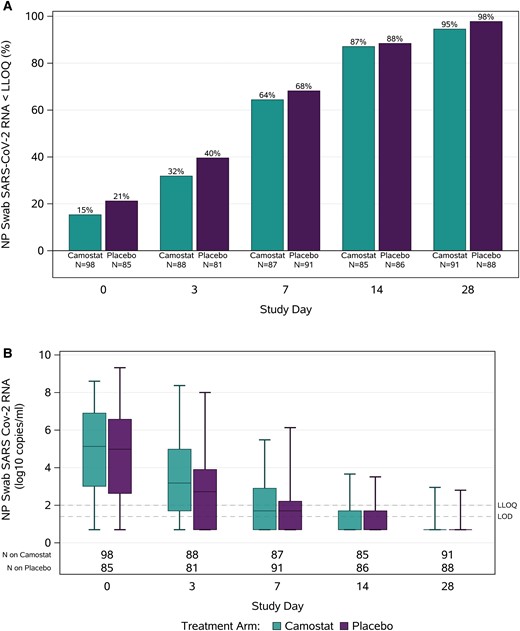
Nasopharyngeal SARS-CoV-2 RNA levels by study visit. Panel A shows the percentages with SARS-CoV-2 RNA less than the LLoQ by day of study visit in the camostat and placebo arms. Panel B summarizes quantitative RNA levels by day of study visit in the camostat and placebo arms—boxes represent interquartile ranges, horizontal lines represent medians, and bars represent minimum and maximum values. Abbreviations: LLoQ, lower limit of quantification (2 log10 copies/mL); LoD, limit of detection (1.4 log10 copies/mL); NP, nasopharyngeal; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Clinical Efficacy
The median (Q1, Q3) time to 2 consecutive days of improvement in all targeted symptoms (primary outcome) was similar in both arms: 9 (5, 19) days with camostat and 9 (6, 19) days with placebo (P = .99) (Figure 3A). The estimated proportion of participants who did not meet the definition of symptom improvement through day 28 was 14% in the camostat arm and 16% in the placebo arm. Time to symptom improvement was also similar between camostat and placebo in subgroup analyses by sex, age, race, ethnicity, risk for progression to severe disease, and time from symptom onset to randomization (Supplementary Figure 3). Eleven participants were hospitalized through day 28, 6 (5.6%) in the camostat arm (4 diagnosed with COVID-19 pneumonia, 1 with appendicitis, 1 with psychotic disorder) and 5 (4.7%) in the placebo arm (all 5 with COVID-19 pneumonia) (Figure 3B) (RR: 1.2; 95% CI: .4, 3.8). There was 1 death in the camostat arm due to complications of COVID-19.
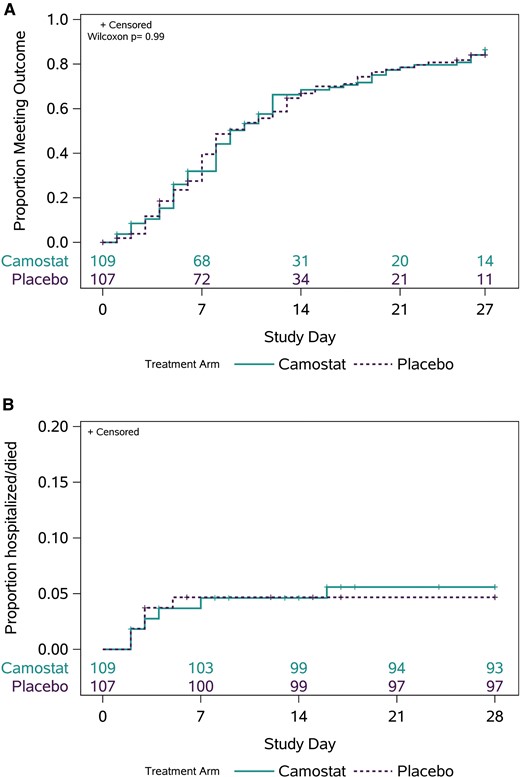
Clinical efficacy. A, Kaplan-Meier curves for improvement in all targeted symptoms for 2 consecutive days in the camostat and placebo arms. B, Kaplan-Meier curves for hospitalization and death by treatment arm through day 28.
There were no differences in other secondary outcomes, including time to all targeted symptoms being absent for 4 consecutive days, time to return to usual health for 2 and for 4 consecutive days, time-averaged total symptom score, or progression of 1 or more COVID-19–associated symptoms to a worse status than at day 0 (Supplementary Figure 4, Supplementary Table 6).
Safety
Eighteen participants reported a grade 3 or higher TEAE through day 28 (Table 2), including 11 (10.1%) in the camostat arm and 7 (6.5%) in the placebo arm (RR: 1.54; 95% CI: .62, 3.83; P = .35). Six participants in the camostat arm and 5 in the placebo arm experienced a serious adverse event through day 28.
