






Abstract
Our objective was to assess the therapeutic noninferiority of dual therapy with darunavir/ritonavir and lamivudine compared to triple therapy with darunavir/ritonavir plus 2 nucleos(t)ides for maintenance of human immunodeficiency virus type 1 (HIV-1) suppression.
This was a multicenter, open-label, noninferiority trial (margin 12%). Patients with HIV-1 RNA <50 copies/mL for 6 months or longer on triple therapy with darunavir/ritonavir and 2 nucleos(t)ides (tenofovir disoproxil fumarate and emtricitabine or abacavir and lamivudine) and with no resistance were randomized to continue therapy (n = 128) or switch to darunavir/ritonavir and lamivudine (n = 129). The primary endpoint was the proportion of participants with HIV-RNA <50 copies/mL after 48 weeks of follow-up according to the snapshot algorithm.
A total of 249 participants received study drugs (intention-to-treat exposed). The proportion of participants with HIV-RNA <50 copies/mL in the dual- and triple-therapy arms was 88.9% (112/126) and 92.7% (114/123; difference, –3.8%; 95% confidence interval, –11.0 to 3.4), respectively. Four participants in the dual-therapy arm and 2 in the triple-therapy arm developed protocol-defined virological failure. Switching to dual therapy was associated with a significant increase in total, low-density lipoprotein, and high-density lipoprotein (HDL) cholesterol, but not in the total-to-HDL cholesterol ratio. Serious adverse events and study drug discontinuations due to adverse events occurred in 4.8% vs 4.9%P = .97) and in 0.8% (1/126) vs 1.6% P = .55) in dual therapy vs triple therapy, respectively.
Dual therapy with darunavir/ritonavir and lamivudine demonstrated noninferior therapeutic efficacy and similar tolerability compared to triple therapy.
NCT02159599.
In patients infected with human immunodeficiency virus type 1 (HIV-1), dual therapy with a boosted protease inhibitor and lamivudine has been studied as initial therapy in antiretroviral-naive patients and as a switch strategy in antiretroviral-experienced patients [1].
In antiretroviral-naive patients, the GARDEL clinical trial [2] demonstrated that a dual-therapy regimen that consisted of lopinavir/ritonavir and lamivudine twice daily was noninferior to a triple-drug regimen of lopinavir/ritonavir and 2 nucleos(t)ide reverse transcriptase inhibitors in achieving and maintaining virologic suppression over 48 weeks of therapy, regardless of baseline viral load.
In antiretroviral-experienced patients with suppressed viremia, the dual-therapy strategy of combining atazanavir/ritonavir with lamivudine has been tested in SALT [3] and ATLAS-M [4], 2 randomized clinical trials. Compared to patients who continued triple therapy, the combination of atazanavir/ritonavir and lamivudine demonstrated noninferior efficacy for maintenance of virological suppression after up to 96 weeks of follow-up. The OLE clinical trial [5] also demonstrated that the dual combination of lopinavir/ritonavir and lamivudine had noninferior therapeutic efficacy for maintenance of virological suppression during 48 weeks compared to continuation of triple therapy with lopinavir/ritonavir and 2 nucleos(t)ides.
Due to its efficacy and safety profile, darunavir boosted with ritonavir or cobicistat is the preferred protease inhibitor in most international guidelines of HIV therapy [6, 7]. To the best of our knowledge, there are no clinical trial data about the efficacy of dual therapy with boosted darunavir and lamivudine.
We performed a randomized, open-label, clinical trial and compared dual therapy with darunavir/ritonavir and lamivudine to continuation of darunavir/ritonavir and 2 nucleos(t)ides. Our objective was to assess the therapeutic noninferiority of the dual-therapy strategy compared to triple therapy over 48 weeks of therapy.
METHODS
Study Design and Participants
The DUAL study (DarUnavir And Lamivudine) is a phase 4, randomized, open-label, multicenter, parallel-group, noninferiority clinical trial that took place at 24 sites in Spain. At recruitment, eligible participants were HIV-infected adults (aged 18 years or older) on a stable antiretroviral regimen that consisted of darunavir/ritonavir 800/100 mg once daily and 2 nucleos(t)ides (tenofovir disoproxil fumarate and emtricitabine or abacavir and lamivudine) for at least 4 weeks. Participants had a plasma HIV-RNA viral load of <50 copies/mL for a minimum of 6 consecutive months. Participants in any line of therapy could be enrolled, after excluding those with prior resistance to darunavir/ritonavir or lamivudine/emtricitabine (confirmed by genotypic tests that showed any of the following resistance mutations: M184V/I, K65R in reverse transcriptase and/or V11I, V32I, L33F, I47V, I50V, I54L/M, G73S, T74P, L76V, I84V, L89V in protease, or suspected by the history of virological failures or discontinuations). Patients with a positive hepatitis B serum surface antigen were also excluded.
The study was performed in accordance with the Declaration of Helsinki. The protocol was reviewed and approved by the Autonomous Community of Madrid Ethics Committee, as the reference committee, and by all committees from all the participating hospitals. Every participant gave written informed consent before undergoing study procedures.
Randomization and Masking
Eligible participants were randomly assigned (1:1) to either switch to lamivudine 300 mg once daily plus darunavir/ritonavir 800/100 mg once daily (dual therapy) or to remain on ongoing triple therapy. Once participants had satisfied all eligibility requirements and were stratified by nucleos(t)ide combination (tenofovir/emtricitabine or abacavir/lamivudine), the treatment arm was assigned using a centralized web-based randomization process. Patients were randomized in a 1:1 ratio to switch the nucleos(t)ides for lamivudine or continue the same regimen. The random number sequence was computer generated. The study design was open label, so participants and investigators were not masked to group allocation.
