





Abstract
An efficient method for the silylation of alcohols was developed using N,O-bis(tert-butyldimethylsilyl)acetamide (BTBSA) and 3 mol % of N,N-dimethyl-4-aminopyridine N-oxide (DMAPO). Monosilylation of 1,2-diols was successfully achieved with high chemoselectivity. In addition, several control experiments revealed that a phenolic hydroxy group was silylated much faster than a primary alcohol in both an inter- and intramolecular manner. The reactivity toward silylation under these systems might stem from the pKa of the hydroxy group.
Silylation of alcohols is a fundamental strategy for protecting hydroxy groups.1 To date, there have been various reports regarding the silylation of alcohols using chiral2 or achiral3 organocatalysts. Of these, the tert-butyldimethylsilyl (TBS) group is widely used for this purpose because it can be easily installed on a hydroxy group and removed under specific conditions.
We have been interested in the chemistry of organocatalysis4 and reported that chiral DMAP derivatives having a 1,1′-binaphthyl unit showed extremely high catalytic activity and enantio- and/or chemoselectivity for enantioselective acyl transfer reactions.5 Examples include O- to C-acyl rearrangements,4f,4m kinetic resolution of secondary,4g tertiary alcohols,4o and d,l-1,2-diols,4h dynamic kinetic resolution,4k and desymmetrization of meso-4i,4n or prochiral molecules.4l,4p Based on our mechanistic studies, hydrogen-bonding interaction between the catalyst and substrate plays an important role in achieving higher levels of enantioselectivity and/or chemoselectivity in an acylation reaction. These results motivated us to develop a catalyst-controlled chemoselective reaction in the silylation of alcohols.
In this communication, we report the details of the chemoselective silylation of various alcohols using BTBSA as a silylation reagent and DMAPO as a catalyst. In addition, several applications of chemoselective silylations are also demonstrated.
Initially, we decided to use meso-hydrobenzoin (1a) as a model substrate to understand the reaction efficiency and chemoselectivity (monosilylate vs disilylate) by using various combinations of a silylation reagent and a pyridine-based achiral catalyst (Table 1). The reaction was carried out with 1.3 equiv. of silylation reagent and 1.3 equiv. of Et3N in the presence of DMAP or DMAPO in dichloromethane at 30 °C for 48 h (Table 1). The use of tert-butyldimethylsilyl chloride (TBSCl) in the presence of DMAP gave the monosilylated product 2a in 25% NMR yield, whereas the use of DMAPO as a catalyst showed much higher catalytic activity to give 2a in 57% NMR yield (Entries 1 and 2). In both cases, disilylated product 3a was not observed. The reaction with tert-butyldimethylsilyl trifluoromethanesulfonate (TBSOTf),6 which is a highly reactive silylation reagent, in the presence of DMAP or DMAPO resulted in a mixture of 2a and 3a (Entries 3 and 4). The results with the use of 1-(tert-butyldimethylsilyl)imidazole (TBSIm)7 or tert-butyldimethylsilyl cyanide (TBSCN)8 in combination with DMAP or DMAPO were inferior to those with the use of TBSCl and TBSOTf (Entries 5–8). We also tested imidate-type silylation reagents, N,O-bis(tert-butyldimethylsilyl)acetamide (BTBSA)9 and N,O-bis(tert-butyldimethylsilyl)trifluoroacetamide (BTBSTFA).10 In both cases, the reaction only proceeded with DMAPO to give the desired monosilylated product 2a in NMR yields of 87% and 82%, respectively (Entries 9–13). Thus, the combination of BTBSA and DMAPO was thought to be optimal for chemoselective transformation in terms of the yield of the desired product 2a. According to the literature, Yoshida and Takao reported the silylation of alcohols using tert-butyldiphenylsilyl chloride (TBDPSCl) and pyridine N-oxide as a catalyst.3b,3d In addition, Johnson reported the use of BTBSA and tetrabutylammonium fluoride as a catalyst for the silylation of alcohols.3a However, to the best of our knowledge, there have been no reports on the use of a combination of BTBSA and DMAPO for the silylation of alcohols. Thus, we pursued this combination for further optimization.
