





Abstract
Selective methane (CH4) oxidation to methanol (CH3OH) is a highly desirable reaction; however, it remains challenging due to the high stability of CH4 and the facile over-oxidation of the products. Recently, Cu-based catalysts have been reported as promising candidates for continuous CH3OH production from CH4. Here, we demonstrated that supported Cu-introduced polyoxometalate (POM) cesium salts catalytically produced CH3OH at 450 °C. Notably, monocopper-introduced POM cesium salts (Cs-Cu1/SiO2) selectively produced CH3OH (>75%), maintaining a stable CH3OH yield over a 24 h time on stream. In contrast, CH3OH was prone to over-oxidation into carbon monoxide (CO) and carbon dioxide (CO2) over Cu(OAc)2/SiO2 or monocopper-introduced POM tetra-n-butylammonium salts (TBA-Cu1/SiO2), due to the aggregation of CuO species. Cu K-edge XANES and Raman spectroscopy revealed that the supported POM cesium salts functioned as molecular templates and potentially prevented the aggregation of Cu active sites.
Selective CH4 oxidation to CH3OH has garnered significant attention for synthesizing chemical raw materials from natural gas over the last decades.1 The current industrial production of CH3OH from CH4 involves several reaction steps via syngas, requiring high-temperature and high-pressure conditions. Although CH4 oxidation to CH3OH is known to be one of the most challenging reactions, several approaches have been reported to achieve selective CH4 oxidation to CH3OH using zeolites,2 metal organic frameworks,3 or polyoxometalates (POMs)4 as catalysts. In particular, the chemical looping process using Cu-zeolite catalysts provides selective CH3OH production5; however, the multistep reactions and the low production rate of CH3OH prevent its practical application in an industrial process. Nowadays, continuous flow reactions using abundant resources of O2 as an oxidant are highly desirable for selective CH4 oxidation to CH3OH,6 but the thermal stability of catalysts and the aggregation of active sites during the reaction remain significant challenges.
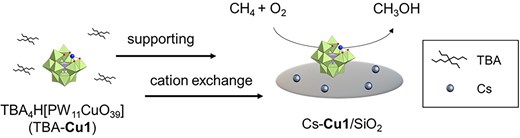
POMs are molecular metal oxide clusters, which can function as molecular templates of catalytic active sites. We have synthesized a wide variety of POM tetra-n-butylammonium (TBA) salts with precisely designed multinuclear structures in organic solvents.7 For example, we designed dinuclear iron or copper cores sandwiched between 2 lacunary POMs of [γ-SiW10O36]8−,8,9 as well as several multinuclear copper-containing ring-shaped POMs.10,11 Recently, we reported selective CH4 oxidation to formaldehyde (HCHO) and CO over thermally stable FeOx subnanoclusters using dinuclear iron-containing POM TBA salts (TBA-Fe2) as precursors of highly dispersed SiO2-supported Fe catalysts.8 In this case, TBA-Fe2 was readily decomposed under high-temperature conditions of CH4 oxidation. POM cesium salts are known for their thermal stability.12 Thus, we have established a preparation method for supported POM cesium salts13 to provide thermal stability to POM-based catalysts. The prepared supported POM cesium salts maintained the well-defined active site structure even under an oxidative atmosphere. Based on this background, we here examined the selective CH4 oxidation to CH3OH using supported Cu-introduced POM cesium salts (Fig. 1). Upon screening POMs with various numbers of Cu cores, we found that a monocopper-introduced POM (Cu1; [PW11CuO39]5−) as a precursor of supported POM cesium salts (Cs-Cu1/SiO2) exhibited high CH3OH selectivity and high durability. In addition, Cu K-edge XANES and Raman spectroscopy revealed the active site structure for the CH4 oxidation.
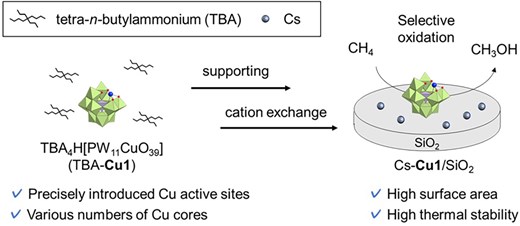
Concept of this study.