Outcomes
The primary endpoint was the proportion of participants with suppressed viral load (HIV-RNA <50 copies/mL) after 48 weeks of follow-up according to the Food and Drug Administration (FDA) snapshot algorithm [8] in the intention-to-treat exposed (ITT-e) population. In this analysis, those participants whose last HIV-RNA result was <50 copies/mL in the analysis window (48 weeks, ±6 weeks) were counted as responders. Patients whose HIV-RNA was not suppressed or who withdrew, changed antiretroviral therapy, or did not have data at the analysis time point were counted as nonresponders.
Secondary endpoints included the proportion of participants with response according to the time to loss of virological response (TLOVR) algorithm [9] and the proportion of participants with virological failure, defined as HIV-RNA >50 copies/mL in the 48-week window or discontinuation before week 48 due to lack of efficacy. Additional secondary endpoints were proportion of participants with emergent mutations of resistance, proportion of participants with 1 or more blips (defined as an unconfirmed episodes of detectable viral load), change in CD4 cell count, safety, and tolerability of the 2 regimens up to week 48.
Procedures
Post-baseline study visits occurred at weeks 4, 12, 24, 36, and 48. We assessed safety with laboratory tests, physical examinations, and reporting of adverse events. Hematology, serum chemistry, urinalysis, CD4 cell count, and fasting lipid concentrations were measured at a designated local laboratory for each study site.
Plasma HIV-RNA concentrations were measured using the techniques available locally at each site, including branched DNA, nucleic acid sequence–based amplification, and real-time polymerase chain reaction. Resistance testing was attempted in every sample with viral load >400 copies/mL.
Statistical Analyses
All analyses were prespecified in the protocol. No interim analysis was performed before week 48.
We assessed virological outcomes in the following 3 populations: the ITT-e population (defined as all participants who received at least 1 dose of study treatment); the per-protocol (PP) population (defined as all participants who received at least 1 dose of study treatment and who had no deviation to the eligibility criteria); and the security, ITT population (all randomized participants).
For the primary endpoint, we analyzed the ITT-e population. For the sensitivity analysis, we analyzed the primary endpoint in the PP and ITT populations. We also analyzed the proportion of participants with response using an observed data approach, censoring participants who discontinued due to nonvirological reasons, and the TLOVR algorithm.
In the primary efficacy analysis, noninferiority was established if the lower bound of a 2-sided 95% confidence interval (CI) for the difference in proportions (dual-therapy group minus triple-therapy group) of participants without failure at week 48 was higher than –12%. A 12% noninferiority margin was chosen based on similar trials of switching antiretrovirals in aviremic HIV-infected participants [4, 5, 10, 11].
The original sample size estimation was 256 participants. It was calculated that this sample size provides 80% power to establish noninferiority for the proportion of participants without therapeutic failure at week 48, with an assumed response rate of 88% in both groups, noninferiority margin of –12%, and a 10% rate of losses to follow-up.
We used an independent-samples t test to compare means for continuous variables and a Wilcoxon test to compare medians for nonnormally distributed continuous variables. Adjusted differences of the mean change in CD4 cell counts, lipids, and renal parameters at week 48 compared to baseline and the test of significance were assessed with a multivariable linear regression using as covariables the group of therapy (dual or triple) and the baseline value (analysis of covariance). We assessed the association between categorical variables using the χ2 test when samples were of sufficient size or with the Fisher exact test when they were not.
RESULTS
Between July 2014 and April 2015, 260 participants were screened and 257 were randomly assigned: 129 to dual therapy and 128 to triple therapy (Figure 1). Of the 257 randomized participants, 249 received at least 1 dose of study medication: 126 in the dual-therapy group and 123 in the triple-therapy group. Baseline characteristics were balanced between study arms (Table 1) except for length of time with suppressed viremia, which was shorter in the dual-therapy group (79.5 vs 113 weeks). Ninety-three (74%) and 33 (26%) participants were previously receiving tenofovir or abacavir, respectively, in the dual-therapy arm. Ninety-three (76%) and 30 (24%) were previously receiving and continued with a tenofovir- or abacavir-containing regimen in the triple-therapy arm.

Trial profile. Dual therapy, switch to darunavir/ritonavir + lamivudine. Triple therapy, maintain darunavir/ritonavir + 2 nucleos(t)ides. Per protocol population excludes patients who received the assigned therapy with protocol deviations. *, Detectable viral load at screening; **, one patient was receiving a single nucleoside at baseline; 1 patient with history of M184V mutation in a previous genotype; ***, 2 erroneously duplicated registers. Abbreviation: IC, informed consent.
