






Abstract
Combining antiviral regimens in the hepatitis C virus (HCV)/human immunodeficiency virus (HIV)–coinfected population can be complex as they share overlapping mechanisms for elimination that may result in drug interactions. The pharmacokinetics, safety, and tolerability of sofosbuvir/velpatasvir (SOF/VEL) with multiple antiretroviral (ARV) regimens were evaluated.
Healthy volunteers were enrolled into 2 phase 1, open-label, randomized, multiple-dose, cross-over studies. SOF/VEL and ARV regimens were administered alone and in combination; ARVs (and pharmacokinetic enhancers) included atazanavir (ATV), cobicistat (COBI), darunavir (DRV), dolutegravir (DTG), efavirenz (EFV), elvitegravir (EVG), emtricitabine (FTC), lopinavir (LPV), raltegravir (RAL), rilpivirine (RPV), ritonavir (RTV), tenofovir alafenamide (TAF), and tenofovir disoproxil fumarate (TDF). Geometric least squares means ratios (coadministration:alone) and 90% confidence intervals were constructed for area under the plasma concentration–time curve over the dosing interval, maximum concentration, and trough, for all analytes. Safety and tolerability were also evaluated.
In total, 237 participants were enrolled. No clinically relevant differences in the pharmacokinetics (PK) of SOF, SOF metabolite GS-331007, or VEL were observed other than an approximate 50% decrease in VEL exposure when administered with EFV/FTC/TDF. No clinically relevant differences in the PK of ARVs were observed when administered with SOF/VEL. Study treatments were well tolerated, including no observed creatinine clearance changes during evaluation of TDF-containing regimens.
SOF/VEL and ARV regimens including ATV, COBI, DRV, DTG, EVG, FTC, LPV, RAL, RPV, RTV, TAF, or TDF may be coadministered without dose adjustment. Use of SOF/VEL with EFV-containing regimens is not recommended due to an approximate 50% reduction in VEL exposure.
Approximately 10% of the 40 million patients worldwide who are chronically infected with human immunodeficiency virus type 1 (HIV-1) are coinfected with the hepatitis C virus (HCV) [1, 2]. In the absence of effective HCV therapy, patients coinfected with HCV/HIV-1 have a higher rate of liver fibrosis progression and higher risk of cirrhosis and hepatic decompensation [3, 4]. As HIV-related morbidity and mortality has decreased, liver-related complications have become a leading cause of death in coinfected patients [5]. The introduction of direct-acting antiviral agents has provided safe and effective combination treatments for HCV infection in patients coinfected with HIV [6–8]. Current treatment guidelines recommend the same regimens for coinfected patients as for those with HCV monoinfection [9].
Sofosbuvir (SOF) is a pangenotypic nucleotide NS5B inhibitor that is approved in combination with other antiviral agents for the treatment of chronic HCV infection of all genotypes. Velpatasvir (VEL) is an HCV NS5A inhibitor with pangenotypic potency [10]. A fixed-dose combination tablet of sofosbuvir 400 mg and velpatasvir 100 mg (SOF/VEL) is approved for the treatment of adults with genotype 1–6 chronic HCV infection [11]. Results from a phase 3 trial demonstrated that SOF/VEL for 12 weeks provides a simple, safe, and highly effective treatment for patients coinfected with HCV and HIV-1 [12]. Its effectiveness in a broad range of patients across a wide range of antiretroviral (ARV) regimens suggests that it could be used by the majority of patients with HCV/HIV coinfection regardless of HCV genotype and presence or absence of compensated or decompensated cirrhosis.
More than 25 ARV drugs in 6 mechanistic classes have been approved for treatment of HIV infection in the United States. These 6 classes include the nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs), nonnucleoside reverse transcriptase inhibitors (NNRTIs), protease inhibitors (PIs), a fusion inhibitor, a CCR5 antagonist, and integrase strand transfer inhibitors (INSTIs). In addition, 2 drugs, cobicistat (COBI) and ritonavir (RTV), are used solely as pharmacokinetic (PK) enhancers (ie, boosters) to improve the PK profile of some ARV drugs (eg, PIs and the INSTI elvitegravir [EVG]). Recommended ARV regimens generally consist of 2 NRTIs plus a third agent—for example, an NNRTI, an INSTI, or a PK-enhanced PI. The choice between components of ARV regimens should be guided by the regimen’s efficacy, genetic barrier to resistance, adverse effects profile, and convenience. The patient’s comorbidities, concomitant medications, and the potential for drug–drug interactions (DDIs) are also important considerations.
