





Abstract
We synthesized the new organic crystalline material ɛ-BETS2(IBr2)2TCE (BETS = bis(ethylenedithio)tetraselenafulvalene, TCE = 1,1,2-trichloroethane) and performed the single-crystal X-ray structural analysis, electrical resistivity measurement, and band calculation to elucidate the electronic properties. We reveal the possibility that the close interactions between cations and anions in the same sheets enable high conductivity. The findings indicate that evidently weak interactions in molecular crystals can almost qualitatively alter the electrical properties.
The metallic and ionic crystals exhibit opposite electronic properties to each other. The metallic crystals include conduction electrons or holes that are responsible for high conductivity.1 In contrast, ionic crystals contain localized electrons, resulting in high resistivity.2 The charge-transfer (CT) complexes can be either metallic or ionic crystals, depending on the crystal structures.3–5 Some possess segregated-type structures, where cations and anions are arranged separately from each other. They become metals and superconductors depending on their crystal structures and thermodynamic conditions.6–9 Others possess mixed-type structures, where cations and anions are in the same columns or sheets in an alternating manner. They often exhibit insulating properties.10–13
The ET-based (ET = bis(ethylenedithio)tetrathiafulvalene, Scheme 1) segregated-type complexes with trihalide anions have been actively studied since the 1980s14–17 and formed a major group of organic CT complexes to produce superconductors,18,19 dimer-Mott insulators,20,21 and Dirac fermion systems.22–24 In this study, we focused on BETS-trihalide CT complexes (BETS = bis(ethylenedithio)tetraselenafulvalene, Scheme 1) to develop new Dirac fermion materials.25–27 BETS is an analogous molecule to ET, in which the inner four sulfur atoms are replaced by selenium. In this letter, we report synthesis, crystal and band structures, and electrical properties of a new mixed-type but highly conducting compound, ɛ-BETS2(IBr2)2TCE (TCE = 1,1,2-trichloroethane), abbreviated as ɛ-IBr2 below.
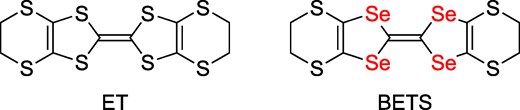
Molecular structures of ET and BETS.
Neutral BETS28,29 and (n-C4H9)4NIBr230 were synthesized by following the reported procedure. The single crystals of ɛ-IBr2 were grown by an electrochemical method. BETS (7 mg, 0.01 mmol) and (n-C4H9)4NIBr2 (70 mg, 0.13 mmol) were added in an H-type cell without a glass filter and dissolved in the distilled 1,1,2-trichloroethane (15 mL) under nitrogen atmosphere. A constant current of 1.0 µA was applied for a week at 27 °C using platinum electrodes (1 mm in diameter). The single-crystal X-ray diffraction of ɛ-IBr2 at 296 K was performed using VariMax Rapid/α (Rigaku) for the data collection. The analysis of the collected data was carried out using CrysAlisPro (Rigaku). The structure was solved and refined using Olex2 (OlexSys) and SHELXTL.31–33 Restrictions on temperature factors were applied to TCE molecules. The crystal data of ɛ-IBr2 is summarized in Supplementary Table S1. Based on the observed crystal structure, the band structure and intermolecular interactions were calculated using CAESAR with an extended Hückel tight-binding (EHTB) method.34 The ac resistivity measurement was performed using the standard four-probe method and the physical property measurement system (Quantum Design PPMS-9) equipment. The square frequency of ac was about 7.5 Hz. The current flowed along the c-axis.
The crystal structure of ɛ-IBr2 (Figs. 1[a]–[b]) belongs to monoclinic P21/c. The asymmetric unit consists of 2 BETS+ cations, 2 IBr2− anions, and 1 TCE molecule. Because IBr2− is a monoanion and TCE is a neutral molecule, ɛ-IBr2 is effectively a 1:1 salt, and the formal charge of BETS molecules is +1 unless there are BETS + -IBr2− CT interactions. One of the 2 IBr2− anions is included in the cation sheets, while the crystal solvent TCE is included in the anion sheets, i.e. mixed-type structures. We call the former cation–anion (CA) sheets and the latter anion–solvent (AS) sheets. The packing pattern of the CA sheets is called ɛ-type.35 To our knowledge, ET2I3(I8)0.5, ET2(IBr2)2(TCE)0.5, and ET2BrI3(diiodoacetylene) are reported as ɛ-type to date.35–37 The CA sheets alternate with the AS sheets along the a-axis. In the CA sheets, the 2 BETS+ cations directly overlap each other (Fig. 1[b]), indicating the dimerization (BETS+)2. The IBr2− anion in the CA sheets is located between the dimers (Fig. 1[b]). Disordered atoms were observed in one of the ethylene groups in BETS+, IBr2−, and TCE at 296 K. The +1 charge of each BETS+ cation and the formation of dimers (BETS+)2 strongly suggest that the band filling of this material is full-filled, suggesting to be an insulating ionic crystal.