Demographic and Baseline Characteristics for the 2 Study Arms
| Characteristic | Dual Therapy (n = 126) | Triple Therapy (n = 123) | Total (n = 249) |
|---|---|---|---|
| Age, y | 44 (36–52) | 43 (37–49) | 43 (36–50) |
| Gender | |||
| Male | 107 (85) | 100 (81) | 207 (83) |
| Mode of transmission | |||
| Intravenous drug use | 19 (15.1) | 15 (12.2) | 34 (13.7) |
| Men who have sex with men | 65 (51.6) | 72 (58.5) | 137 (55) |
| Heterosexual | 34 (27) | 32 (26) | 66 (26.5) |
| Hepatitis C | 32 (25.4) | 28 (22.8) | 60 (24.1) |
| Baseline CD4 count (cells/µL) | 596 (433–810) | 568 (451–739) | 589 (443–762) |
| Nadir CD4 count (cells/µL) | 253 (122–367) | 240 (117–328) | 246 (120–327) |
| Weeks since undetectable viral load (<50 copies/mL) | 79.5 (38–157) | 113 (57–184) | 100 (45–166) |
| Previous nucleos(t)ide | |||
| Tenofovir | 93 (74) | 93 (76) | 186 (75) |
| Abacavir | 33 (26) | 30 (24) | 63 (25) |
| Characteristic | Dual Therapy (n = 126) | Triple Therapy (n = 123) | Total (n = 249) |
|---|---|---|---|
| Age, y | 44 (36–52) | 43 (37–49) | 43 (36–50) |
| Gender | |||
| Male | 107 (85) | 100 (81) | 207 (83) |
| Mode of transmission | |||
| Intravenous drug use | 19 (15.1) | 15 (12.2) | 34 (13.7) |
| Men who have sex with men | 65 (51.6) | 72 (58.5) | 137 (55) |
| Heterosexual | 34 (27) | 32 (26) | 66 (26.5) |
| Hepatitis C | 32 (25.4) | 28 (22.8) | 60 (24.1) |
| Baseline CD4 count (cells/µL) | 596 (433–810) | 568 (451–739) | 589 (443–762) |
| Nadir CD4 count (cells/µL) | 253 (122–367) | 240 (117–328) | 246 (120–327) |
| Weeks since undetectable viral load (<50 copies/mL) | 79.5 (38–157) | 113 (57–184) | 100 (45–166) |
| Previous nucleos(t)ide | |||
| Tenofovir | 93 (74) | 93 (76) | 186 (75) |
| Abacavir | 33 (26) | 30 (24) | 63 (25) |
Dual therapy = switching to darunavir/r + lamivudine. Triple therapy = maintain triple therapy with darunavir/r + 2 nucleos(t)ide reverse transcriptase inhibitor. Data are expressed as median (interquartile range) or n (%).
Demographic and Baseline Characteristics for the 2 Study Arms
| Characteristic | Dual Therapy (n = 126) | Triple Therapy (n = 123) | Total (n = 249) |
|---|---|---|---|
| Age, y | 44 (36–52) | 43 (37–49) | 43 (36–50) |
| Gender | |||
| Male | 107 (85) | 100 (81) | 207 (83) |
| Mode of transmission | |||
| Intravenous drug use | 19 (15.1) | 15 (12.2) | 34 (13.7) |
| Men who have sex with men | 65 (51.6) | 72 (58.5) | 137 (55) |
| Heterosexual | 34 (27) | 32 (26) | 66 (26.5) |
| Hepatitis C | 32 (25.4) | 28 (22.8) | 60 (24.1) |
| Baseline CD4 count (cells/µL) | 596 (433–810) | 568 (451–739) | 589 (443–762) |
| Nadir CD4 count (cells/µL) | 253 (122–367) | 240 (117–328) | 246 (120–327) |
| Weeks since undetectable viral load (<50 copies/mL) | 79.5 (38–157) | 113 (57–184) | 100 (45–166) |
| Previous nucleos(t)ide | |||
| Tenofovir | 93 (74) | 93 (76) | 186 (75) |
| Abacavir | 33 (26) | 30 (24) | 63 (25) |
| Characteristic | Dual Therapy (n = 126) | Triple Therapy (n = 123) | Total (n = 249) |
|---|---|---|---|
| Age, y | 44 (36–52) | 43 (37–49) | 43 (36–50) |
| Gender | |||
| Male | 107 (85) | 100 (81) | 207 (83) |
| Mode of transmission | |||
| Intravenous drug use | 19 (15.1) | 15 (12.2) | 34 (13.7) |
| Men who have sex with men | 65 (51.6) | 72 (58.5) | 137 (55) |
| Heterosexual | 34 (27) | 32 (26) | 66 (26.5) |
| Hepatitis C | 32 (25.4) | 28 (22.8) | 60 (24.1) |
| Baseline CD4 count (cells/µL) | 596 (433–810) | 568 (451–739) | 589 (443–762) |
| Nadir CD4 count (cells/µL) | 253 (122–367) | 240 (117–328) | 246 (120–327) |
| Weeks since undetectable viral load (<50 copies/mL) | 79.5 (38–157) | 113 (57–184) | 100 (45–166) |
| Previous nucleos(t)ide | |||
| Tenofovir | 93 (74) | 93 (76) | 186 (75) |
| Abacavir | 33 (26) | 30 (24) | 63 (25) |
Dual therapy = switching to darunavir/r + lamivudine. Triple therapy = maintain triple therapy with darunavir/r + 2 nucleos(t)ide reverse transcriptase inhibitor. Data are expressed as median (interquartile range) or n (%).
At week 48, proportions of participants with HIV-RNA viral loads <50 copies/mL in the dual- and triple-therapy arms, respectively (ITT-e population, snapshot analysis), were 88.9% (112/126) vs 92.7% (114/123; difference, –3.8%; 95% CI, –11.0 to 3.4), meeting a priori criteria for noninferiority (Figure 2A). Sensitivity analysis in the PP and ITT population and the analysis using the observed data approach or TLOVR algorithm confirmed these findings (Figure 2B, Supplementary Table S1). Mean changes in CD4 count from baseline to week 48 were +33 cells/µL in the dual-therapy arm and +25 cells/µL in the triple-therapy arm (adjusted difference, 8 cells/µL; 95% CI, –38 to 54).