Sofosbuvir is a substrate of P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) but is not a substrate of cytochrome P450 (CYP) or uridine diphosphate glycosyltransferase 1A1 (UGT). The primary circulating SOF inactive metabolite GS-331007 is not a substrate of P-gp, BCRP, renal organic anion transporter (OAT) 1, OAT3, or organic cation transporter 2. Sofosbuvir does not inhibit or induce drug transporters or metabolizing enzymes. Velpatasvir is a substrate of P-gp, BCRP, and organic anion transport polypeptide (OATP) 1B, and is a minor substrate of CYP2B6, CYP2C8, and CYP3A4. Velpatasvir inhibits P-gp, BCRP, and OATP1B, and does not inhibit or induce metabolizing enzymes such as CYPs or UGT1A1. Because many ARVs may be a substrate of 1 or more drug transporters that SOF/VEL inhibits, or may inhibit or induce drug transporters or metabolizing enzymes involved in the disposition of SOF/VEL, potential for DDIs between SOF/VEL and ARV regimens exist (Supplementary Table 1). Accordingly, DDI studies were designed to evaluate the potential for DDIs between SOF/VEL (400/100 mg) and commonly prescribed HIV ARV regimens.
The pharmacokinetics, safety, and tolerability of SOF/VEL and boosted or unboosted ARV regimens administered alone and in combination are presented herein.
METHODS
Study Population and Design
Two phase 1, open-label, randomized, multiple-dose, cross-over studies were conducted, consisting of 9 cohorts (studies GS-US-342-1167 and GS-US-342-1326; Supplementary Table 2). Healthy men and nonpregnant, nonlactating women between 18 and 45 years of age, inclusive were enrolled, with a body mass index of 19–30 kg/m2, electrocardiogram (ECG) with normal PR and QT intervals corrected for heart rate using the Fridericia formula (QTcF), normal renal function (creatinine clearance ≥90 mL/min), no significant medical history, and in good general health as determined by the investigator at the time of screening. All cohorts were conducted as Williams design cross-overs except for cohort 1 (SOF/VEL with or without efavirenz [EFV]/emtricitabine [FTC]/tenofovir disoproxil fumarate [TDF]), which was conducted as a parallel crossover given the long terminal half-life of EFV. Treatment duration and washouts within each cohort were determined based on the half-lives of the compounds and are presented in Supplementary Table 2.
Safety Assessments
Safety was evaluated by periodic physical examinations including vital signs, assessment of 12-lead ECGs, clinical laboratory tests at baseline and at various time points during the studies, and by documentation of adverse events (AEs) and use of concomitant medications.
Pharmacokinetic Sampling and Bioanalytical Procedures
Samples for PK analysis were collected predose (≤5 minutes) and at ≥13 selected time points over the course of the dosing interval (24 hours for all compounds except raltegravir [RAL; 12 hours]) following administration of each treatment. Blood samples were centrifuged for collection of plasma and frozen throughout shipment and storage. Plasma concentrations were determined by validated high-performance liquid chromatography–tandem mass spectroscopy methods performed at QPS, Inc (Newark, Delaware) or Tandem Labs (VEL only; Salt Lake City, Utah). Validated linear calibration ranges (including lower limits of quantitation), interassay precision ranges (percentage coefficient of variation), and accuracy ranges (expressed as the percentage relative error) are shown in Supplementary Table 3.
Pharmacokinetic Analyses
Pharmacokinetic parameters were estimated by application of standard noncompartmental methods (Phoenix WinNonlin software, Pharsight Corporation, Princeton, New Jersey). The linear up/log down trapezoidal rule was applied in conjunction with the appropriate noncompartmental model, with input values for dose, time of dose, plasma concentration, and corresponding real times relative to drug dosing times. Primary PK endpoints for all analytes were area under the plasma concentration–time curve over the dosing interval (AUCtau), maximum observed plasma concentration (Cmax), and drug concentration at the end of the dosing interval (Ctau).