Summary of Treatment-Emergent Adverse Events Through Day 28
| Camostat (n = 109) | Placebo (n = 107) | RR [95% CI] | |
|---|---|---|---|
| TEAEs, n (%) | 44 (40) | 39 (36) | … |
| Not gradable | 0 (0) | 2 (2) | … |
| Grade 1 | 14 (13) | 13 (12) | … |
| Grade 2 | 19 (17) | 17 (16) | … |
| Grade 3 | 10 (9) | 7 (7) | … |
| Grade 4 | 0 (0) | 0 (0) | … |
| Grade 5 (death) | 1 (1)a | 0 (0) | … |
| ≥ Grade 3 | 11 (10.1) | 7 (6.5) | 1.54 [.62, 3.83], P = .35 |
| SAEs, n (%) | 6 (5.5) | 5 (4.7) | … |
| Camostat (n = 109) | Placebo (n = 107) | RR [95% CI] | |
|---|---|---|---|
| TEAEs, n (%) | 44 (40) | 39 (36) | … |
| Not gradable | 0 (0) | 2 (2) | … |
| Grade 1 | 14 (13) | 13 (12) | … |
| Grade 2 | 19 (17) | 17 (16) | … |
| Grade 3 | 10 (9) | 7 (7) | … |
| Grade 4 | 0 (0) | 0 (0) | … |
| Grade 5 (death) | 1 (1)a | 0 (0) | … |
| ≥ Grade 3 | 11 (10.1) | 7 (6.5) | 1.54 [.62, 3.83], P = .35 |
| SAEs, n (%) | 6 (5.5) | 5 (4.7) | … |
The table summarizes the number of participants with at least 1 TEAE through day 28. Summaries by TEAE grade are based on the highest adverse event grade through day 28 for each participant.
Abbreviations: CI, confidence interval; COVID-19, coronavirus disease 2019; RR, relative risk; SAE, serious adverse event; TEAE, treatment-emergent serious adverse event.
One death after hospitalization related to COVID-19.
Summary of Treatment-Emergent Adverse Events Through Day 28
| Camostat (n = 109) | Placebo (n = 107) | RR [95% CI] | |
|---|---|---|---|
| TEAEs, n (%) | 44 (40) | 39 (36) | … |
| Not gradable | 0 (0) | 2 (2) | … |
| Grade 1 | 14 (13) | 13 (12) | … |
| Grade 2 | 19 (17) | 17 (16) | … |
| Grade 3 | 10 (9) | 7 (7) | … |
| Grade 4 | 0 (0) | 0 (0) | … |
| Grade 5 (death) | 1 (1)a | 0 (0) | … |
| ≥ Grade 3 | 11 (10.1) | 7 (6.5) | 1.54 [.62, 3.83], P = .35 |
| SAEs, n (%) | 6 (5.5) | 5 (4.7) | … |
| Camostat (n = 109) | Placebo (n = 107) | RR [95% CI] | |
|---|---|---|---|
| TEAEs, n (%) | 44 (40) | 39 (36) | … |
| Not gradable | 0 (0) | 2 (2) | … |
| Grade 1 | 14 (13) | 13 (12) | … |
| Grade 2 | 19 (17) | 17 (16) | … |
| Grade 3 | 10 (9) | 7 (7) | … |
| Grade 4 | 0 (0) | 0 (0) | … |
| Grade 5 (death) | 1 (1)a | 0 (0) | … |
| ≥ Grade 3 | 11 (10.1) | 7 (6.5) | 1.54 [.62, 3.83], P = .35 |
| SAEs, n (%) | 6 (5.5) | 5 (4.7) | … |
The table summarizes the number of participants with at least 1 TEAE through day 28. Summaries by TEAE grade are based on the highest adverse event grade through day 28 for each participant.
Abbreviations: CI, confidence interval; COVID-19, coronavirus disease 2019; RR, relative risk; SAE, serious adverse event; TEAE, treatment-emergent serious adverse event.
One death after hospitalization related to COVID-19.
DISCUSSION
We explored a drug-repurposing strategy for COVID-19 treatment after in vitro data suggested that camostat interfered with cell entry and replication of SARS-CoV, SARS-CoV-2, and Middle East respiratory syndrome coronavirus (MERS-CoV) and reduced mortality in a mouse model of SARS-CoV infection [8, 13].
In this randomized, placebo-controlled phase 2 study of 216 nonhospitalized adults with mild-to-moderate COVID-19, camostat was safe but no different than placebo in reducing detection rates or quantitative levels of NP SARS-CoV-2 RNA or symptom duration. No signal of clinical efficacy was observed in any subgroup, including those treated earlier in the disease course and those at higher risk for progression to severe COVID-19.