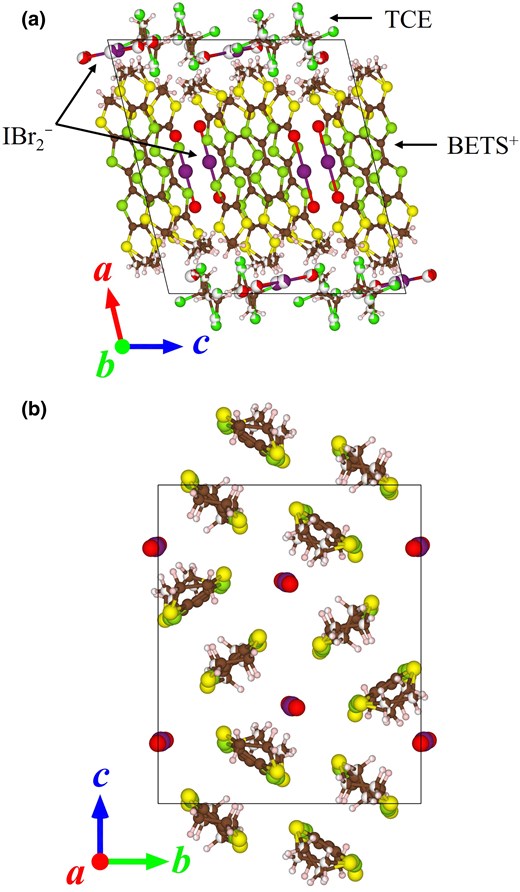
a) Crystal structure of ɛ-IBr2 (296 K) viewed along the b-axis. b) Cation-anion-mixed sheets viewed along the a-axis.
To elucidate the electronic properties of ɛ-IBr2, we calculated the band structure based on the EHTB method.34 We also performed similar calculations based on the density functional theory, yet could not find appropriate functional and basis sets to yield meaningful results (see SI for details). The band structure and Fermi surface of ɛ-IBr2 at 296 K are shown in Figs. 2[a], [b]. The hole Fermi surface from the valence band of ɛ-IBr2 is observed around the M point (Fig. 2[b]). Despite the formal integral charges in the CA sheets, the band calculation indicated small but finite CT between cations and anions in the CA sheets. The projected density of states at EF from the IBr2− anion in the CA sheets is estimated as 0.1044 states eV−1 cell−1 (1.17%; Supplementary Figs. S1 and S2). Although such a small contribution based on EHTB could not exclude other possibilities of doping origin like lattice defects at the anion sites, the Fermi surface should originate from the spontaneous hole doping from (BETS+)2 to IBr2−. Details of the intermolecular interactions in the CA sheets of ɛ-IBr2 at 296 K are summarized in Supplementary Tables S3 and S4. Regarding the BETS+ (singly occupied molecular orbital [SOMO])-BETS+ (SOMO) interactions (Supplementary Table S3), transfer integral t of the pair A (tA) is several times as large as others, being consistent with the strong dimerization observed by the X-ray structural analysis. Regarding the BETS+ (SOMO)-IBr2− interactions (Supplementary Table S4), the absolute values of the transfer integrals of the pair I and the pair II are ca. a half of or comparable to those of the BETS+ (SOMO)-BETS+ (SOMO) interactions except for tA, suggesting that the conduction pathways include the IBr2− species. As a result, the SOMOs in ɛ-IBr2 are delocalized not only on the BETS+ cations but also on the IBr2− anions (Fig. 2[c]). The Fermi surface and carriers should originate from the underlying CT interactions between BETS+ and IBr2−, leading to the high conductivity of ɛ-IBr2.
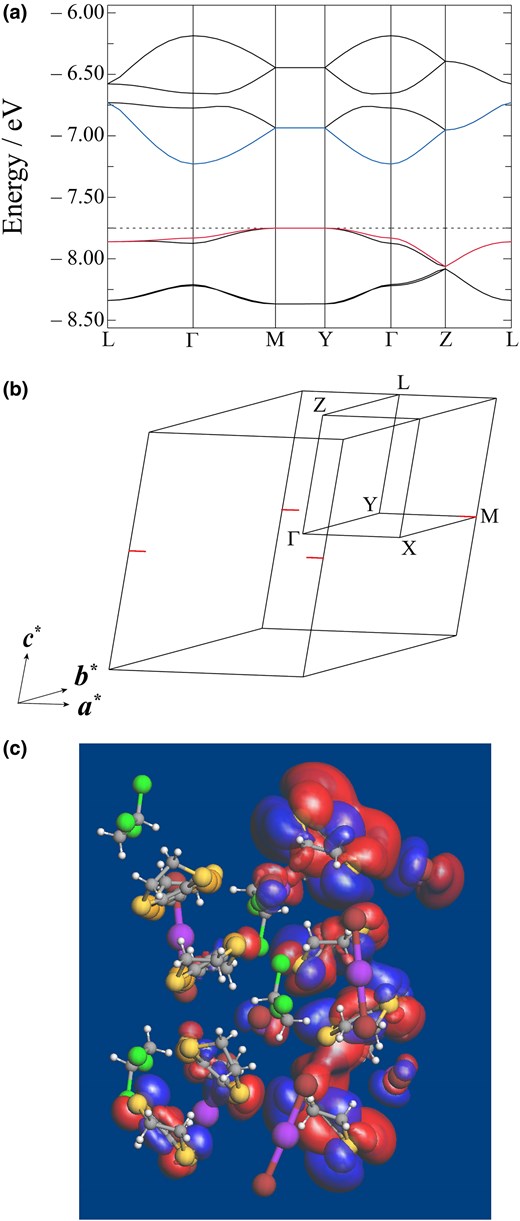
a) Calculated band structure of ɛ-IBr2 at 296 K, where Γ = (0, 0, 0), Y = (0, 0.5, 0), Z = (0, 0.0, 0.5), M = (0.5, 0.5, 0), L = (0, 0.5, 0.5) in the reciprocal space. The broken line denotes the Fermi level EF. b) Fermi surface of ɛ-IBr2 at 296 K, where the linear hole surfaces from valence bands are observed around M point. c) SOMO of ɛ-IBr2 at 296 K.