Statistical Analyses
A parametric (normal theory) analysis of variance using a mixed-effects model appropriate for a cross-over design was fit to the natural logarithmic transformation of AUCtau, Cmax, and Ctau of each analyte. Sample size for each cohort was based on variability estimates of AUCtau, Cmax, and Ctau for respective analytes and accounted for potential dropouts. The final sample size provided at least 90% power to conclude lack of PK alteration based on the expected ratio of geometric least squares means (%GLSM) of treatments (coadministered:alone) of 100% and associated 90% confidence interval (CI) range of 70%–143% for all analytes except RAL (50%–200%). The fixed and random effects were study treatment and subject, respectively.
Descriptive statistics were used for the safety analysis set, which included all subjects who received at least 1 dose of study drug. Adverse events were coded with the Medical Dictionary for Regulatory Activities (version 16.1 or 17.1).
RESULTS
Subject Demographics and Disposition
A total of 237 participants were randomized, and all were included in the safety analysis set. Seven participants discontinued prematurely: 5 due to withdrawn consent, 1 due to pregnancy, and 1 due to an AE (described below). Baseline characteristics are shown in Supplementary Table 4. The majority of participants were white and male; ages ranged from 19 to 45 years.
Safety
Study drug treatments were generally well tolerated. All AEs were grade 1 or 2 in severity (Table 1). No grade 3 or 4 AEs, serious AEs, or deaths were reported. One AE leading to premature study drug discontinuation occurred (grade 1 urticaria, which resolved with intervention 5 days after the event’s onset and was considered related to study treatment [SOF/VEL administered alone]). Of note, urticaria was not a commonly reported AE for SOF/VEL across phase 3 studies [11]. The most frequently reported AEs reported across all cohorts were headache (n = 28) and constipation (n = 10). Thirty subjects had AEs that were considered by the investigator to be related to study treatment; the most frequently reported treatment-related AEs were headache (n = 10), nausea (n = 10), and vomiting (n = 6). There were no clinically significant trends in laboratory abnormalities, vital sign measurements, or ECG findings. One pregnancy was reported in a study subject who was subsequently withdrawn from the study.
Safety Summary
| Adverse Event | Cohort 1 | Cohort 2 | Cohort 3 | Cohort 4 | Cohort 5 | Cohort 6 | Cohort 7 | Cohort 8 | Cohort 9 |
|---|---|---|---|---|---|---|---|---|---|
| SOF/VEL With or Without | |||||||||
| EFV/FTC/ TDF (n = 30) | FTC/RPV/ TDF (n = 24) | DTG (n = 24) | FTC/TDF + RAL (n = 30) | EVG/COBI/ FTC/ TAF (n = 23) | EVG/COBI/ FTC/ TDF (n = 24) | ATV + RTV + FTC/TDF (n = 24) | DRV + RTV + FTC/TDF (n = 30) | LPV/RTV + FTC/TDF (n = 23) | |
| AEs | 10 (33) | 14 (58) | 11 (46) | 10 (33) | 7 (29) | 10 (39) | 10 (42) | 14 (45) | 7 (29) |
| Grade 3–4 AEs | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 | 0 | 0 | 0 | 0 |
| Serious AEs | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 | 0 | 0 | 0 | 0 |