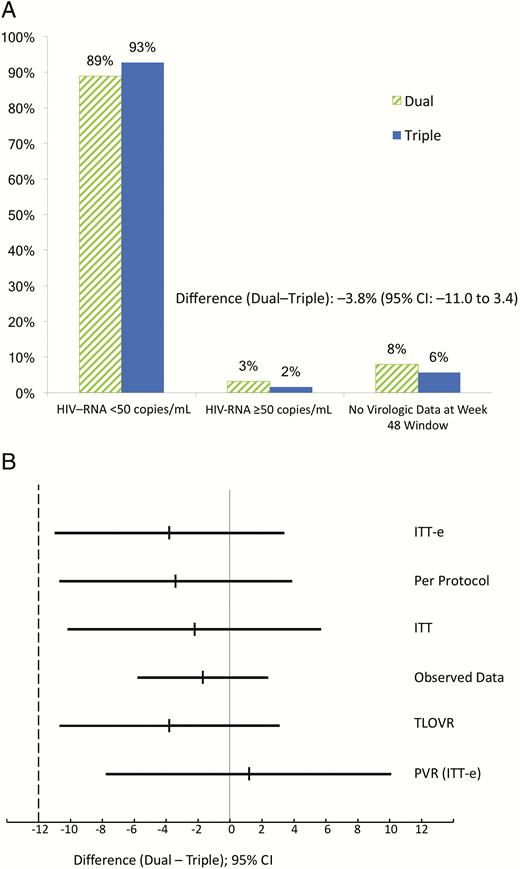
Outcome at 48 weeks. A, Primary endpoint. Snapshot analysis in the intention-to-treat exposed (ITT-e) population. B, Secondary and sensitivity analyses (differences and 95% confidence intervals). Per protocol, snapshot analysis in the per protocol population; ITT, snapshot analysis in the ITT population; observed data, excluding discontinuations due to nonvirological reasons; PVR (no virological failure, no blips) in the ITT-e population. Abbreviations: CI, confidence interval; HIV, human immunodeficiency virus; ITT-e, intention-to-treat exposed; PVR, persistent virological response; TLOVR, time to loss of virological response (Food and Drug Administration algorithm).
Four participants in the dual-therapy group and 2 in the triple-therapy group developed protocol-defined virological failure. Two participants in each arm had virological failure due to low-level, detectable viral load in the 48-week window (triple, 130 and 68; dual, 116 and 79 HIV-RNA copies/mL). One additional participant randomized to the dual-therapy group had a detectable viral load (80 copies/mL) at the baseline determination; he was discontinued 2 weeks later when failure was confirmed. The other participant considered a virological failure in the dual arm was discontinued at week 24 after confirmed virological rebound (988 and 259 copies/mL). The proportions of participants without virological failure in the observed data analysis were 96.6% and 98.3% (112/116 in dual and 114/116 in triple; difference, –1.7%; 95% CI, –5.8% to 2.4%).
There were 14 participants with viral load blips in the dual-therapy group and 17 in the triple-therapy group. Persistent virological response (defined as absence of protocol-defined virological failure or any blip; ITT-e population) was 85.7% (108/126) in the dual-therapy arm and 84.5% (104/123) in the triple-therapy arm (difference, 1.2%; 95% CI, –7.8 to 10.1; (Figure 2B, Supplementary Figure S1).
Three participants in the dual arm had a viral load >400 copies/mL during the study (2 of them with protocol-defined virological failure). Samples from 2 of the 3 participants could be amplified showing wild-type virus in both cases. Two participants had a viral load >400 copies/mL in the triple-therapy arm (none of them with protocol-defined virological failure). One sample from these 2 participants could be amplified showing the L10I, A71T, and L76W mutations in the protease gene, with the virus remaining fully susceptible to darunavir. None of the 3 available resistance tests showed the M184V mutation to lamivudine.
Most participants experienced at least 1 adverse event; 69.8% (88/126) in the dual-therapy arm and 75.6% (93/123) in the triple-therapy arm (Table 2), of whom 4.8% (6/126) and 4.9% (6/123), respectively, experienced serious adverse events (P = .97). Study drug discontinuations due to adverse events occurred in 0.8% (1/126) vs 1.6% (2/123; P = .55) in the dual- and triple-therapy arms, respectively.
Adverse Events Overview
| Event | Dual Therapy (n = 126) | Triple Therapy (n = 123) | P Value |
|---|---|---|---|
| Summary of AEs | |||
| Patients with ≥1 AE Total | 88 (70) 197 | 93 (76) 207 | .307 |
| Patients with ≥1 grade 2 to 4 AE Total | 15 (12) 17 | 18 (15) 30 | .525 |
| Patients with ≥1 serious AE Total | 6 (5) 6 | 6 (5) 7 | .966 |
| Discontinuation due to AEs | 1 (1)a | 2 (2)b | .547 |
| Deaths | 0 | 0 | NA |
| Adverse events that occurred in at least 5% of participants in either group | |||
| Respiratory: patients with ≥1 AE Total | 31 (25) 42 | 29 (24) 36 | .850 |
| Infections: patients with ≥1 AE Total | 22 (17) 26 | 18 (15) 23 | .544 |
| Digestive: patients with ≥1 AE Total | 18 (14) 22 | 22 (18) 27 | .439 |
| Muscular or skeletal: patients with ≥1 AE Total | 16 (13) 17 | 22 (18) 27 | .255 |
| Neuropsychiatric: patients with ≥1 AE Total | 12 (10) 15 | 12 (10) 15 | .950 |
| Metabolic: patients with ≥1 AE Total | 13 (10) 15 | 8 (7) 9 | .279 |
| Genitourinary: patients with ≥1 AE Total | 8 (6) 9 | 6 (5) 6 | .614 |
| General disorders: patients with ≥1 AE Total | 7 (6) 8 | 7 (6) 8 | .963 |
| Ear, nose, throat: patients with ≥1 AE Total | 7 (6) 7 | 8 (7) 8 | .753 |
| Oral cavity: patients with ≥1 AE Total | 3 (2) 5 | 7 (6) 7 | .184 |
| Event | Dual Therapy (n = 126) | Triple Therapy (n = 123) | P Value |
|---|---|---|---|
| Summary of AEs | |||
| Patients with ≥1 AE Total | 88 (70) 197 | 93 (76) 207 | .307 |
| Patients with ≥1 grade 2 to 4 AE Total | 15 (12) 17 | 18 (15) 30 | .525 |
| Patients with ≥1 serious AE Total | 6 (5) 6 | 6 (5) 7 | .966 |
| Discontinuation due to AEs | 1 (1)a | 2 (2)b | .547 |
| Deaths | 0 | 0 | NA |
| Adverse events that occurred in at least 5% of participants in either group | |||
| Respiratory: patients with ≥1 AE Total | 31 (25) 42 | 29 (24) 36 | .850 |
| Infections: patients with ≥1 AE Total | 22 (17) 26 | 18 (15) 23 | .544 |
| Digestive: patients with ≥1 AE Total | 18 (14) 22 | 22 (18) 27 | .439 |
| Muscular or skeletal: patients with ≥1 AE Total | 16 (13) 17 | 22 (18) 27 | .255 |
| Neuropsychiatric: patients with ≥1 AE Total | 12 (10) 15 | 12 (10) 15 | .950 |
| Metabolic: patients with ≥1 AE Total | 13 (10) 15 | 8 (7) 9 | .279 |
| Genitourinary: patients with ≥1 AE Total | 8 (6) 9 | 6 (5) 6 | .614 |
| General disorders: patients with ≥1 AE Total | 7 (6) 8 | 7 (6) 8 | .963 |
| Ear, nose, throat: patients with ≥1 AE Total | 7 (6) 7 | 8 (7) 8 | .753 |
| Oral cavity: patients with ≥1 AE Total | 3 (2) 5 | 7 (6) 7 | .184 |
Data are n (%). Dual therapy = switching to darunavir/r + lamivudine. Triple therapy = maintain triple therapy with darunavir/r + 2 nucleos(t)ide reverse transcriptase inhibitors.