Screening of silylation reagents and catalysts for 1aa
| Entry | Silylation reagent | Catalyst | 2a (%)b | 3a (%)b | 1a (%)b |
|---|---|---|---|---|---|
| 1 | TBSCl | DMAP | 25 | N.D.c | 75 |
| 2 | DMAPO | 57 | N.D.c | 43 | |
| 3 | TBSOTf | DMAP | 72 | 11 | 17 |
| 4 | DMAPO | 67 | 5 | 27 | |
| 5 | TBSIm | DMAP | N.D.c | N.D.c | 100 |
| 6 | DMAPO | N.D.c | N.D.c | 100 | |
| 7 | TBSCN | DMAP | 38 | N.D.c | 62 |
| 8 | DMAPO | 17 | N.D.c | 83 | |
| 9 | BTBSA | DMAP | N.D.c | N.D.c | 100 |
| 10 | DMAPO | 87d | N.D.c | 13d | |
| 11 | none | N.D.c | N.D.c | 100 | |
| 12 | BTBSTFA | DMAP | N.D.c | N.D.c | 100 |
| 13 | DMAPO | 82d | N.D.c | 18d |
| Entry | Silylation reagent | Catalyst | 2a (%)b | 3a (%)b | 1a (%)b |
|---|---|---|---|---|---|
| 1 | TBSCl | DMAP | 25 | N.D.c | 75 |
| 2 | DMAPO | 57 | N.D.c | 43 | |
| 3 | TBSOTf | DMAP | 72 | 11 | 17 |
| 4 | DMAPO | 67 | 5 | 27 | |
| 5 | TBSIm | DMAP | N.D.c | N.D.c | 100 |
| 6 | DMAPO | N.D.c | N.D.c | 100 | |
| 7 | TBSCN | DMAP | 38 | N.D.c | 62 |
| 8 | DMAPO | 17 | N.D.c | 83 | |
| 9 | BTBSA | DMAP | N.D.c | N.D.c | 100 |
| 10 | DMAPO | 87d | N.D.c | 13d | |
| 11 | none | N.D.c | N.D.c | 100 | |
| 12 | BTBSTFA | DMAP | N.D.c | N.D.c | 100 |
| 13 | DMAPO | 82d | N.D.c | 18d |
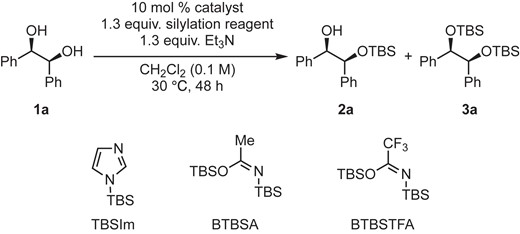
aReactions were performed on a 0.3 mmol scale under an argon atmosphere. bNMR yields were determined by 1H NMR analysis using 2-methoxynaphthalene as an internal standard. cNot detected. dAverage of two runs.
