





Abstract
Convergent synthesis of the BCDEF ring system of maitotoxin was achieved in 10 steps via Suzuki–Miyaura coupling of an enol phosphate corresponding to the B ring derived from L-ribose in 6 steps and an exo-olefin prepared from the DEF ring in 5 steps. Although initial attempts to obtain an O,S-acetal were unsuccessful, giving a bicyclic O,S-acetal as a byproduct, protecting group manipulation and optimization of reaction conditions solved the problem to afford the desired O,S-acetal as the major product, which was converted to the BCDEF ring via introduction of the angular methyl group.
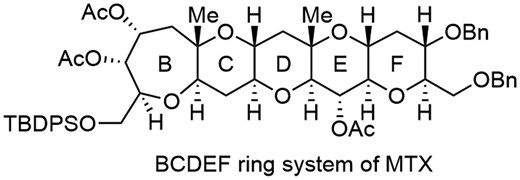
Maitotoxin (MTX) is a secondary metabolite produced by the epiphytic dinoflagellate Gambierdiscus toxicus (Fig. 1), which elicits potent Ca2+ influx activity. The structure was determined by the Yasumoto group to be a ladder-shaped polycyclic ether comprised of 32 cyclic ethers including 98 asymmetric carbons.1 The complete stereochemistry of MTX was determined by synthesis of partial structures and degradation of the natural product by the Kishi2 and Tachibana3 groups. Because of its unique structure and potent biological activity, MTX has attracted a great deal of interest among synthetic chemists.4–6 During the course of our structure–activity relationship studies based on chemical synthesis, partial structures of MTX were synthesized, including the DEF, GHI, LMNO, QRS, and WXYZA’B’C’D’E’F’ ring systems.6 Herein, we report convergent synthesis of the BCDEF ring system of MTX, whereas syntheses of the BCDE and ABCDEFG rings of MTX were reported by the Nakata4d and Nicolaou5c groups, respectively.
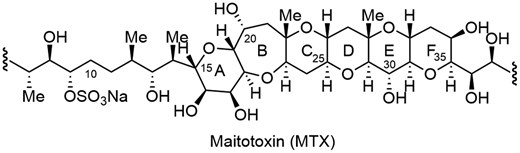
Partial structure of MTX corresponding to A–F ring system.
A retrosynthetic analysis of the BCDEF ring system 1 of MTX is shown in Scheme 1. The pentacyclic ether 1 would be obtained via Suzuki–Miyaura coupling of enol phosphate 2 (B ring) and exo-olefin 3 (DEF ring) followed by methylation of the O,S-acetal. The enol phosphate 2 could be obtained from L-ribose via the corresponding seven-membered ring lactone. The exo-olefin 3 is to be obtained from 56f which has already been synthesized via our furan-based strategy.6c,6f,7 Although successful applications of Suzuki–Miyaura coupling8 to total syntheses of ladder-shaped polyethers by the Sasaki and Fuwa group9–16 and the Jamison group17 are remarkable, and it was utilized for the synthesis of the ABCDEFG ring of MTX by the Nicolaou group,5c it was uncertain whether this strategy could be applied to the synthesis of 1.
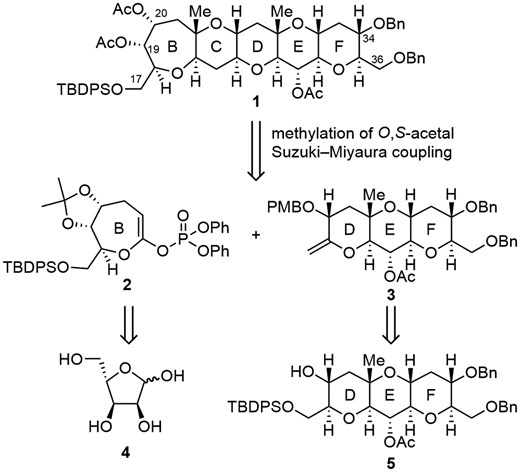
Retrosynthetic analysis of BCDEF ring system 1 of MTX.