Abbreviation: AE, adverse event.
aOne case of hyperlipidemia.
bOne case of diarrhea, one case of Hodgkin’s lymphoma.
Adverse Events Overview
| Event | Dual Therapy (n = 126) | Triple Therapy (n = 123) | P Value |
|---|---|---|---|
| Summary of AEs | |||
| Patients with ≥1 AE Total | 88 (70) 197 | 93 (76) 207 | .307 |
| Patients with ≥1 grade 2 to 4 AE Total | 15 (12) 17 | 18 (15) 30 | .525 |
| Patients with ≥1 serious AE Total | 6 (5) 6 | 6 (5) 7 | .966 |
| Discontinuation due to AEs | 1 (1)a | 2 (2)b | .547 |
| Deaths | 0 | 0 | NA |
| Adverse events that occurred in at least 5% of participants in either group | |||
| Respiratory: patients with ≥1 AE Total | 31 (25) 42 | 29 (24) 36 | .850 |
| Infections: patients with ≥1 AE Total | 22 (17) 26 | 18 (15) 23 | .544 |
| Digestive: patients with ≥1 AE Total | 18 (14) 22 | 22 (18) 27 | .439 |
| Muscular or skeletal: patients with ≥1 AE Total | 16 (13) 17 | 22 (18) 27 | .255 |
| Neuropsychiatric: patients with ≥1 AE Total | 12 (10) 15 | 12 (10) 15 | .950 |
| Metabolic: patients with ≥1 AE Total | 13 (10) 15 | 8 (7) 9 | .279 |
| Genitourinary: patients with ≥1 AE Total | 8 (6) 9 | 6 (5) 6 | .614 |
| General disorders: patients with ≥1 AE Total | 7 (6) 8 | 7 (6) 8 | .963 |
| Ear, nose, throat: patients with ≥1 AE Total | 7 (6) 7 | 8 (7) 8 | .753 |
| Oral cavity: patients with ≥1 AE Total | 3 (2) 5 | 7 (6) 7 | .184 |
| Event | Dual Therapy (n = 126) | Triple Therapy (n = 123) | P Value |
|---|---|---|---|
| Summary of AEs | |||
| Patients with ≥1 AE Total | 88 (70) 197 | 93 (76) 207 | .307 |
| Patients with ≥1 grade 2 to 4 AE Total | 15 (12) 17 | 18 (15) 30 | .525 |
| Patients with ≥1 serious AE Total | 6 (5) 6 | 6 (5) 7 | .966 |
| Discontinuation due to AEs | 1 (1)a | 2 (2)b | .547 |
| Deaths | 0 | 0 | NA |
| Adverse events that occurred in at least 5% of participants in either group | |||
| Respiratory: patients with ≥1 AE Total | 31 (25) 42 | 29 (24) 36 | .850 |
| Infections: patients with ≥1 AE Total | 22 (17) 26 | 18 (15) 23 | .544 |
| Digestive: patients with ≥1 AE Total | 18 (14) 22 | 22 (18) 27 | .439 |
| Muscular or skeletal: patients with ≥1 AE Total | 16 (13) 17 | 22 (18) 27 | .255 |
| Neuropsychiatric: patients with ≥1 AE Total | 12 (10) 15 | 12 (10) 15 | .950 |
| Metabolic: patients with ≥1 AE Total | 13 (10) 15 | 8 (7) 9 | .279 |
| Genitourinary: patients with ≥1 AE Total | 8 (6) 9 | 6 (5) 6 | .614 |
| General disorders: patients with ≥1 AE Total | 7 (6) 8 | 7 (6) 8 | .963 |
| Ear, nose, throat: patients with ≥1 AE Total | 7 (6) 7 | 8 (7) 8 | .753 |
| Oral cavity: patients with ≥1 AE Total | 3 (2) 5 | 7 (6) 7 | .184 |
Data are n (%). Dual therapy = switching to darunavir/r + lamivudine. Triple therapy = maintain triple therapy with darunavir/r + 2 nucleos(t)ide reverse transcriptase inhibitors.
Abbreviation: AE, adverse event.
aOne case of hyperlipidemia.
bOne case of diarrhea, one case of Hodgkin’s lymphoma.