Consistent with our results, in another recently completed randomized controlled trial of 295 outpatients with a high risk of progression to severe COVID-19 who were enrolled within 72 hours from receiving a positive test result, there was no reduction in hospitalizations or deaths with camostat 200 mg 4 times daily for 14 days [24]. Additionally, a randomized controlled trial in 205 hospitalized patients with COVID-19 found no advantage of camostat treatment for clinical improvement and time to discharge [19].
Several other agents including hydroxychloroquine [25–27] and nitazoxanide [28] eventually failed to demonstrate clinical efficacy after promising in vitro results and despite anecdotal clinical benefit, underlining the critical role of rigorous clinical trials.
Possible explanations why the antiviral activity of camostat observed in preclinical studies did not translate to clinical efficacy are as follows. First, most antiviral medications for acute infections must be administered early during infection, and 55% of the participants on camostat were enrolled more than 5 days from symptom onset. While we cannot exclude that camostat would be effective if given considerably earlier, this appears unlikely as subgroup analyses even in the participants who were enrolled at 5 days or fewer failed to produce any noticeable signal. Similarly, a trial enrolling outpatients within 3 days from a positive test result (albeit irrespective of the presence of symptoms) did not show efficacy of camostat in preventing hospitalizations, but substantially fewer participants than anticipated reached this endpoint [24]. Second, blocking the TMPRSS-2–dependent pathway for infection may not be sufficient as there are alternative routes of viral entry: SARS-CoV-2 spike can alternatively be primed by cathepsin B/L in endolysosomes, although TMPRSS-2–mediated entry via the cell surface was identified as the predominant entry mechanism [13, 29, 30]. More recent in vitro results suggest that the use of TMPRSS-2 as an entry factor may change as SARS-CoV-2 evolves and may be less important for entry of the Omicron versus the Delta variant, but the participants of our trial were enrolled before the emergence of Omicron [31]. Strategies to specifically inhibit the endosomal pathway of SARS-CoV-2, which is TMPRSS-2–independent, eg, with hydroxychloroquine, did not prove effective in clinical trials for COVID-19 [25, 26]. Blocking both endosomal and cell surface–based entry via a combination of camostat with a cathepsin B/L inhibitor failed to increase survival compared with camostat alone in a mouse model of lethal SARS-CoV infection but was not adequately tested in humans [13]. Third, oral dosing of camostat in our trial may not achieve concentrations sufficient for viral inhibition in the target respiratory epithelia; a recent study demonstrated that topical application of camostat inhibits viral infection on human airway cells, reaching considerably higher local concentrations than with systemic treatment [32]. Moreover, while overall self-reported adherence appeared adequate, some participants may have had lower drug exposure due to suboptimal adherence.
Among the strengths of our trial are the placebo-controlled, double-blinded, multicenter study design, a diverse population of participants from 54 centers across the United States, intensive monitoring with rigorous biomarker sampling, and symptom measures well suited to accurately reflect the course of illness and viral parameters.
There are several limitations. Only symptomatic individuals were included; hence, our data are not applicable to people with asymptomatic COVID-19. As enrollment occurred in the first half of 2021, most participants were unvaccinated, and it is unclear how well our results represent the potential activity of camostat on more recently emerging SARS-CoV-2 variants [31]. The study allowed enrollment for 10 days after diagnosis and symptom onset, a relatively long time compared with other studies, which could skew the results to individuals who are already improving.
In summary, while camostat was safe, it did not demonstrate any virological or clinical efficacy in symptomatic, nonhospitalized adults with mild-to-moderate COVID-19. Based on these outcomes, the agent did not qualify for advancement to a phase 3 trial within ACTIV-2/A5401. These results highlight the critical importance of randomized controlled trials in the evaluation of therapies with promising preclinical efficacy, even during a global public health emergency.
Supplementary Data
Supplementary materials are available at Clinical Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Notes
Author Contributions. N. J. wrote the first draft of the manuscript with input from K. W. C., D. M. S., J. Z. L., and M. J. G. M. J. G., A. K., C. M., and M. D. H. verified the underlying data reported in the manuscript and performed the statistical analysis. All authors contributed to the study design and/or conduct, and all authors helped revising the manuscript for important intellectual content. All authors have seen and approved the final text and gave final approval of the version to be published.
Acknowledgments. The authors thank the study participants, site staff, site investigators, and the entire ACTIV-2/A5401 study team; the AIDS Clinical Trials Group, including Lara Hosey, Jhoanna Roa, and Nilam Patel; the UW Virology Specialty Laboratory, including Emily Degli-Angeli, Erin Goecker, Glenda Daza, Socorro Harb, and Joan Dragavon; the ACTG Laboratory Center, including Grace Aldrovandi and William Murtaugh; Frontier Science, including Marlene Cooper, Howard Gutzman, and Kevin Knowles; the Harvard Center for Biostatistics in AIDS Research (CBAR) and ACTG Statistical and Data Analysis Center (SDAC); the National Institute of Allergy and Infectious Diseases (NIAID)/Division of AIDS (DAIDS), including Peter Kim, Bill Erhardt, Lorraine Waring, and Diane Hessinger; the Foundation for the National Institutes of Health and the Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV) partnership, including Stacey Adams; and the PPD clinical research services of ThermoFisher Scientific.