| Treatment discontinuation due to AEs | 1 (3)a | 0 (0) | 0 (0) | 0 (0) | 0 | 0 | 0 | 0 | 0 |
| AEs >10% | 5 (17)b | 3 (13)c | 6 (25)c | 5 (17)c | 0 | 0 | 5 (21)c | 0 | 0 |
| 5 (21)d | 5 (17)e | ||||||||
| 3 (13)b | 3 (10)f | ||||||||
| Grade 3–4 laboratory abnormality | 3 (10)g | 1 (4)g | 2 (8)g | 2 (7)g | 1 (4)g | 5 (19)g | 18 (75)h | 3 (10)g | 2 (8)i |
| Adverse Event | Cohort 1 | Cohort 2 | Cohort 3 | Cohort 4 | Cohort 5 | Cohort 6 | Cohort 7 | Cohort 8 | Cohort 9 |
|---|---|---|---|---|---|---|---|---|---|
| SOF/VEL With or Without | |||||||||
| EFV/FTC/ TDF (n = 30) | FTC/RPV/ TDF (n = 24) | DTG (n = 24) | FTC/TDF + RAL (n = 30) | EVG/COBI/ FTC/ TAF (n = 23) | EVG/COBI/ FTC/ TDF (n = 24) | ATV + RTV + FTC/TDF (n = 24) | DRV + RTV + FTC/TDF (n = 30) | LPV/RTV + FTC/TDF (n = 23) | |
| AEs | 10 (33) | 14 (58) | 11 (46) | 10 (33) | 7 (29) | 10 (39) | 10 (42) | 14 (45) | 7 (29) |
| Grade 3–4 AEs | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 | 0 | 0 | 0 | 0 |
| Serious AEs | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 | 0 | 0 | 0 | 0 |
| Treatment discontinuation due to AEs | 1 (3)a | 0 (0) | 0 (0) | 0 (0) | 0 | 0 | 0 | 0 | 0 |
| AEs >10% | 5 (17)b | 3 (13)c | 6 (25)c | 5 (17)c | 0 | 0 | 5 (21)c | 0 | 0 |
| 5 (21)d | 5 (17)e | ||||||||
| 3 (13)b | 3 (10)f | ||||||||
| Grade 3–4 laboratory abnormality | 3 (10)g | 1 (4)g | 2 (8)g | 2 (7)g | 1 (4)g | 5 (19)g | 18 (75)h | 3 (10)g | 2 (8)i |
Data are presented as No. (%).
Abbreviations: AE, adverse event; ATV, atazanavir; COBI, cobicistat; DRV, darunavir; DTG, dolutegravir; EFV, efavirenz; EVG, elvitegravir; FTC, emtricitabine; LPV, lopinavir; RAL, raltegravir; RPV, rilpivirine; RTV, ritonavir; SOF, sofosbuvir; TAF, tenofovir alafenamide; TDF, tenofovir disoproxil fumarate; VEL, velpatasvir.
aUrticaria.
bConstipation.
cHeadache.
dChapped lips.
eNausea.
fVomiting.
gBlood in urine.
hBlood in urine (n = 1), hyperbilirubinemia (n = 17).
iBlood in urine (n = 1), elevated low-density lipoprotein (n = 1).
Safety Summary
| Adverse Event | Cohort 1 | Cohort 2 | Cohort 3 | Cohort 4 | Cohort 5 | Cohort 6 | Cohort 7 | Cohort 8 | Cohort 9 |
|---|---|---|---|---|---|---|---|---|---|
| SOF/VEL With or Without | |||||||||
| EFV/FTC/ TDF (n = 30) | FTC/RPV/ TDF (n = 24) | DTG (n = 24) | FTC/TDF + RAL (n = 30) | EVG/COBI/ FTC/ TAF (n = 23) | EVG/COBI/ FTC/ TDF (n = 24) | ATV + RTV + FTC/TDF (n = 24) | DRV + RTV + FTC/TDF (n = 30) | LPV/RTV + FTC/TDF (n = 23) | |
| AEs | 10 (33) | 14 (58) | 11 (46) | 10 (33) | 7 (29) | 10 (39) | 10 (42) | 14 (45) | 7 (29) |
| Grade 3–4 AEs | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 | 0 | 0 | 0 | 0 |
| Serious AEs | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 | 0 | 0 | 0 | 0 |
| Treatment discontinuation due to AEs | 1 (3)a | 0 (0) | 0 (0) | 0 (0) | 0 | 0 | 0 | 0 | 0 |
| AEs >10% | 5 (17)b | 3 (13)c | 6 (25)c | 5 (17)c | 0 | 0 | 5 (21)c | 0 | 0 |
| 5 (21)d | 5 (17)e | ||||||||
| 3 (13)b | 3 (10)f | ||||||||
| Grade 3–4 laboratory abnormality | 3 (10)g | 1 (4)g | 2 (8)g | 2 (7)g | 1 (4)g | 5 (19)g | 18 (75)h | 3 (10)g | 2 (8)i |