A series of Cu-introduced POM TBA salts was synthesized according to literature procedures (anionic structure of each Cu-introduced POM is shown in Supplementary Fig. S1).9–11,14 Supported Cu-introduced POM cesium salts were prepared by stepwise support on SiO2 and cation exchange from TBA+ to Cs+ using cesium trifluoromethanesulfonate (CsOTf) as a cesium source.13 Cs-Cs1/SiO2 was supposed to be highly dispersed on SiO2 without decomposition of the POM structure (Supplementary Fig. S2). Additionally, Cs-Cu2/SiO2 was especially characterized by XRD, Raman spectroscopy, Cu K-edge XANES, and W LIII-edge XANES (Supplementary Figs. S3 to S6) to reveal that precisely designed the Cu-based active sites incorporated in POMs were maintained after support and cation exchange (see the Supplementary Information).
Catalytic CH4 oxidation reactions were conducted in a fixed-bed flow-type reactor at 450 °C, with water vapor flow to extract CH3OH from the active sites.15,16 To begin with, the optimal number of Cu cores introduced in POMs was investigated to achieve selective CH3OH production. Supported POM cesium salts with 8, 12, or 16 Cu cores (Cs-Cu8/SiO2, Cs-Cu12/SiO2, Cs-Cu16/SiO2, respectively) tended to promote the over-oxidation of CH4, resulting in low CH3OH selectivity (Table 1, entries 4 to 6). This result is consistent with the previous reports that aggregated CuO species do not promote selective CH3OH production.17 Supported POM cesium salts with 1, 2, or 4 Cu cores (Cs-Cu1/SiO2, Cs-Cu2/SiO2, Cs-Cu4/SiO2, respectively) exhibited relatively low CO2 yield (Table 1, entries 1 to 3), suggesting that tetranuclear or lower Cu active sites are favorable for the selective oxidation of CH4. Especially, Cs-Cu1/SiO2 exhibited the highest CH3OH selectivity (>75%) with a high CH3OH turnover frequency (TOF) of 0.62 mol h−1 as its initial activity. Also, the hourly CH3OH productivity (21 μmol g−1-cat h−1) of the catalytic system developed in this study is not inferior to those of the chemical looping systems using Cu zeolites reported so far, as summarized in Supplementary Table S2.
Optimization of the number of Cu-introduced in POM templates.
| Entry | Catalyst | Cu [μmol] | Conv. [%] | Yield [μmol h−1] | CH3OH TOF [mol h−1] | |||
|---|---|---|---|---|---|---|---|---|
| CH4 | CH3OH | HCHO | CO | CO2 | ||||
| 1a | Cs-Cu1/SiO2 | 1.7 | 0.003 | 1.0 | 0.03 | <0.14 | <0.17 | 0.62 |
| 2 | Cs-Cu2/SiO2 | 2.7 | 0.007 | 0.92 | 0.16 | 0.37 | 1.8 | 0.34 |
| 3 | Cs-Cu4/SiO2 | 2.4 | 0.003 | 1.2 | 0.13 | <0.14 | <0.05 | 0.52 |
| 4 | Cs-Cu8/SiO2 | 3.5 | 0.014 | 1.4 | 0.08 | 3.2 | 2.4 | 0.40 |
| 5 | Cs-Cu12/SiO2 | 3.2 | 0.024 | 1.6 | 0.27 | 5.1 | 5.2 | 0.51 |
| 6 | Cs-Cu16/SiO2 | 2.4 | 0.043 | 1.9 | 0.33 | 9.9 | 9.1 | 0.82 |
| Entry | Catalyst | Cu [μmol] | Conv. [%] | Yield [μmol h−1] | CH3OH TOF [mol h−1] | |||
|---|---|---|---|---|---|---|---|---|
| CH4 | CH3OH | HCHO | CO | CO2 | ||||
| 1a | Cs-Cu1/SiO2 | 1.7 | 0.003 | 1.0 | 0.03 | <0.14 | <0.17 | 0.62 |
| 2 | Cs-Cu2/SiO2 | 2.7 | 0.007 | 0.92 | 0.16 | 0.37 | 1.8 | 0.34 |
| 3 | Cs-Cu4/SiO2 | 2.4 | 0.003 | 1.2 | 0.13 | <0.14 | <0.05 | 0.52 |
| 4 | Cs-Cu8/SiO2 | 3.5 | 0.014 | 1.4 | 0.08 | 3.2 | 2.4 | 0.40 |
| 5 | Cs-Cu12/SiO2 | 3.2 | 0.024 | 1.6 | 0.27 | 5.1 | 5.2 | 0.51 |
| 6 | Cs-Cu16/SiO2 | 2.4 | 0.043 | 1.9 | 0.33 | 9.9 | 9.1 | 0.82 |
Reaction conditions: CH4/O2/Ar = 20/0.1/9.9, total flow rate of 30 mL min−1, 450 °C, water bubbling (0 °C, 0.6 kPa), catalyst 100 mg, time on stream = 1 h.