Screening of silylation reagents and catalysts for 1aa
| Entry | Silylation reagent | Catalyst | 2a (%)b | 3a (%)b | 1a (%)b |
|---|---|---|---|---|---|
| 1 | TBSCl | DMAP | 25 | N.D.c | 75 |
| 2 | DMAPO | 57 | N.D.c | 43 | |
| 3 | TBSOTf | DMAP | 72 | 11 | 17 |
| 4 | DMAPO | 67 | 5 | 27 | |
| 5 | TBSIm | DMAP | N.D.c | N.D.c | 100 |
| 6 | DMAPO | N.D.c | N.D.c | 100 | |
| 7 | TBSCN | DMAP | 38 | N.D.c | 62 |
| 8 | DMAPO | 17 | N.D.c | 83 | |
| 9 | BTBSA | DMAP | N.D.c | N.D.c | 100 |
| 10 | DMAPO | 87d | N.D.c | 13d | |
| 11 | none | N.D.c | N.D.c | 100 | |
| 12 | BTBSTFA | DMAP | N.D.c | N.D.c | 100 |
| 13 | DMAPO | 82d | N.D.c | 18d |
| Entry | Silylation reagent | Catalyst | 2a (%)b | 3a (%)b | 1a (%)b |
|---|---|---|---|---|---|
| 1 | TBSCl | DMAP | 25 | N.D.c | 75 |
| 2 | DMAPO | 57 | N.D.c | 43 | |
| 3 | TBSOTf | DMAP | 72 | 11 | 17 |
| 4 | DMAPO | 67 | 5 | 27 | |
| 5 | TBSIm | DMAP | N.D.c | N.D.c | 100 |
| 6 | DMAPO | N.D.c | N.D.c | 100 | |
| 7 | TBSCN | DMAP | 38 | N.D.c | 62 |
| 8 | DMAPO | 17 | N.D.c | 83 | |
| 9 | BTBSA | DMAP | N.D.c | N.D.c | 100 |
| 10 | DMAPO | 87d | N.D.c | 13d | |
| 11 | none | N.D.c | N.D.c | 100 | |
| 12 | BTBSTFA | DMAP | N.D.c | N.D.c | 100 |
| 13 | DMAPO | 82d | N.D.c | 18d |
aReactions were performed on a 0.3 mmol scale under an argon atmosphere. bNMR yields were determined by 1H NMR analysis using 2-methoxynaphthalene as an internal standard. cNot detected. dAverage of two runs.
The reaction was carried out in various solvent with 3 mol % DMAPO and 1.3 equiv. BTBSA in 30 °C for 24 h after tentative optimization11 of catalyst loading (10 to 3 mol % of DMAPO), base (triethylamine was not necessary in terms of the yield of 2a), and reaction time (48 to 24 h) (Table 2).
Screening of solventsa
| Entry | Solvent | 2a (%)b | 3a (%)b |
|---|---|---|---|
| 1 | CH2Cl2 | 55 | N.D.c |
| 2 | hexane | 0 | N.D.c |
| 3 | toluene | 12 | N.D.c |
| 4 | Et2O | 18 | N.D.c |
| 5 | THF | 20 | N.D.c |
| 6 | DMF | 13 | N.D.c |
| 7 | AcOEt | 10 | N.D.c |
| 8 | CH3CN | 46 | N.D.c |
| 9 | tert-amyl alcohol | 0 | N.D.c |
| 10d | CH3CN | 85 | N.D.c |
| Entry | Solvent | 2a (%)b | 3a (%)b |
|---|---|---|---|
| 1 | CH2Cl2 | 55 | N.D.c |
| 2 | hexane | 0 | N.D.c |
| 3 | toluene | 12 | N.D.c |
| 4 | Et2O | 18 | N.D.c |
| 5 | THF | 20 | N.D.c |
| 6 | DMF | 13 | N.D.c |
| 7 | AcOEt | 10 | N.D.c |
| 8 | CH3CN | 46 | N.D.c |
| 9 | tert-amyl alcohol | 0 | N.D.c |
| 10d | CH3CN | 85 | N.D.c |
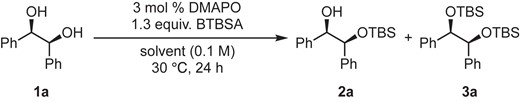
aReactions were performed on a 0.3 mmol scale under an argon atmosphere. bDetermined by 1H NMR using 2-methoxynaphthalene as an internal standard. cNot detected. dThe reaction was performed at 60 °C.