The most frequent adverse events, occurring in at least 5% of participants, were respiratory infections and gastrointestinal and muscular or skeletal events; these were comparable between arms (Table 2). No deaths or AIDS-defining events occurred during the trial. Numbers of grade 3 or 4 laboratory adverse events were small and comparable in both arms (Table 3).
Laboratory Abnormalities: Grade 3 or 4
| Laboratory Abnormality | Dual Therapy (n = 126) | Triple Therapy (n = 123) | P Value |
|---|---|---|---|
| Any grade 3 or 4 laboratory abnormality | 4 (3) | 4 (3) | .972 |
| Alanine aminotransferase (>5 ×ULN) | 0 | 0 | |
| Aspartate aminotransferase (>5 ×ULN) | 0 | 1 (1) | .310 |
| Total cholesterol (>300 mg/dL) | 4 (3) | 3 (2) | .725 |
| Triglycerides (>750 mg/dL) | 0 | 0 |
| Laboratory Abnormality | Dual Therapy (n = 126) | Triple Therapy (n = 123) | P Value |
|---|---|---|---|
| Any grade 3 or 4 laboratory abnormality | 4 (3) | 4 (3) | .972 |
| Alanine aminotransferase (>5 ×ULN) | 0 | 0 | |
| Aspartate aminotransferase (>5 ×ULN) | 0 | 1 (1) | .310 |
| Total cholesterol (>300 mg/dL) | 4 (3) | 3 (2) | .725 |
| Triglycerides (>750 mg/dL) | 0 | 0 |
Data are number of patients with 1 or more adverse events (%).. Dual therapy = switching to darunavir/r + lamivudine. Triple therapy = maintain triple therapy with darunavir/r + 2 nucleos(t)ide reverse transcriptase inhibitors.
Abbreviation: ULN, upper limit of normal.
Laboratory Abnormalities: Grade 3 or 4
| Laboratory Abnormality | Dual Therapy (n = 126) | Triple Therapy (n = 123) | P Value |
|---|---|---|---|
| Any grade 3 or 4 laboratory abnormality | 4 (3) | 4 (3) | .972 |
| Alanine aminotransferase (>5 ×ULN) | 0 | 0 | |
| Aspartate aminotransferase (>5 ×ULN) | 0 | 1 (1) | .310 |
| Total cholesterol (>300 mg/dL) | 4 (3) | 3 (2) | .725 |
| Triglycerides (>750 mg/dL) | 0 | 0 |
| Laboratory Abnormality | Dual Therapy (n = 126) | Triple Therapy (n = 123) | P Value |
|---|---|---|---|
| Any grade 3 or 4 laboratory abnormality | 4 (3) | 4 (3) | .972 |
| Alanine aminotransferase (>5 ×ULN) | 0 | 0 | |
| Aspartate aminotransferase (>5 ×ULN) | 0 | 1 (1) | .310 |
| Total cholesterol (>300 mg/dL) | 4 (3) | 3 (2) | .725 |
| Triglycerides (>750 mg/dL) | 0 | 0 |
Data are number of patients with 1 or more adverse events (%).. Dual therapy = switching to darunavir/r + lamivudine. Triple therapy = maintain triple therapy with darunavir/r + 2 nucleos(t)ide reverse transcriptase inhibitors.
Abbreviation: ULN, upper limit of normal.
Switching to dual therapy was associated with a significant increase in total, low-density lipoprotein (LDL) and high-density lipoprotein (HDL) cholesterol, but not in the total-to-HDL cholesterol ratio, in those participants who received tenofovir disoproxil fumarate at baseline (Figure 3A). There was no significant change in lipids in participants who received abacavir at baseline (Figure 3A). A nonsignificant improvement in estimated creatinine clearance was observed in participants on tenofovir disoproxil fumarate therapy after switching to dual therapy compared to those who were maintained on triple therapy (Figure 3B).
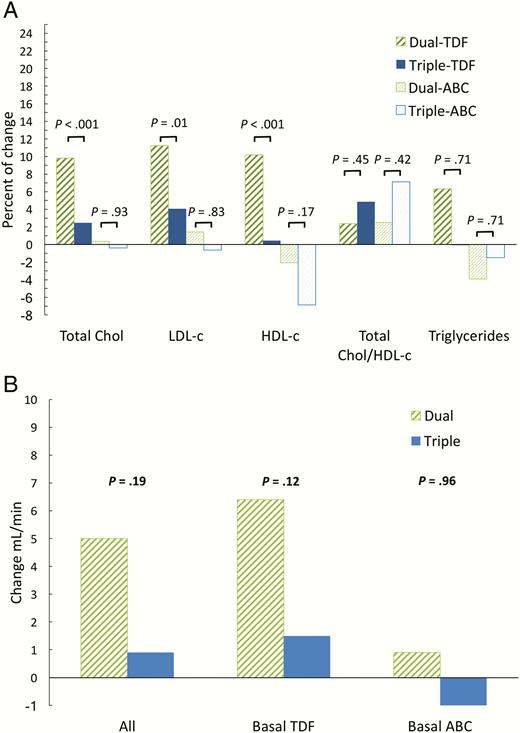
Changes at 48 weeks. A, Percent change (mean) in total cholesterol, cholesterol fractions, and triglycerides by nucleos(t)ide at baseline. B, Change in estimated creatinine clearance (mean) (mL/min; Cockroft–Gault equation) according to the nucleos(t)ides at baseline. Abbreviations: ABC, Abacavir; Chol, cholesterol; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; TDF, tenofovir disoproxil fumarate.
DISCUSSION
The results of the DUAL trial indicate that the combination of darunavir/ritonavir and lamivudine is as efficacious as triple therapy with darunavir/ritonavir and 2 nucleos(t)ides for maintenance of virological suppression in HIV-infected participants without resistance to darunavir or lamivudine. Our primary analysis and 5 sensitivity analyses support the noninferiority of the dual-therapy strategy. In the primary analysis, the small and nonsignificant difference in favor of triple therapy (3.8%) in the proportion of participants with HIV-RNA <50 copies/mL at week 48 was motivated mainly by nonvirological reasons.