aCatalyst 50 mg.
Optimization of the number of Cu-introduced in POM templates.
| Entry | Catalyst | Cu [μmol] | Conv. [%] | Yield [μmol h−1] | CH3OH TOF [mol h−1] | |||
|---|---|---|---|---|---|---|---|---|
| CH4 | CH3OH | HCHO | CO | CO2 | ||||
| 1a | Cs-Cu1/SiO2 | 1.7 | 0.003 | 1.0 | 0.03 | <0.14 | <0.17 | 0.62 |
| 2 | Cs-Cu2/SiO2 | 2.7 | 0.007 | 0.92 | 0.16 | 0.37 | 1.8 | 0.34 |
| 3 | Cs-Cu4/SiO2 | 2.4 | 0.003 | 1.2 | 0.13 | <0.14 | <0.05 | 0.52 |
| 4 | Cs-Cu8/SiO2 | 3.5 | 0.014 | 1.4 | 0.08 | 3.2 | 2.4 | 0.40 |
| 5 | Cs-Cu12/SiO2 | 3.2 | 0.024 | 1.6 | 0.27 | 5.1 | 5.2 | 0.51 |
| 6 | Cs-Cu16/SiO2 | 2.4 | 0.043 | 1.9 | 0.33 | 9.9 | 9.1 | 0.82 |
| Entry | Catalyst | Cu [μmol] | Conv. [%] | Yield [μmol h−1] | CH3OH TOF [mol h−1] | |||
|---|---|---|---|---|---|---|---|---|
| CH4 | CH3OH | HCHO | CO | CO2 | ||||
| 1a | Cs-Cu1/SiO2 | 1.7 | 0.003 | 1.0 | 0.03 | <0.14 | <0.17 | 0.62 |
| 2 | Cs-Cu2/SiO2 | 2.7 | 0.007 | 0.92 | 0.16 | 0.37 | 1.8 | 0.34 |
| 3 | Cs-Cu4/SiO2 | 2.4 | 0.003 | 1.2 | 0.13 | <0.14 | <0.05 | 0.52 |
| 4 | Cs-Cu8/SiO2 | 3.5 | 0.014 | 1.4 | 0.08 | 3.2 | 2.4 | 0.40 |
| 5 | Cs-Cu12/SiO2 | 3.2 | 0.024 | 1.6 | 0.27 | 5.1 | 5.2 | 0.51 |
| 6 | Cs-Cu16/SiO2 | 2.4 | 0.043 | 1.9 | 0.33 | 9.9 | 9.1 | 0.82 |
Reaction conditions: CH4/O2/Ar = 20/0.1/9.9, total flow rate of 30 mL min−1, 450 °C, water bubbling (0 °C, 0.6 kPa), catalyst 100 mg, time on stream = 1 h.
aCatalyst 50 mg.