Screening of solventsa
| Entry | Solvent | 2a (%)b | 3a (%)b |
|---|---|---|---|
| 1 | CH2Cl2 | 55 | N.D.c |
| 2 | hexane | 0 | N.D.c |
| 3 | toluene | 12 | N.D.c |
| 4 | Et2O | 18 | N.D.c |
| 5 | THF | 20 | N.D.c |
| 6 | DMF | 13 | N.D.c |
| 7 | AcOEt | 10 | N.D.c |
| 8 | CH3CN | 46 | N.D.c |
| 9 | tert-amyl alcohol | 0 | N.D.c |
| 10d | CH3CN | 85 | N.D.c |
| Entry | Solvent | 2a (%)b | 3a (%)b |
|---|---|---|---|
| 1 | CH2Cl2 | 55 | N.D.c |
| 2 | hexane | 0 | N.D.c |
| 3 | toluene | 12 | N.D.c |
| 4 | Et2O | 18 | N.D.c |
| 5 | THF | 20 | N.D.c |
| 6 | DMF | 13 | N.D.c |
| 7 | AcOEt | 10 | N.D.c |
| 8 | CH3CN | 46 | N.D.c |
| 9 | tert-amyl alcohol | 0 | N.D.c |
| 10d | CH3CN | 85 | N.D.c |
aReactions were performed on a 0.3 mmol scale under an argon atmosphere. bDetermined by 1H NMR using 2-methoxynaphthalene as an internal standard. cNot detected. dThe reaction was performed at 60 °C.
Although the reaction in the selected solvent resulted in a lower yield of 2a at 30 °C (Entries 2–9 vs 1), that in CH2Cl2 was still moderate and had room for improvement. Thus, we decided to run the reaction at a higher reaction temperature in CH3CN because the boiling point of MeCN (82 °C) was higher than that of CH2Cl2 (40 °C). Finally, we found that the reaction in MeCN at 60 °C gave the desired product 2a in 85% NMR yield under these conditions (Entry 10).
After further screening of the reaction conditions (see the Supporting Information for details), the following reaction conditions were selected: 3 mol % of DMAPO, 1.3 equiv. of BTBSA in MeCN (0.5 M) at 60 °C for 16 h. With the optimal conditions in hand, the silylation of an array of diols was examined (Figure 1). The reaction of 1a afforded monosilylate 2a in 98% isolated yield with high chemoselectivity (disilylate 3a was not observed). The reaction of hydrobenzoins having 4-substituted aryls 1b and 1c also proceeded efficiently to give 2b and 2c in yield of 86% and 93%, respectively. In contrast, the use of cyclic meso-diols 1d–f gave the monosilylates in moderate yield (63%, 54%, and 43% isolated yields, respectively) along with disilylates or recovery of the starting material. The reason for these results with cyclic substrates under these conditions is not yet clear. Furthermore, d,l-hydrobenzoin series 1g–j were also subjected to this reaction, and desired monosilylates 2g–j were obtained in high yields with high chemoselectivity. The reaction with cyclic d,l-diol 1k delivered monosilylate 2k in 49% yield along with recovery of 1k (38% recovery). This reaction showed a similar tendency to the reaction of 1f (meso-diol). Based on the results obtained with diols, acyclic 1,2-diols gave monosilylates in high yield with high chemoselectivity.
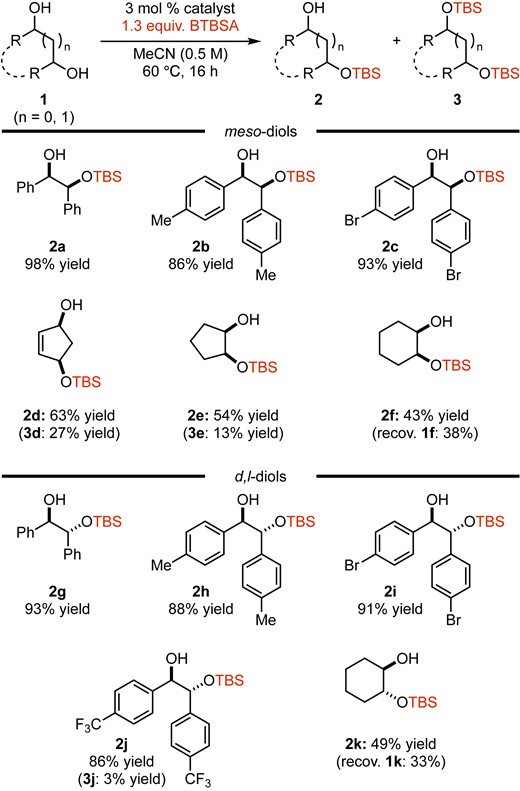
Chemoselective silylation of diols.