In noninferiority studies it is important to include a control arm that represents standard clinical practice and to show that the active control arm performs in the new trial as well as in prior trials. This is the case with DUAL. The combination of tenofovir disoproxil fumarate/emtricitabine or abacavir/lamivudine and darunavir/ritonavir is a standard regimen in clinical practice and was maintained at a high rate (93%) of virological suppression at week 48. This rate of suppression is even higher than the one expected in our original assumptions and strengthens our claim of noninferiority of the dual-therapy combination.
We were interested in determining whether the dual combination could ensure a maintained viral load suppression as well as triple therapy. This is a relevant issue because other simplification strategies such as boosted protease inhibitor monotherapy had a higher risk of episodes of low-level viremia. In DUAL we analyzed the proportion of participants who had suppressed viremia at all visits that included a viral load measurement. The proportion of participants with persistent virological response was not lower for the dual-therapy arm (85.7%) than for the triple-therapy arm (84.5%). In addition, we did not find differences in the proportion of participants with blips. We believe these are important results that challenge the need for using a nucleos(t)ide other than lamivudine in participants treated with fully active darunavir maintaining virological suppression. Similar results were seen in the OLE clinical trial [5].
Virological failure was very rare in both arms. Only 5 participants had viral rebounds >400 HIV-RNA copies/mL and none had virological failure associated with the development of the M184V mutation. Our results add to prior studies of dual therapy with a boosted protease inhibitor and lamivudine that have not shown a higher risk of developing lamivudine resistance [2–5].
Both treatments were well tolerated, with a very low rate of discontinuation due to adverse events or intolerance. This is an expected result because the DUAL trial already included participants who were tolerating darunavir/ritonavir and 2 nucleos(t)ides. In a 48-week trial it is not possible to estimate the long-term safety advantage of using only lamivudine as a single nucleoside. We believe that in the long term it is very likely that avoiding tenofovir disoproxil fumarate or abacavir would translate to better safety outcomes since both drugs have been associated with short- and long-term adverse events [12–14].
We found small differences between treatment groups in safety parameters. Total, LDL and HDL cholesterol significantly increased in the dual-therapy arm compared to the triple-therapy arm but with no significant difference in the total cholesterol-to-HDL ratio. These results were, as expected, more pronounced in participants who stopped tenofovir. It is well known that tenofovir has a lipid-lowering effect [15]. Although differences were small, we found a nonstatistically significant increase in the estimated creatinine clearance in participants who stopped tenofovir in the dual-therapy arm.
One important limitation of our study results is that noninferiority of the dual-therapy strategy can be proven with only a margin of 12%. The same noninferiority margin has been used in prior registrational trials in which a switch in strategies in virologically suppressed patients was evaluated [16–18]. However, since November 2015, after the DUAL trial had finished enrollment, FDA has recommended that trials that evaluate switch strategies should select a primary endpoint that is focused not on the proportions of suppressed patients but on the rate of virological failure in each treatment group. For this different endpoint, the FDA advises use of a noninferiority margin of 4% [8]. DUAL does not have enough statistical power to prove noninferiority with this new endpoint. However, virological failures were very rare in DUAL and not associated with the development of resistance.
A total of 1051 patients have been included in DUAL, OLE, SALT, and ATLAS-M [3–5]. In each trial the dual-therapy combination of a boosted protease inhibitor and lamivudine was noninferior compared with triple therapy with a boosted protease inhibitor and 2 nucleos(t)ides using the same noninferiority margin of 12%. It would be interesting to perform a metaanalysis of these 4 trials to determine if the dual-therapy strategy is also noninferior to triple therapy using the current endpoint recommended by FDA.
The other limitation of DUAL is its open-label design, which could lead to bias. However, one advantage is that the daily number of pills did not change after randomization because the only intervention was to change coformulated nucleos(t)ides for lamivudine.
We believe that the results of DUAL, OLE, SALT, and ATLAS-M strongly support the strategy of using a fully active boosted protease inhibitor and lamivudine as a therapeutic option in virologically suppressed patients. Currently, results of DUAL are probably the most relevant of all of these trials because darunavir is the only protease inhibitor recommended as a preferred or alternative regimen in all international expert guidelines [6, 7, 19]. One additional benefit of the dual-therapy strategy is a better cost-efficacy profile since the cost of lamivudine, a generic drug, is lower than the cost of any combination of 2 nucleosides.
In summary, results of the DUAL study support the use of the combination of darunavir/ritonavir and lamivudine as a switch strategy in HIV-1–infected patients who are virologically suppressed while receiving darunavir/ritonavir and 2 nucleos(t)ides. This strategy has the benefit of using darunavir, a boosted protease inhibitor with a good efficacy and safety profile, and lamivudine, a nucleoside with an excellent long-term safety profile.
Supplementary Data
Supplementary materials are available at Clinical Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Footnotes
Presented in part: International Congress of Drug Therapy in HIV Infection. Glasgow, Scotland, 23–26 October 2016. Abstract O331.
Notes
Author contributions. J. A., F. P., and E. R. reviewed, analyzed, and interpreted the data and designed and oversaw the study. J. A. and F. P. wrote the first draft. B. A. performed the statistical analysis, amended the draft report, and approved the final version. All the other authors enrolled patients, acquired data, amended the draft report, and approved the final version.
Acknowledgments. We thank the DUAL-GESIDA 8014-RIS-EST45 trial study participants for participation in the study.