Using Cs-Cu1/SiO2 as a model catalyst, kinetic studies of the CH4 consumption rate (reaction rate) were performed. Based on the kinetic analysis, the CH4 consumption rate exhibited approximately first-order dependence on the amount of Cu in the catalysts (Supplementary Fig. S7a). The product distribution is shown in Supplementary Table S3, suggesting that CH3OH may serve as an intermediate in CH4 oxidation. A positive CH4 partial pressure dependence (Supplementary Fig. S7b) and almost no dependence on O2 partial pressure were also observed (Supplementary Fig. S7c), which is consistent with previous reports.16 The low dependence of the CH4 consumption rate on O2 partial pressure indicated that the reoxidation step of the catalyst was not the rate-limiting step. The temperature dependence of CH4 conversion and product yields was investigated in the range of 350 °C to 500 °C (Supplementary Fig. S7d).
The highest yield of CH3OH was obtained at 450 °C, while over-oxidation occurred more readily at 500 °C. Tao et al. reviewed a series of CH4 oxidation to CH3OH over Cu zeolites and demonstrates that temperatures of approximately 450 °C are required for the formation of Cu active species.18
Next, catalytic activities of various Cu-based supported catalysts were compared. CH3OH TOF was calculated based on the loading amount of Cu in each catalyst (Supplementary Table S1). As expected from literature,17 bulk CuO promoted the complete oxidation of CH4 to CO2 (Table 2, entry 1). Similarly, over-oxidation to CO and CO2 occurred on Cu(OAc)2/SiO2 (OAc = acetate) and CsOTf–Cu(OAc)2/SiO2 (Table 2, entries 4 and 5). Although the XRD patterns of Cu(OAc)2/SiO2 show no obvious peaks (Supplementary Fig. S8a), the peak intensities in the Raman spectra that can be attributed to CuO19 were increased after 1 h of the reaction (Cu(OAc)2/SiO2–1 h) (Supplementary Fig. S8b). High CO and CO2 yields were also observed on TBA-Cu1/SiO2 (Table 2, entry 2). According to our previous report,8 TBA salts of Cu1 were also assumed to decompose into CuOx subnanoclusters, possibly promoting the sequential oxidation of products. Indeed, both the XRD patterns and the Raman spectra of TBA-Cu1/SiO2 after the reaction (TBA-Cu1/SiO2–1 h) revealed the formation of WO3, which is the decomposed product of the Cu1 anion structure (Supplementary Fig. S9).
Control experiments of various Cu-introduced supported catalysts.
| Entry | Catalyst | Cu [μmol] | Conv. [%] | Yield [μmol h−1] | CH3OH TOF [mol h−1] | |||
|---|---|---|---|---|---|---|---|---|
| CH4 | CH3OH | HCHO | CO | CO2 | ||||
| 1a | CuO | 50 | 0.082 | 0.80 | ND | 0.40 | 40 | 0.02 |
| 2 | TBA-Cu1/SiO2 | 2.7 | 0.016 | 0.87 | 0.35 | 4.1 | 2.4 | 0.32 |
| 3b | Cs-Cu1/SiO2 | 1.7 | 0.003 | 1.0 | 0.03 | <0.14 | <0.17 | 0.62 |
| 4 | Cu(OAc)2/SiO2 | 1.6 | 0.020 | 0.47 | 0.42 | 3.2 | 5.7 | 0.29 |
| 5 | CsOTf–Cu(OAc)2/SiO2 | 2.0 | 0.021 | 1.1 | 0.16 | 3.3 | 5.9 | 0.57 |
| 6 | Cs-SiW10/SiO2 | … | <0.001 | 0.12 | 0.03 | ND | 0.10 | … |
| 7 | Cu(OAc)2–Cs-SiW10/SiO2 | 1.8 | 0.006 | 1.1 | 0.09 | <0.27 | 1.4 | 0.61 |
| Entry | Catalyst | Cu [μmol] | Conv. [%] | Yield [μmol h−1] | CH3OH TOF [mol h−1] | |||
|---|---|---|---|---|---|---|---|---|
| CH4 | CH3OH | HCHO | CO | CO2 | ||||
| 1a | CuO | 50 | 0.082 | 0.80 | ND | 0.40 | 40 | 0.02 |
| 2 | TBA-Cu1/SiO2 | 2.7 | 0.016 | 0.87 | 0.35 | 4.1 | 2.4 | 0.32 |
| 3b | Cs-Cu1/SiO2 | 1.7 | 0.003 | 1.0 | 0.03 | <0.14 | <0.17 | 0.62 |
| 4 | Cu(OAc)2/SiO2 | 1.6 | 0.020 | 0.47 | 0.42 | 3.2 | 5.7 | 0.29 |
| 5 | CsOTf–Cu(OAc)2/SiO2 | 2.0 | 0.021 | 1.1 | 0.16 | 3.3 | 5.9 | 0.57 |
| 6 | Cs-SiW10/SiO2 | … | <0.001 | 0.12 | 0.03 | ND | 0.10 | … |
| 7 | Cu(OAc)2–Cs-SiW10/SiO2 | 1.8 | 0.006 | 1.1 | 0.09 | <0.27 | 1.4 | 0.61 |
Reaction conditions: CH4/O2/Ar = 20/0.1/9.9, total flow rate of 30 mL min−1, 450 °C, water bubbling (0 °C, 0.6 kPa), catalyst 100 mg, time on stream = 1 h.