Synthesis of the enol phosphate 2 is shown in Scheme 2. According to the literature, starting from D-ribose,18 L-ribose 4 was converted to an acetonide (86%), which was followed by protection of the primary alcohol as a TBDPS ether (90%) to give 6. Wittig olefination of the hemiacetal 6 with stabilized ylide in the presence of benzoic acid furnished an α,β-unsaturated ester (90%, E:Z = 1.5:1),19 and hydrogenation of the olefin with concomitant hydrogenolysis of the benzyl ester using Pearlman catalyst afforded hydroxy carboxylic acid 7 (90%). Yamaguchi lactonization of 7 resulted in the formation of seven-membered ring lactone 8 (93%),20 which was treated with (PhO)2P(O)Cl in the presence of KHMDS to afford enol phosphate 2 (88%).9–17,21
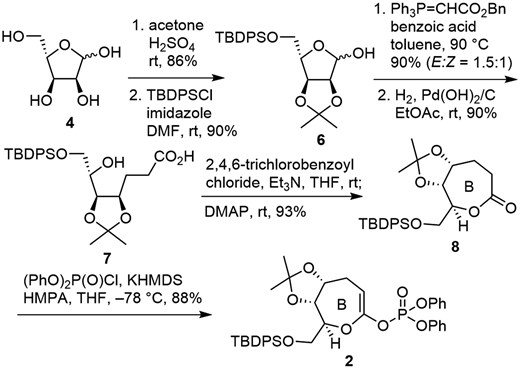
Synthesis of enol phosphate 2 corresponding to B ring of MTX.
Synthesis of the exo-olefin 3 is shown in Scheme 3. The DEF ring 56f was synthesized from furan derivative 9 and terminal olefin 10 via our furan-based strategy in 9 steps.6c,6f,7 The secondary alcohol of 5 was protected as a PMB ether 11 with PMBO(CF3)C=NPh (PMB-NPTFA)22 in the presence of TfOH, which was a cleaner reaction compared with that with PMBO(CCl3)C=NH.23 Removal of the TBDPS group of 11 with TBAF gave alcohol 12 (91%, 2 steps). The resulting primary alcohol was treated with Tf2O in the presence of 2,6-lutidine to furnish triflate 13 (67%). After conversion to iodide 14 by treatment with NaI (95%), β-elimination of 14 with t-BuOK was accompanied by concomitant removal of the acetyl group to afford exo-olefin 3 (96%). Incidentally, direct conversion of alcohol 12 to iodide 14, and elimination of triflate 13 were unsuccessful.
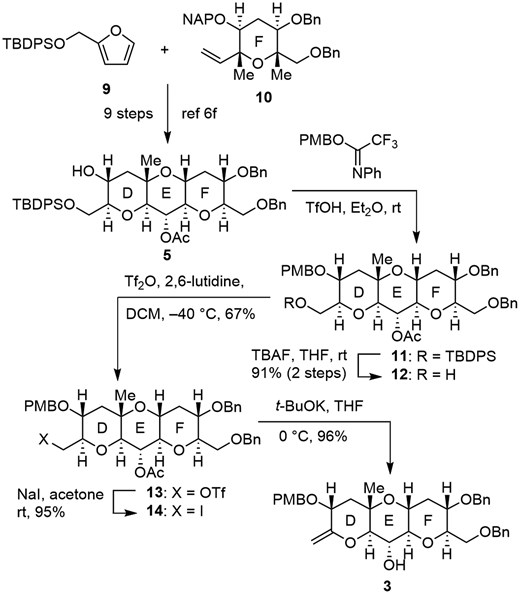
Synthesis of exo-olefin 3 corresponding to DEF ring of MTX.
Having synthesized the B ring 2 and the DEF ring 3, we moved on to the coupling of these fragments (Scheme 4). Hydroboration of the exo-olefin 3 with 9-BBN to give alkylborane 15, followed by Suzuki–Miyaura coupling with enol phosphate 2 using PdCl2(dppf)‧CH2Cl2 in the presence of aqueous NaHCO3 in DMF at room temperature, resulted in the formation of desired enol ether 16 (78%).11d After protection of the hydroxy group as an acetate (94%), hydroboration of 17 with BH3·SMe2 followed by oxidative workup with aqueous NaBO3 furnished 18 (76%) as a single isomer.