Next, we tested selected mono-ols 4a–f to better understand the current reaction (Figure 2). Silylation of primary alcohols proceeded smoothly to give corresponding silyl ether 5a–c in 80–91% yield.
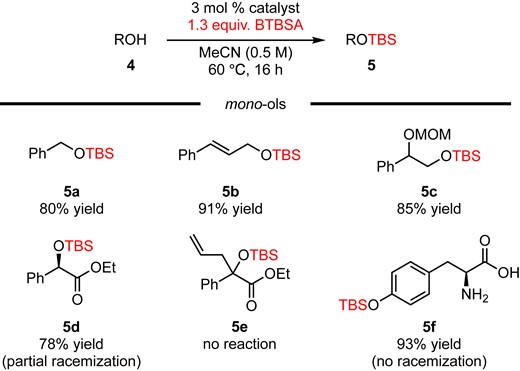
Silylation of mono-ols.
On the other hand, ethyl d-(−)-mandelate (4d) gave 5d in 78% yield with partial racemization (>99:1 er of 4d was converted to 87:13 er of 5d). The reaction of tertiary alcohol 4e did not proceed, which means that the current system is not effective for sterically demanding alcohols. Furthermore, the reaction of l-(−)-tyrosine (4f) proceeded smoothly to give 5f in 93% yield as a sole product, albeit it has reactive functionalities (-COOH and -NH2) toward silylation. These findings motivated us to further examine the chemoselective silylation of a phenolic hydroxy group in the presence of highly reactive functionalities.
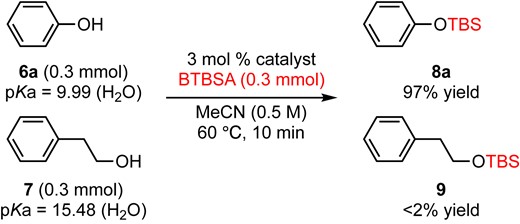
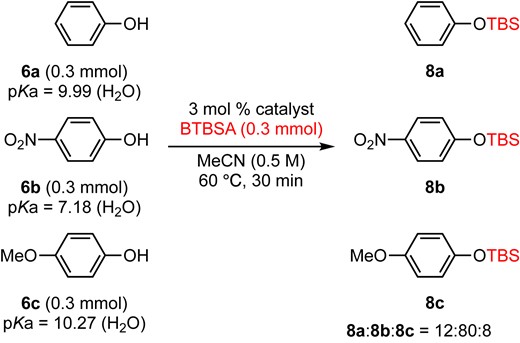
An intermolecular version of a chemoselective transformation (a phenolic vs a primary hydroxy group) was examined (eq 1). A 1:1 mixture of phenol (6a) and 2-phenylethanol (7) was subjected to the reaction conditions for 10 min, and only the phenolic hydroxy group was silylated selectively to give 8a in 97% GC yield. According to the pKa values (9.9912 for 6a, 15.4813 for 7), silylation of a more acidic phenolic hydroxy group was much faster than that of a primary alcohol under the BTBSA and DMAPO system. We also examined the competitive silylation reaction of phenol derivatives having different acidities (eq 2). A 1:1:1 mixture of phenol (6a, pKa = 9.99), p-nitrophenol (6b, pKa = 7.18), and p-methoxy phenol (6c, pKa = 10.27) was subjected to the BTBSA and DMAPO system for 30 min to give 12:80:8 of its silylated product 8a, 8b, and 8c, respectively, by crude 1H NMR analysis. These results clearly indicated that an acidic hydroxy group (lowest pKa value) is readily silylated by the BTBSA and DMAPO system.