aCatalyst 4 mg.
bCatalyst 50 mg.
Control experiments of various Cu-introduced supported catalysts.
| Entry | Catalyst | Cu [μmol] | Conv. [%] | Yield [μmol h−1] | CH3OH TOF [mol h−1] | |||
|---|---|---|---|---|---|---|---|---|
| CH4 | CH3OH | HCHO | CO | CO2 | ||||
| 1a | CuO | 50 | 0.082 | 0.80 | ND | 0.40 | 40 | 0.02 |
| 2 | TBA-Cu1/SiO2 | 2.7 | 0.016 | 0.87 | 0.35 | 4.1 | 2.4 | 0.32 |
| 3b | Cs-Cu1/SiO2 | 1.7 | 0.003 | 1.0 | 0.03 | <0.14 | <0.17 | 0.62 |
| 4 | Cu(OAc)2/SiO2 | 1.6 | 0.020 | 0.47 | 0.42 | 3.2 | 5.7 | 0.29 |
| 5 | CsOTf–Cu(OAc)2/SiO2 | 2.0 | 0.021 | 1.1 | 0.16 | 3.3 | 5.9 | 0.57 |
| 6 | Cs-SiW10/SiO2 | … | <0.001 | 0.12 | 0.03 | ND | 0.10 | … |
| 7 | Cu(OAc)2–Cs-SiW10/SiO2 | 1.8 | 0.006 | 1.1 | 0.09 | <0.27 | 1.4 | 0.61 |
| Entry | Catalyst | Cu [μmol] | Conv. [%] | Yield [μmol h−1] | CH3OH TOF [mol h−1] | |||
|---|---|---|---|---|---|---|---|---|
| CH4 | CH3OH | HCHO | CO | CO2 | ||||
| 1a | CuO | 50 | 0.082 | 0.80 | ND | 0.40 | 40 | 0.02 |
| 2 | TBA-Cu1/SiO2 | 2.7 | 0.016 | 0.87 | 0.35 | 4.1 | 2.4 | 0.32 |
| 3b | Cs-Cu1/SiO2 | 1.7 | 0.003 | 1.0 | 0.03 | <0.14 | <0.17 | 0.62 |
| 4 | Cu(OAc)2/SiO2 | 1.6 | 0.020 | 0.47 | 0.42 | 3.2 | 5.7 | 0.29 |
| 5 | CsOTf–Cu(OAc)2/SiO2 | 2.0 | 0.021 | 1.1 | 0.16 | 3.3 | 5.9 | 0.57 |
| 6 | Cs-SiW10/SiO2 | … | <0.001 | 0.12 | 0.03 | ND | 0.10 | … |
| 7 | Cu(OAc)2–Cs-SiW10/SiO2 | 1.8 | 0.006 | 1.1 | 0.09 | <0.27 | 1.4 | 0.61 |
Reaction conditions: CH4/O2/Ar = 20/0.1/9.9, total flow rate of 30 mL min−1, 450 °C, water bubbling (0 °C, 0.6 kPa), catalyst 100 mg, time on stream = 1 h.
aCatalyst 4 mg.
bCatalyst 50 mg.