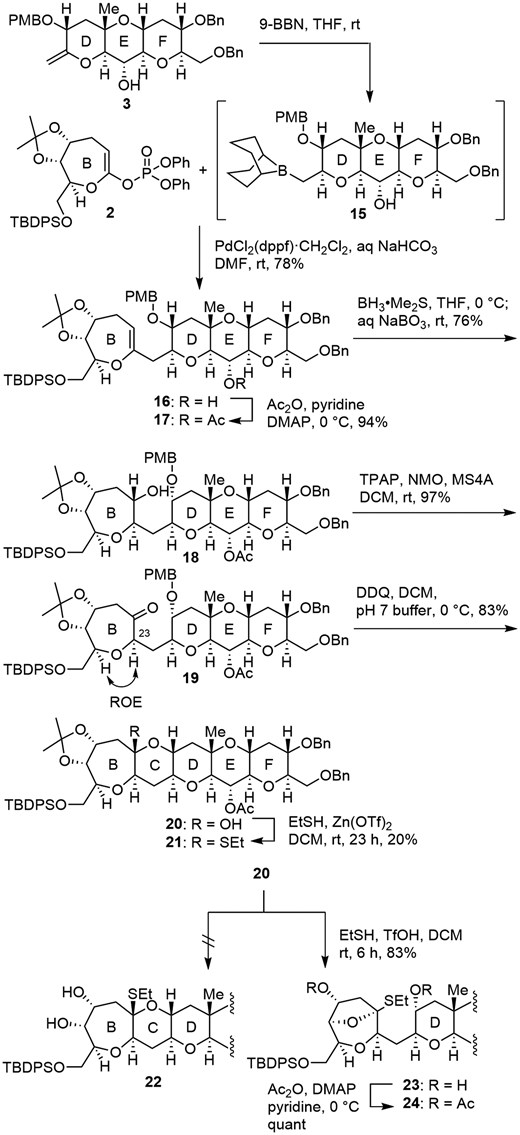
Synthesis of hemiacetal 20 and attempts at O,S-acetal formation.
Ley–Griffith oxidation of the alcohol 18 gave ketone 19 (97%),24 and the configuration at C23 was determined by ROE experiments. Removal of the PMB group of 19 with DDQ gave hemiacetal 20 (83%) as a single isomer. Although the stereochemistry of the hemiacetal moiety could not be determined by NMR analysis, the structure shown in Scheme 4 is based on the thermodynamic stability obtained by calculations (see Supplementary material). The next task, conversion of hemiacetal 20 to the corresponding O,S-acetal was not an easy task in spite of a number of precedents (Scheme 4).12a,12b,25 O,S-acetal formation by treatment with EtSH in the presence of Zn(OTf)226 was sluggish to furnish 21 in only 20% yield after 23 h, presumably because of the high ring strain of the oxocarbenium ion (see Supplementary material). Therefore, more reactive TfOH6e was used for O,S-acetal formation. Although the reaction proceeded smoothly with concomitant removal of the acetonide, desired 22 was not obtained, and undesired bicyclic O,S-acetal 23 was formed (83%), whose structure was determined after conversion to acetate 24. A similar example of bicyclic acetal formation was reported by the Hirama group in the synthesis of the 7-membered M ring of Caribbean ciguatoxin C-CTX-1.27
Plausible reaction pathways for the O,S-acetal are shown in Scheme 5. Removal of the acetonide of 20 would furnish intermediates, hemiacetal A and hydroxy ketone B, as an equilibrium mixture. There are 2 possibilities to afford the desired O,S-acetal 22: from A via oxocarbenium ion C, or from B via thiocarbenium ion D.
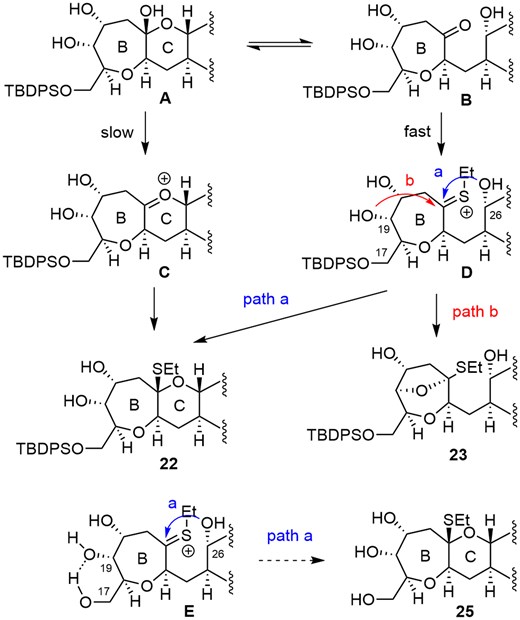
Plausible reaction pathways for O,S-acetal formation.