Disclaimer. Janssen did not participate in the study design, management of the trial, or writing of the report but received the final draft of the manuscript for review and comments. All operational aspects of the study, including study design, monitoring, data collection, data analysis, and writing of the report were managed by Fundación SEIMC-Gesida. All authors had full access to all the data in the study and are responsible for the veracity and completeness of the data reported. The corresponding authors had final responsibility for the decision to submit for publication.
Financial support. This work was supported in part by research grants from Red Temática Cooperativa de Investigación en SIDA G03/173 (RIS-EST45). Fundación SEIMC-Gesida sponsored the study. Janssen Pharmaceutical and Red Española de Investigación en Sida (RIS) provided trial funding.
Potential conflicts of interest. All authors: F.P. has received personal fees from Abbvie, Gilead, Janssen, MSD and Viiv, outside the submitted work. E.R. has received personal fees from Abbvie, BMS, Gilead, Janssen, MSD and Viiv, outside the submitted work. M.L. has received personal fees from Gilead, Janssen, MSD and ViiV, outside the submitted work. I.P. has received personal fees from Viiv, Janssen, Gilead, MSD, outside the submitted work. J.S. has received personal fees from ViiV, Janssen, Gilead, MSD, and BMS, outside the submitted work. J.A.I. has received research grants from Abbvie, BMS, Gobierno Vasco, FIPSE and FISS; personal fees from Abbvie and Janssen, outside the submited work. A.P. has received personal fees from Abbvie, Gilead, Janssen and Viiv, outside the submitted work. P.D. has received honoraria for lectures or advisory boards and his institution research grant from ViiV Healthcare, Gilead Sciences, MSD and Janssen. R.P. has received personal fees from Gilead, Janssen, MSD and ViiV, outisde the submitted work. M.C. has received personal fees from Abbvie, Gilead, Janssen, MSD and Viiv, outside the submitted work. A.C. has received personal fees from Gilead, Janssen, MSD and Viiv, outside the submitted work. F.R. has received personal fees from Gilead, Janssen, MSD and ViiV, outside the submitted work. M.J.T. has received personal fees from Gilead, BMS and MSD, outside the submitted work. P.R. has received personal fees from Gilead, Janssen, MSD and ViiV, outside the submitted work. P.B. has received personal fees from Gilead, Janssen, MSD and Viiv, outside the submitted work. H.K. has received personal fees from Abbvie, BMS, Gilead, Janssen, MSD and ViiV, outside the submitted work. A.R. has received grants and personal fees from BMS, Gilead, VIIV, MSD, Jansen, Abbvie, outside the submitted work. B.A. no reported conflicts of interest. M.Y. no reported conflicts of interest. J.R.A. has received personal fees from Viiv, Janssen, Gilead, MSD, outside the submitted work. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
DUAL-GESIDA 8014-RIS-EST45 trial study team include the following:
H. Univ. La Paz: JR Arribas (protocol co-chair), R. Montejano, J.J. González-García, M.L. Montes, J.I. Bernardino, I. Pérez-Valero, J.M. Castro, M. Mayoral; H. Univ. 12 de Octubre: F. Pulido (protocol cochair), M. Lagarde, O. Bisbal, M. Matarranz, A. Hernando, L. Domínguez-Domínguez, R. Rubio; H. Univ. Vall d’Hebron: E. Ribera (protocol cochair), A. Curran, J. Navarro, J. Burgos, I. Ocaña, V. Falco; H. Clín. Univ. Virgen de la Victoria: J. Santos, R. Palacios, I. Pérez, C.M. González-Doménech; H. Univ. Donostia: M. Ibarguren, M.A. Goenaga, F. Rodriguez-Arrondo, M.A. von Wichmann, X. Kortajarena, M.P. Carmona; H. Son Llatzer: A. Payeras, M. Raya, A. Salom; H. Santa Creu i Sant Pau: P. Domingo, M.M. Gutierrez, M.G. Mateo, M.A. Sambeat; H. Príncipe de Asturias: J. Sanz, J. de Miguel, E. Casas, A. Arranz; H. Univ. Severo Ochoa: M. Cervero, R. Torres; H. Infanta Elena: F.J. Rodríguez-Gómez, J.M. Fajardó, F.J. Martínez-Marcos, M.D. Merino, M. Raffo, I. Suárez-Lozano; H. Clínico: M.J. Tellez, J. Vergas, V. Estrada; H. Infanta Leonor: P. Ryan, J. Troya, G. Cuevas, V. Diez-Viñas, T. Talaván, F.J. Solís; H. de Mataró: P. Barrufet, L. Force; H. del Mar: H. Knobel, A. González, E. Lerma, J. Villar; H. Reina Sofía: A. Rivero, A. Camacho, I. Machuca, T. Brieva, A. Rivero-Juárez; H. Clinic i Provincial: J.M. Gatell, J. Rojas; H. Univ. de Bellvitge: D. Podzamczer; E. Van Den Eynde, L. Acerete, A. Navarro, M.S. Diyacovo; H. Virgen de las Nieves: J. Pasquau, C. García; H. Fundación de Alcorcón: J.E. Losa, C.A.J. Henriquez; H. General Universitario de Alicante: J. Portilla, L. Giner, I. Portilla, M. Pampliega, V. Boix, E. Merino, S. Reus, D. Torrus; H. Germans Trias i Pujol: B. Clotet, E. Negredo, A. Chamarro, P. Corbasi; H. Univ. Miguel Servet: D. Gil-Pérez, P. Arazo; H. Univ. Gregorio Marañón: F. Parras, M. Ramírez, I. Gutiérrez-Cuéllar; H. Son Espases: M. Riera, L. Gil-Alonso, H. Vilchez; and Fundación SEIMC-GESIDA: M. Yllescas, B. Alejos, E. Aznar, H. Esteban, P. González, S. González, M. de Miguel.
References
Author notes
F. P. and J. R. A. contributed equally to this manuscript.


{kind=link}
{kind=link}
{kind=link}