The CH4 conversion was relatively low over Cs-SiW10/SiO2 which does not contain Cu sites (Table 2, entry 6), indicating that the molecular template of Cs-SiW10 is not the active species for CH4 oxidation. Cu(OAc)2–Cs-SiW10/SiO2, which is synthesized via stepwise addition of Cu(OAc)2 on Cs-SiW10/SiO2 followed by calcination, exhibited similar initial activity to Cs-Cu1/SiO2 except for its high CO2 yield (Table 2, entry 7). The high CO2 production is probably due to partially aggregated CuO species on SiO2; however, highly dispersed Cu species can catalyze the selective CH4 oxidation into CH3OH.
To reveal the difference between Cs-Cu1/SiO2 and Cu(OAc)2–Cs-SiW10/SiO2, a time course examination was conducted. Figure 2 shows the change in CH3OH TOF for 24 h of each catalyst. Cs-Cu1/SiO2 approximately maintained its catalytic activity for 24 h. Although the CO2 yield was approximately 0.11 mol h−1 in the initial activity, it gradually decreased to below the quantification limit after 6 h of the reaction (Supplementary Table S4). The turnover number (TON) of CH3OH integrated over the 24 h reaction was 10.1 mol and that of HCHO was 2.0 mol , indicating that Cs-Cu1/SiO2 catalytically produced the partial oxidation products of CH4. However, Cu(OAc)2–Cs-SiW10/SiO2 gradually deactivated, especially after 18 h of the reaction. CH3OH TON for 24 h was 9.2 mol and that of HCHO was 0.4 mol , which were lower than those of Cs-Cu1/SiO2. Additionally, a slightly higher CO2 yield was observed over Cu(OAc)2–Cs-SiW10/SiO2 throughout the 24 h reaction compared to Cs-Cu1/SiO2 (Supplementary Table S5).
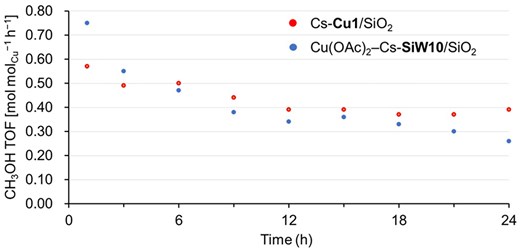
Time course examination for Cs-Cu1/SiO2 (red) and Cu(OAc)2–Cs-SiW10/SiO2 (blue) under the reaction conditions described in Table 2.
The structure of Cs-Cu1/SiO2 after the reaction was analyzed using Raman spectroscopy. A comparison of the Raman spectra of Cs-Cu1/SiO2 before and after the reaction revealed no signals corresponding to WO3, while slight shifts in the peaks were observed. This result indicates that the anion structure of Cu1 was maintained even after CH4 oxidation at 450 °C (Fig. 3a). However, when comparing Cu(OAc)2–Cs-SiW10/SiO2 before and after the 24 h reaction, an increase in the peak intensity around 1,120 cm−1 was observed after the reaction (Fig. 3b). This can be attributed to CuO,19 although it should be noted that one of the peaks of SiO2 is also broadly observed around 1,080 cm−1. Based on these results, over-oxidation of the products may be favored on Cu(OAc)2–Cs-SiW10/SiO2 due to the aggregation of CuO species.
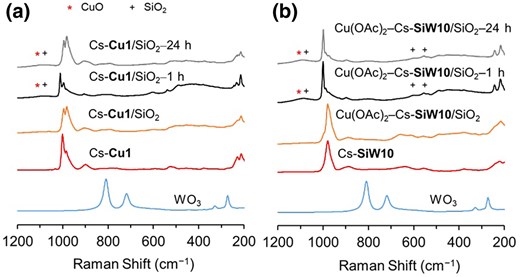
Raman spectra of a) Cs-Cu1/SiO2 before and after reaction, b) Cu(OAc)2–Cs-SiW10/SiO2 before and after the reaction. Catalyst–t h represents the catalyst after t h of the reaction.