Formation of oxocarbenium ion C would likely be impeded by ring strain; therefore, formation of thiocarbenium ion D would be faster. Nucleophilic attack of the hydroxy group at C19 might be favored (5-membered ring formation) over that at C26 (six-membered ring formation) to form undesired O,S-acetal 23. We expected that removal of the TBDPS group at C17 from D would cause a conformational change by intramolecular hydrogen bonding between hydroxy groups at C17 and C19 of thiocarbenium ion E, thus suppressing undesired nucleophilic attack of the hydroxy group at C19 to afford desired O,S-acetal 25. (For conformations of D and E, see Supplementary material).
Therefore, as shown in Scheme 6, both acetonide and TBDPS group of 20 were removed with HF·Py. Subsequent treatment with EtSH in the presence of Zn(OTf)2 for 3.7 h afforded desired O,S-acetal 25 as the major product as expected, with concomitant formation of undesired bicyclic O,S-acetal 26 in a 5.6:1 ratio. Prolonged reaction time (23 h) caused isomerization of 25 to 26 (25:26 = 1:1), suggesting that 26 is the more stable isomer (for comparison of energies of 25 and 26, see Supplementary material).
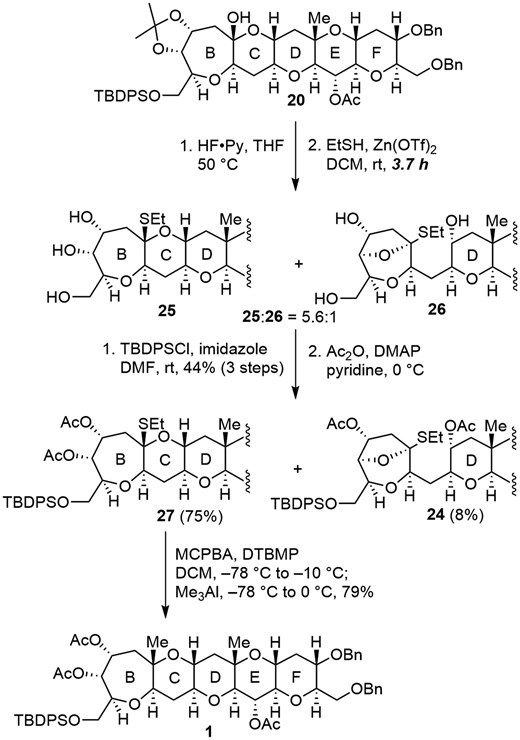
Synthesis of BCDEF ring system 1 of MTX.
Selective protection of the primary alcohol as a TBDPS ether (44%, 3 steps) followed by acetylation of the remaining secondary alcohols afforded 27 (75%) and 24 (8%) after separation by silica gel column chromatography. Finally, methylation of the O,S-acetal 27 by the improved method reported by Nicolaou5h by successive treatment with MCPBA in the presence of 2,6-di-tert-butyl-4-methylpyridine (DTBMP) and then Me3Al resulted in the formation of the BCDEF ring 1 of MTX as a single isomer (79%). The configuration at C22 was determined by ROE experiments (see Supplementary material).
In conclusion, convergent synthesis of the BCDEF ring system 1 of MTX has been achieved via Suzuki–Miyaura coupling in 10 steps from the enol phosphate 2 prepared from L-ribose in 6 steps and the exo-olefin 3 derived from the DEF ring in 5 steps. Although initial attempts to obtain the desired O,S-acetal were unsuccessful, giving the bicyclic O,S-acetal as a byproduct, protecting group manipulation solved this problem to obtain desired O,S-acetal as the major product, which was converted to the BCDEF ring 1 via introduction of the angular methyl group. The longest linear sequence from 9 and 10 is 24 steps with 30 total steps. Further synthetic studies of MTX are currently underway in our laboratory.
Supplementary data
Supplementary material is available at Chemistry Letters online.
Funding
This work was supported by Japan Society for the Promotion of Science (JSPS) grant numbers JP19H02720, JP23H01962, JP23K26655, the Asahi Glass Foundation, and the Nagase Science and Technology Foundation.
References
Author notes
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}