| Adverse Event | Cohort 1 | Cohort 2 | Cohort 3 | Cohort 4 | Cohort 5 | Cohort 6 | Cohort 7 | Cohort 8 | Cohort 9 |
|---|---|---|---|---|---|---|---|---|---|
| SOF/VEL With or Without | |||||||||
| EFV/FTC/ TDF (n = 30) | FTC/RPV/ TDF (n = 24) | DTG (n = 24) | FTC/TDF + RAL (n = 30) | EVG/COBI/ FTC/ TAF (n = 23) | EVG/COBI/ FTC/ TDF (n = 24) | ATV + RTV + FTC/TDF (n = 24) | DRV + RTV + FTC/TDF (n = 30) | LPV/RTV + FTC/TDF (n = 23) | |
| AEs | 10 (33) | 14 (58) | 11 (46) | 10 (33) | 7 (29) | 10 (39) | 10 (42) | 14 (45) | 7 (29) |
| Grade 3–4 AEs | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 | 0 | 0 | 0 | 0 |
| Serious AEs | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 | 0 | 0 | 0 | 0 |
| Treatment discontinuation due to AEs | 1 (3)a | 0 (0) | 0 (0) | 0 (0) | 0 | 0 | 0 | 0 | 0 |
| AEs >10% | 5 (17)b | 3 (13)c | 6 (25)c | 5 (17)c | 0 | 0 | 5 (21)c | 0 | 0 |
| 5 (21)d | 5 (17)e | ||||||||
| 3 (13)b | 3 (10)f | ||||||||
| Grade 3–4 laboratory abnormality | 3 (10)g | 1 (4)g | 2 (8)g | 2 (7)g | 1 (4)g | 5 (19)g | 18 (75)h | 3 (10)g | 2 (8)i |
Data are presented as No. (%).
Abbreviations: AE, adverse event; ATV, atazanavir; COBI, cobicistat; DRV, darunavir; DTG, dolutegravir; EFV, efavirenz; EVG, elvitegravir; FTC, emtricitabine; LPV, lopinavir; RAL, raltegravir; RPV, rilpivirine; RTV, ritonavir; SOF, sofosbuvir; TAF, tenofovir alafenamide; TDF, tenofovir disoproxil fumarate; VEL, velpatasvir.
aUrticaria.
bConstipation.
cHeadache.
dChapped lips.
eNausea.
fVomiting.
gBlood in urine.
hBlood in urine (n = 1), hyperbilirubinemia (n = 17).
iBlood in urine (n = 1), elevated low-density lipoprotein (n = 1).
Most laboratory abnormalities were grade 1 or 2 in severity. Grade 3 blood in urine was reported in 21 subjects (20 females and 1 male) and were transient. Grade 3 or 4 hyperbilirubinemia was reported in subjects receiving treatment with atazanavir (ATV) and is a known side effect of ATV due to inhibition of uridine glucuronosyl diphosphate transferase; incidence and severity of hyperbilirubinemia was similar with or without SOF/VEL. No other grade 3 or 4 laboratory abnormalities were reported.
Pharmacokinetic Results
Effect of ARVs on the PK of SOF, GS-331007, or VEL
Pharmacokinetic parameters for SOF, its metabolite GS-331007, and VEL when SOF/VEL was administered alone compared to coadministration with ARVs are shown in Supplementary Table 5 and depicted in Figure 1.
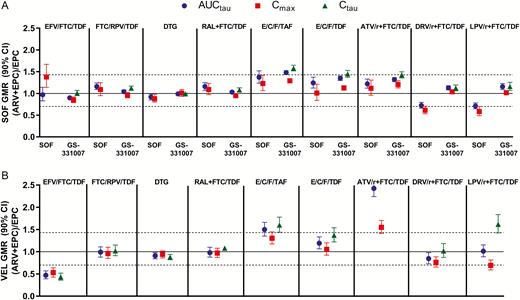
Effect of antiretrovirals on sofosbuvir and GS-331007 (A) and velpatasvir (B). Abbreviations: ARV, antiretroviral; ATV/r, atazanavir plus ritonavir; AUCtau, area under the plasma concentration–time curve over the dosing interval; CI, confidence interval; Cmax, maximum observed plasma concentration; Ctau, drug concentration at the end of the dosing interval; DRV/r, darunavir plus ritonavir; DTG, dolutegravir; E/C/F, elvitegravir/cobicistat/emtricitabine; EFV, efavirenz; EPC, Epclusa (SOF/VEL); FTC, emtricitabine; GMR, geometric mean ratio; LPV/r, lopinavir plus ritonavir; RAL, raltegravir; RPV, rilpivirine; SOF, sofosbuvir; TAF, tenofovir alafenamide; TDF, tenofovir disoproxil fumarate; VEL, velpatasvir.