The XRD patterns of Cs-Cu1/SiO2 and Cu(OAc)2–Cs-SiW10/SiO2 after the 24 h reaction revealed that Cs-Cu1 or Cs-SiW10 had aggregated on the SiO2; however, no WO3-derived peaks were observed (Supplementary Fig. S10).
To clarify the chemical states of the Cu species, Cu K-edge XANES spectra were measured (Fig. 4). CuO exhibited characteristic features in the shoulder peak region (8,980 to 8,985 eV), corresponding to the Cu 1 s → Cu 4p–O 2p transition. Additionally, the intensity of the shoulder peak region for Cu(OAc)2/SiO2 after 1 h of use (Cu(OAc)2/SiO2–1 h) exhibited the Cu 1 s → Cu 4p–O 2p transitions corresponding to Cu+ and Cu2+ at 8,982 to 8,984 eV and 8,985 eV, respectively (Fig. 4b).3,20 These features are similar to those of CuO, suggesting the formation of CuOx species on SiO2, which may promote the over-oxidation of the products. Cu(OAc)2–Cs-SiW10/SiO2 after the 24 h reaction exhibited a higher intensity in the shoulder peak region than Cs-Cu1/SiO2 after the 24 h reaction, which is consistent with the higher yield of CO2 and the lower CH3OH TON observed in the time course examination. This suggests that in Cs-Cu1/SiO2, lacunary POMs (molecular templates) protected the Cu-based active site from aggregation under the reaction conditions, thereby enabling continuous and selective oxidation of CH4.
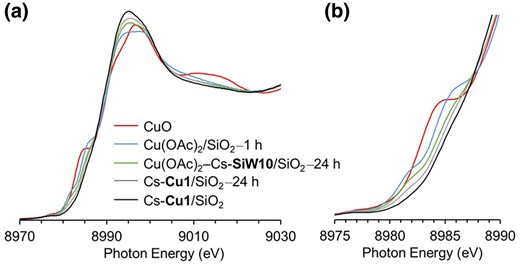
a) Cu K-edge XANES spectra and b) a magnification of the shoulder peak region in a).
In conclusion, we have developed a novel approach for the catalytic oxidation of CH4 into CH3OH using supported Cu-introduced POM cesium salts. Supported mononuclear Cu-introduced POM (Cs-Cu1/SiO2) exhibited high CH3OH selectivity (>75%) and catalytic durability for 24 h time on stream. Compared with supported POM TBA salts (TBA-Cu1/SiO2) or supported Cu oxide (Cu(OAc)2/SiO2), which were prone to over-oxidizing CH3OH, the POM templates maintained their structure under the reaction conditions and inhibited CuO aggregation. Additionally, Cs-Cu1/SiO2 exhibited superior catalytic durability compared to Cu(OAc)2–Cs-SiW10/SiO2 (stepwise addition), maintaining a stable TOF for CH3OH. Cu K-edge XANES spectra and Raman spectra indicated that POM cesium salts functioned as molecular templates, effectively suppressing the aggregation of Cu-based active sites.
In the future, increasing the reaction rate will be essential for the industrial production of CH3OH from natural gas. Therefore, we expect that our findings will contribute to the development of more effective Cu-based catalysts using supported POM cesium salts, which function as molecular templates.
Supplementary data
Supplementary material is available at Chemistry Letters online.
Funding
This work was financially supported by Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Nos. 22H04971 and 24K08157. T. S. was supported by the JSPS for the Research Fellowship for Young Scientists (Grant No. 24KJ0685). We greatly appreciate Dr. Tetsuo Nagami (Aichi Synchrotron Radiation Center, AichiSR) for the support for XAFS measurements in BL5S1 (Proposal No. 202405084) and Dr. Hironori Ofuchi (Japan Synchrotron Radiation Research Institute, SPring-8) for the support of XAFS measurements at BL14B2 (Proposal No. 2024A1546). We also appreciate Mr. Kohei Kuyama for his assistance in the initial study.
References
Author notes
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}