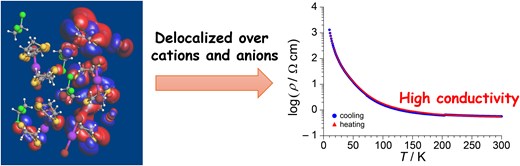
The temperature dependence of the resistivity of ɛ-IBr2 along the c-axis is shown in Fig. 3. The resistivity of this material exhibits semiconducting behavior in the temperature range of measurement. However, the resistivity ρRT at 300 K is 0.56 Ω cm, which is ca. 10−3 to 10−4 times as small as those of related organic ionic crystals (Table 1).36,38,39 Sample dependence of ɛ-IBr2 is shown in Supplementary Fig. S7. A rather wide range of resistivity behavior suggests that different factors simultaneously dominate the behavior. Figure 3 also shows the temperature dependence of the charge gap Eg of ɛ-IBr2 obtained from the resistivity measurement. The values of Eg are also so small at all temperatures of measurement that it does not appear to be an organic ionic crystal. As discussed above, the band calculation suggested that ɛ-IBr2 should be narrowly metallic. Therefore, considering the accuracy of EHTB calculation, the results of the resistivity measurements and band calculations are largely consistent: both indicated that ɛ-IBr2 should be located at the border of metals and insulators. It is also worth noting that ɛ-IBr2 retains its high conductivity down to low temperatures like molecular crystals with metallic conductivity: ρ(50 K) ∼ 101 Ω cm. Generally, weak but isotropic inter-site interactions are favorable for metallic crystals to occur, leading to high-dimensional partially occupied bands. The CA sheets of ɛ-IBr2 satisfy this requirement. The HOMOs between BETS (−8.15 eV, SOMO of BETS+) and IBr2− (−9.62 eV) are relatively close to each other in their energies, enabling weak but rather isotropic CT interactions between them (Supplementary Fig. S4).
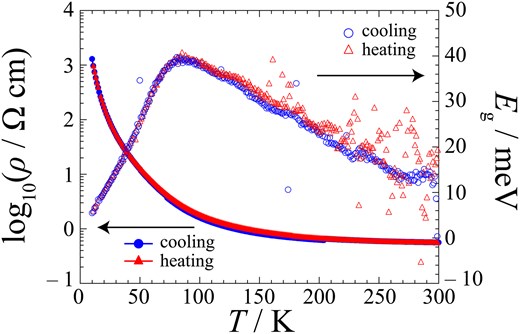
Temperature dependences of electrical resistivity (left axis) and charge gap (right axis) of ɛ-IBr2. The circles and triangles distinguish the heating and cooling runs, respectively.
In conclusion, we have found that the new organic crystalline material ɛ-BETS2(IBr2)2TCE exhibits high conductivity comparable to those of the molecular metallic crystals. Regarding the possible conduction mechanism, the CT interactions between cations and anions in the same sheets govern the electronic properties of this material. Generally, in low-dimensional materials containing both strong and weak interactions, the latter is often ignored in the discussion of their physical properties. However, ɛ-BETS2(IBr2)2TCE indicates that weak interactions can almost qualitatively alter their electrical properties, which will provide a different view of molecular functional materials.
Acknowledgments
We thank S. Yamamoto for the useful discussion. We thank S. Mori and R. Konishi (ADRES, Ehime University) for their assistance in the single-crystal X-ray structural analyses, and K. Konishi (Department of Physics, Faculty of Science, Ehime University) for his assistance on the resistivity measurements.
Supplementary data
Supplementary material is available at Chemistry Letters online.
Funding
This work was partially funded by a Grant-in-Aid for Scientific Research (22H02034, 24K21755, 23H03964, and 24K01470) from the JSPS and the Canon Foundation (Science and Technology that Achieves a Good Future).
References
Author notes
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}