Sofosbuvir and GS-331007
Other than higher SOF Cmax (38%) when SOF/VEL was administered with EFV/FTC/TDF, the 90% CIs for the %GLSM ratios for all primary PK parameters (AUCtau, Cmax, and Ctau [if measurable]) were within predetermined lack of PK alteration boundaries of 70%–143% following when SOF/VEL was administered with EFV/FTC/TDF, FTC/rilpivirine [RPV]/TDF, dolutegravir [DTG], RAL + FTC/TDF, EVG/COBI/FTC/TDF, or ATV + RTV + FTC/TDF (Supplementary Table 5). Higher SOF AUC (37%) was observed when administered with EVG/COBI/FTC/tenofovir alafenamide (TAF), and lower SOF AUC (28% and 29%) was observed when administered with darunavir (DRV) + RTV + FTC/TDF and lopinavir (LPV)/RTV + FTC/TDF, respectively. No changes in GS-331007 exposure (AUC) were observed following administration of SOF/VEL with any ARV regimen except EVG/COBI/FTC/TAF, where 48% higher GS-331007 AUC was observed.
Velpatasvir
Overall VEL exposure (AUC) was not altered by FTC/RPV/TDF, DTG, RAL + FTC/TDF, EVG/COBI/FTC/TDF, DRV + RTV + FTC/TDF, or LPV/RTV + FTC/TDF. Approximately 50% lower VEL exposure was observed following administration of SOF/VEL with EFV/FTC/TDF, and administration of SOF/VEL with EVG/COBI/FTC/TAF or ATV + RTV + FTC/TDF resulted in higher VEL AUC (50% and 142%, respectively).
Effect of SOF/VEL on the PK of ARVs
Pharmacokinetic parameters for ARVs within regimens administered alone compared to coadministration with SOF/VEL are shown in Supplementary Tables 6 and 7 and depicted in Figure 2.
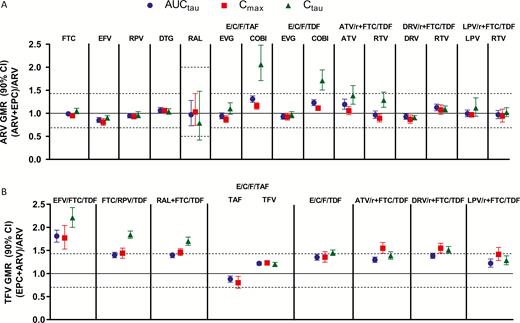
Effect of sofosbuvir/velpatasvir on antiretrovirals. Abbreviations: ARV, antiretroviral; ATV/r, atazanavir plus ritonavir; AUCtau, area under the plasma concentration–time curve over the dosing interval; CI, confidence interval; COBI, cobicistat; Cmax, maximum observed plasma concentration; Ctau, drug concentration at the end of the dosing interval; DRV/r, darunavir plus ritonavir; DTG, dolutegravir; E/C/F, elvitegravir/cobicistat/emtricitabine; EFV, efavirenz; EPC, Epclusa (SOF/VEL); FTC, emtricitabine; GMR, geometric mean ratio; LPV/r, lopinavir plus ritonavir; RAL, raltegravir; RPV, rilpivirine; SOF, sofosbuvir; TAF, tenofovir alafenamide; TDF, tenofovir disoproxil fumarate; VEL, velpatasvir.
No change in the overall exposure (AUC) of FTC, EFV, RPV, DTG, RAL, EVG, COBI, ATV, DRV, LPV, RTV, TAF, or tenofovir (TFV) derived from TAF was observed following administration with SOF/VEL. Higher COBI Ctau (71%–103%) was observed but, given the short COBI half-life, did not result in significant changes to AUC. Higher Ctau of ATV (~39%) and RTV (~29%) when administered as part of ATV + RTV + FTC/TDF and slightly lower RAL Ctau (~21%) was observed following coadministration with SOF/VEL; no change in the AUC or Cmax was observed following coadministration with SOF/VEL for any of these agents.