The authors also thank the members of the ACTIV-2/A5401 Data and Safety Monitoring Board: Graeme A. Meintjes, PhD, MBChB (Chair), Barbara E. Murray, MD, Stuart Campbell Ray, MD, Valeria Cavalcanti Rolla, MD, PhD, Haroon Saloojee, MB.BCh, FCPaed, MSc, Anastasios A. Tsiatis, PhD, Paul A. Volberding, MD, Jonathan Kimmelman, PhD, David Glidden, PhD, and Sally Hunsberger, PhD (Executive Secretary).
Disclaimer. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Financial support. This work was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award numbers UM1AI068636 (J. S. C. and R. W. C.), UM1AI068634/-15S1 (M. D. H., A. K., and C. M.), UM1AI106701 (A. L. G., and R. W. C.), and 1K08AI155173-01A1 (N. J.), and the AIDS Clinical Trials Group (ACTG) (N. J.), as well as by Harvard University Development Core Award number 5P30AI060354-16 (N. J.). Study medication was donated by Sagent Pharmaceuticals, Schaumburg, Illinois.
Data availability. The authors confirm that all data underlying the findings are fully available. Data are available under restricted access due to ethical restrictions. Access can be requested by submitting a data request at https://submit.mis.s-3.net/ and will require the written agreement of the AIDS Clinical Trials Group (ACTG) and the manufacturer of the investigational product. Requests will be addressed as per ACTG standard operating procedures. Completion of an ACTG Data Use Agreement may be required.
Code availability. All analyses were performed using code available in standard software packages. No new code was developed for this manuscript. Specifics on programs used are available upon request from [email protected].
References
Author notes
N. J., K. W. C., D. M. S. and J. Z. L. contributed equally to this work.
Study team members are listed in the Supplementary Appendix.
Potential conflicts of interest. N. J.: research funding to the institution from Sagent Pharmaceuticals as Principal Investigator of a phase II trial (CAMELOT). K. W. C.: research funding to the institution from Merck, Sharp & Dohme, consultant fees to the author from Pardes Biosciences, honoraria from the International Antiviral Society (IAS)–USA for presentations; K.W.C. also reports serving as Chair of a Safety Monitoring Committee for an investigator-initiated study through the University of California San Francisco (UCSF). E. S. D.: receives consulting fees from Gilead Sciences, Merck, Theratechnologies, and GSK/ViiV and research support through the institution from Gilead Sciences and GSK/ViiV. D. A. W.: has received funding to the institution to support research and honoraria for advisory boards and consulting from Gilead Sciences. D. A. W. also reports grants or contracts from Eli Lilly and Merck. P. A. H.: received research funding from the National Institutes of Health (NIH) and consulting fees paid to the institution from Sagent Pharmaceuticals. A. L. G.: reports contract testing from Abbott, Cepheid, Novavax, Pfizer, Janssen, and Hologic and research support from the Bill and Melinda Gates Foundation, Gilead, and Merck, outside of the described work. J. J. E.: reports institutional research funding from Gilead Sciences; is an ad hoc consultant to GSK/Vir Biotechnology, Gilead Sciences, and Merck; and Data Monitoring Committee (DMC) Chair for Invivyd (formerly Adagio) phase III studies. J. S. C.: has consulted for Merck and Company's advisory board. D. M. S.: has consulted for the following companies: Fluxergy, Kiadis, Linear Therapies, Evidera, VxBiosciences, Model Medicines, Bayer Pharmaceuticals, Lucira, and Pharma Holdings. D. M. S. also reports honoraria for presentations or speaking engagements from IAS USA, and stock options from Fluxergy, Linear Therapies, and Model Medicines. J. Z. L.: has consulted for AbbVie and reports grants or contracts from Merck. C. M.: reports unpaid participation on the BONE STAR Data and Safety Monitoring Board (DSMB). M. D. H.: reports NIH research and training grants related to infectious diseases (paid to institution); travel support as an invited speaker at the Conference on Retroviruses and Opportunistic Infections (CROI 2023); unpaid participation in a COVID-related data committee for World Health Organization; board membership (as part of employment with Harvard) on the Botswana Harvard Partnership; and miscellaneous interests via their spouse's research grants from NIH. A. J. was an employee and B. S. is an employee of Sagent Pharmaceuticals. All other authors report no potential conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.


{kind=link}
{kind=link}
{kind=link}