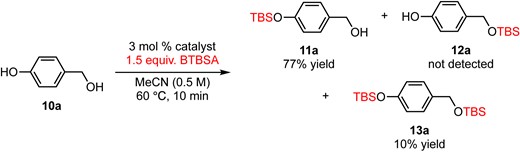
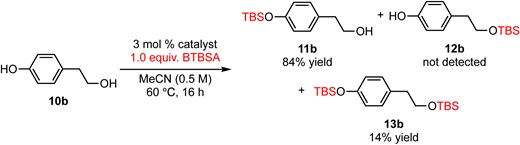
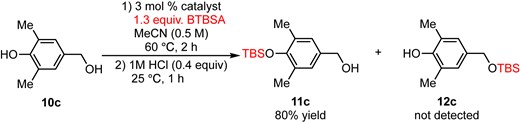
Further applications of chemoselective silylation involving a phenolic hydroxy group and a primary hydroxy group within the same molecule are demonstrated in eqs 3–5. The reaction of 4-hydroxybenzyl alcohol (10a) with 3 mol % DMAPO and 1.5 equiv. of BTBSA for 10 min afforded 11a in 77% yield along with 10% of disilylate 13a (eq 3). Surprisingly, a phenolic hydroxy group was silylated selectively to give 11a and no monosilylate 12a was observed. In the case of tyrosol (10b), a phenolic hydroxy group was selectively silylated to give 11b in 84% yield along with disilylate 13b in 14% yield (eq 4). Yoshida and Takao’s method for the silylation of 10b using TBDPSCl and N-oxide catalyst only gave TBDPS ether of a primary alcohol (TBDPS instead of TBS in compound 12b).3d The most notable point is that the current method (BTBSA and DMAPO) offers direct access to 11b without any protecting group manipulations. Finally, a phenolic hydroxy group with a sterically demanding ortho-dimethyl group was also silylated selectively to give 11c in 80% yield (eq 5). Although the reaction of those substrates using typical conditions (imidazole and TBSCl) afforded the silylated product of a primary alcohol selectively (e.g., products 12a–c, see the Supporting Information for details), these reactions (eqs 3–5) demonstrated the utility of the current method for direct access to a silylated product of a phenolic hydroxy group in the presence of a highly reactive primary alcohol without cumbersome protecting group manipulations.
In conclusion, we developed an efficient method for the silylation of alcohols using BTBSA and 3 mol % of DMAPO. The reaction proceeded smoothly for an array of alcohols including diols and mono-ols. The high chemoselectivity of monosilylate was observed, and second silylation was unlikely to occur due to the steric hindrance of first TBS group. Monosilylation of a phenolic hydroxy group in the presence of a primary alcohol within the same molecule was also achieved with high chemoselectivity. Several applications revealed that a phenolic hydroxy group was silylated much faster than a primary alcohol and its reactivity toward silylation might stem from the acidity of a hydroxy group. Applications of these methods to the enantioselective silylation of alcohols are now in progress.
Acknowledgment
This research was partially supported by JSPS Kakenhi Grant Number JP19K05456. The authors also thank DAICEL corporation for analyzing an enantiomeric ratio of 5f.
Supporting Information is available on https://doi.org/10.1246/cl.220281.
References and Notes
Protection for the Hydroxyl Group, Including 1,2- and 1,3-Diols in Greene's Protective Groups in Organic Synthesis, 5th ed, ed by P. G. M. Wuts, John Wiley & Sons, Inc., Hoboken, 2014, pp. 201.
a)
b)
c)
d)
e)
f)
g)
i)
j)
k)
l)
m)
a)
d)
a)
b)
c)
d)
e)
f)
g)
h)
i)
j)
k)
l)
m)
n)
o)
p)
b)

{kind=link}
{kind=link}