Approximately 40% higher TFV AUC was observed following administration of TDF-based regimens with SOF/VEL except for EFV/FTC/TDF, where an 81% higher TFV exposure was observed. The resulting TFV exposure for EFV/FTC/TDF in the presence of SOF/VEL (TFV AUC 3920 h × ng/mL) was consistent with that of other TDF-containing regimens (TFV AUC range, 4140 to 5330 h × ng/mL).
DISCUSSION
A significant proportion of individuals infected with HIV are also chronically infected with HCV. As treatment of both diseases requires multiple drug regimens and many of these drugs act as victims and/or perpetrators of metabolizing enzyme- and/or drug transport-mediated DDIs, understanding DDI potential is important for proper care of HCV/HIV-coinfected patients. Specifically, because DDIs exist within some regimens (eg, COBI boosting TAF and EVG in EVG/COBI/FTC/TAF), DDI studies with components of a regimen may not accurately reflect DDI potential, suggesting that a regimen–regimen approach be most appropriate to provide direct clinical guidance, particularly where intraregimen DDIs exist. Accordingly, DDI studies between SOF/VEL and boosted and unboosted ARV regimens were conducted in healthy adult volunteers. The mechanisms underlying the observed interactions are detailed below to identify perpetrating agents and enable clinicians to apply the findings of these studies to clinical situations where regimens other than those evaluated herein are in use or being considered.
Sofosbuvir is a substrate for efflux transporters such as P-gp, particularly intestinally, and higher exposure was observed when SOF/VEL was administered with P-gp inhibitors such as COBI. Although EFV is considered an inducer of P-gp, no change in SOF exposure was observed, suggesting that EFV induces hepatic, but not intestinal, P-gp. Modestly lower SOF exposure was, however, observed following administration with DRV + RTV and LPV/RTV, suggesting weak induction of P-gp by DRV and LPV, which are known activators of pregnane X receptor [13]. Collectively, no observed interactions with SOF warranted dose adjustment or preclude the use of SOF/VEL with ARV regimens.
Velpatasvir is a substrate of efflux transporters (eg, P-gp), OATP1B, and, to a lesser extent, CYP enzymes, mainly CYP3A. Administration of SOF/VEL with EFV/FTC/TDF resulted in approximate 50% lower VEL exposure, attributable to induction of CYP3A and P-gp. Based on this result, SOF/VEL is not recommended for use with EFV-containing ARV regimens. Higher VEL exposure (50%) was observed following administration with EVG/COBI/FTC/TAF; COBI is an inhibitor of CYP3A, P-gp, and OATP1B, and is responsible for the observed increases in VEL exposure. Higher VEL exposure (142%) was also observed following administration of SOF/VEL with ATV + RTV + FTC/TDF, attributable to strong inhibition of efflux transport, OATP1B, and CYP enzymes by ATV + RTV. Based on the lack of observed relationships between VEL exposure and safety findings in phase 3 studies, along with lack of safety findings in phase 1 studies where high exposures were observed (eg, thorough QT study), no VEL dose adjustment is required when administered with COBI-containing regimens or with boosted regimens including ATV. No other significant changes in VEL PK were observed with other ARVs.
For ARVs, administration of SOF/VEL resulted in no change to the PK of EFV, RPV, DTG, RAL, EVG, FTC, DRV, or LPV. Modest increases in ATV, COBI, and RTV Ctau were observed when administered with SOF/VEL, though none were significant enough as to impact overall exposure (AUC) and corresponding safety or efficacy profiles of the respective agents. As it pertains to ATV specifically, based on the ATV US prescribing information, dose adjustment of ATV was not warranted following a similar increase in ATV Ctau (85% higher) when administered with telaprevir, and incidence and severity in hyperbilirubinemia were similar in the presence or absence of SOF/VEL [14].
TAF and TFV exposure were both unchanged following administration of EVG/COBI/FTC/TAF with SOF/VEL. Velpatasvir has been shown to inhibit P-gp, of which TAF is a substrate; however, because TAF is already administered with COBI, a P-gp inhibitor, as part of the ARV regimen, the addition of VEL did not result in further increase in TAF or TFV (from TAF) exposure.
Administration of SOF/VEL with TDF-containing ARV regimens results in modestly higher TFV exposure (AUC) of approximately 40%. The comparatively larger increase in TFV exposure following administration of SOF/VEL with EFV/FTC/TDF is attributable to administration of EFV/FTC/TDF under fasting conditions, resulting in lower reference TFV exposure [15, 16]. Overall, similar absolute TFV exposures were achieved following administration of SOF/VEL and any TDF-containing regimens, none of which requiring dose adjustment [17, 18]. Given the roughly proportionate increase in AUC, Cmax, and Ctau and lack of change in half-life (t1/2) (data not shown), the likely mechanism is of interaction is presystemic inhibition of P-gp and/or BCRP by VEL, resulting in an increase in tenofovir disoproxil bioavailability, rather than inhibition of active tubular secretion of TFV, as in vitro data suggest that VEL is not expected to inhibit renal transporters (eg, OAT1/3 or multidrug resistance-associated protein 2 [MRP2]) at observed clinical plasma concentrations. Of note, no changes in creatinine clearance were observed in subjects receiving TDF-containing regimens alone or with SOF/VEL across the phase 1 study cohorts.
The clinical implications of coadministering SOF/VEL and boosted and unboosted HIV ARV regimens were further informed by a phase 3 study (ASTRAL-5), which evaluated the safety and efficacy of SOF/VEL in HCV/HIV-coinfected patients [12]. All boosted and unboosted ARVs evaluated herein were permitted during the phase 3 study with the exception of EFV, due to lower VEL exposure, and TAF-containing regimens due to lack of market accessibility at the time of study conduct [12]. Given the association between TDF and renal events, and in light of the observed DDI with VEL in phase 1 studies, the exposure of TFV was evaluated in this study for all patients on TDF-containing regimens (n = 91 of 106 total subjects). Data showed similar TFV exposure for those receiving SOF/VEL with boosted (TFV AUC: 3740 h × ng/mL; n = 56) or unboosted (TFV AUC: 3590 h × ng/mL; n = 35) TDF-containing regimens [19]. These TFV exposures are also consistent with those observed in HIV-infected patients on boosted regimens in the absence of SOF/VEL and do not require TDF dose adjustment, though monitoring for tenofovir-related AEs during administration with SOF/VEL is prudent [19]. Wyles et al present additional details regarding renal safety for patients on TDF-containing regimens throughout a full 12-week treatment course with SOF/VEL in the phase 3 study [12].
In summary, phase 1 studies were conducted evaluating an array of DDIs between SOF/VEL and boosted and unboosted ARV regimens. Mechanisms underlying the observed interactions have been detailed to enable clinicians to extend the findings to other clinical situations where regimens other than those evaluated herein are in use or being considered. Collectively, the data indicate that SOF/VEL and ARV regimens including ATV, COBI, DRV, DTG, EVG, FTC, LPV, RAL, RPV, RTV, TAF, or TDF may be administered together without dose adjustment. Use of SOF/VEL with EFV-containing regimens is not recommended due to an approximate 50% reduction in VEL exposure following coadministration with EFV/FTC/TDF.
Supplementary Data
Supplementary materials are available at Clinical Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Notes
Acknowledgments. The authors acknowledge the individuals who participated in these studies and the sites and study management staff whose efforts made this work possible. The authors acknowledge Anna Kido for contribution to the study design, implementation, interpretation, and/or editorial assistance during the development of the manuscript.
Financial support. This study was sponsored by Gilead Sciences, Inc.
Potential conflicts of interest. All authors are employees of Gilead and hold stock interest in the company. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
Presented in part: The Liver Meeting, American Association for the Study of Liver Diseases, San Francisco, California, 2015 (poster presentation); Conference on Retroviruses and Opportunistic Infections, Boston, Massachusetts, 2016 (oral presentation); International AIDS Conference, Durban South Africa 2016 (oral presentation).


{kind=link}
{kind=link}