





Abstract
Amlexanox (ALX) is a small-molecule drug for the treatment of inflammatory, autoimmune, metabolic, and tumor diseases. At present, there are no studies on whether ALX has a therapeutic effect on inflammatory bowel disease (IBD). In this study, we used a mouse model of dextran sulfate sodium-induced colitis to investigate the effect of ALX-targeted inhibition of TBK1 on colitis. We found that the severity of colitis in mice was correlated with TBK1 expression. Notably, although ALX inhibited the activation of the TBK1-NF-κB/TBK1-IRF3 pro-inflammatory signaling pathway, it exacerbated colitis and reduced survival in mice. The results of drug safety experiments ruled out a relationship between this exacerbating effect and drug toxicity. In addition, ELISA results showed that ALX promoted the secretion of IL-1β and IFN-α, and inhibited the production of cytokines IL-6, TNF-α, IL-10, TGF-β, and secretory IgA. Flow cytometry results further showed that ALX promoted T-cell proliferation, activation, and differentiation, and thus played a pro-inflammatory role; also, ALX inhibited the generation of dendritic cells and the polarization of macrophages to M1 type, thus exerting anti-inflammatory effect. These data suggest that the regulation of ALX on the function of different immune cells is different, so the effect on the inflammatory response is bidirectional. In conclusion, our study demonstrates that simply inhibiting TBK1 in all immune cells is not effective for the treatment of colitis. Further investigation of the anti-inflammatory mechanism of ALX on dendritic cells and macrophages may provide a new strategy for the treatment of IBD.
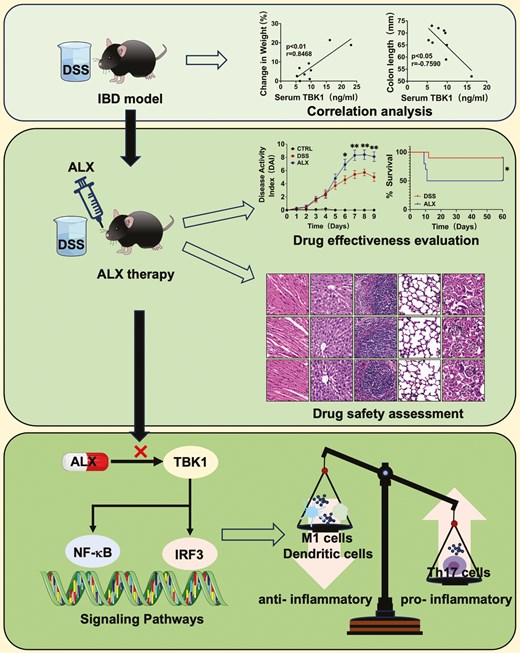
Introduction
Inflammatory bowel disease (IBD) is a chronic, progressive gastrointestinal disorder that includes ulcerative colitis (UC) and Crohn’s disease (CD) [1, 2]. IBD has gradually become a global disease and its prevalence has risen steadily in Western countries and soared in newly industrialized countries [3–6]. In addition, weight loss, diarrhea, rectal bleeding, and fatigue are typical clinical symptoms of IBD patients, which can lead to work disability, depression, and anxiety [7, 8]. Because of the characteristics of alternating episodes of relapse and remission, patients experience complications, such as perforation, uncontrolled hemorrhage, intestinal stricture, toxic megacolon, fulminant colitis, dysplasia, and colorectal cancer [9–12]. Furthermore, the currently available treatments for IBD do not provide a complete cure for patients. Conventional drug therapies, such as aminosalicylates (5-ASA), glucocorticoids, and immunosuppressants are often discontinued because of severe side effects, including nephritis, osteoporosis, and malignancy [13–15]. Therefore, the increasing prevalence, and characteristics of the disease course with alternating relapsing and remitting episodes, severe complications, and suboptimal treatments, have caused a significant burden to the healthcare system and highlight the need for new therapies for IBD.
TANK-binding kinase 1 (TBK1) is a serine/threonine protein kinase that is ubiquitously expressed in all tissues. Current studies suggest that TBK1 is a key mediator of the inflammatory response, autophagy, proliferation, survival, insulin signal transduction, and metabolism [16–19]. An animal study on autoimmune encephalomyelitis (EAE) revealed that conditional knockout of TBK1 in T cells alleviates the progression of EAE, primarily by reducing the effusion of T cells in the draining lymph nodes and reducing T-cell infiltration in the central nervous system [20]. Another study on IBD demonstrated that rebamipide, a drug used for the treatment of UC, directly inhibited TBK1 in human colonic epithelial cells, thus providing an anti-inflammatory effect [21]. These studies highlight the potential of TBK1 as a therapeutic target for inflammatory and autoimmune diseases.
Amlexanox (ALX) is a small molecule inhibitor that targets TBK1 and IKKε. Previous studies have confirmed the efficacy and safety of ALX for treating recurrent oral ulcers (aphthous ulcers) and asthma [22, 23]. ALX inhibits the release of inflammatory mediators, such as histamine and leukotrienes from mast cells, neutrophils and monocytes, which attenuates delayed allergic reactions and inflammatory effects [24, 25]. Recently, ALX has also shown significant anti-inflammatory effects in animal models and clinical trials of type II diabetes, nonalcoholic fatty liver disease, and obesity [26–29]. The mechanism involves the inhibition of TBK1 and IKKε by competing with ATP binding to the enzyme, thus blocking the phosphorylation of various proteins in the downstream signaling pathway. Based on these studies, ALX is currently considered a promising drug for inflammatory diseases, autoimmune diseases, metabolic diseases, and tumors [16, 30–33]. Using a mouse model, Quan et al. reported that ALX inhibited the progression of EAE by attenuating the activation of the TBK1/AKT and TBK1/IRF3 signaling pathways, therefore inhibiting dendritic cell maturation, reducing T-cell response, and attenuating the inflammatory response [34]. In clinical trials, ALX improved glycemic control in some patients with type 2 diabetes, and its efficacy is primarily associated with differences in the expression of inflammation-related genes in patients [27]. These findings suggest that ALX exhibits anti-inflammatory effects by targeting the inhibition of TBK1, modulating the activation of downstream signaling pathways, and regulating immune cell function [35]. However, there are no studies on whether ALX exerts a therapeutic effect on colonic inflammation during IBD.
In this study, we investigated the correlation between TBK1 and disease severity of IBD in dextran sulfate sodium (DSS)-induced colitis model, and further explored the efficacy, safety, and mechanism of ALX treatment in IBD. The implementation of this project is expected to provide a new strategy for the treatment of IBD.
Materials and methods
Animals
Female C57BL/6 mice (6–8 weeks, 17–18 g) were purchased from Chengdu Dashuo Laboratory (Chengdu, China). All mice were bred locally in the same dedicated pathogen-free facility with five mice per cage. All studies were approved and supervised by the Medical Ethics Committee of the Second West China Hospital of Sichuan University (No. 2,023,065).
DSS-induced colitis
The mice in the DSS and ALX groups were fed 3.5% dextran sodium sulfate (DSS, MW 36–50 kDa, MP Biomedicals, OH, USA) for five days, followed by distilled water (dH2O). During the experimental period, body weights were recorded daily and the disease activity index (DAI), including general appearance, weight loss, diarrhea and rectal bleeding, was calculated [36, 37]. A general appearance scoring rule was established as follows: normal = 0; piloerection = 1; lethargy and piloerection = 2; motionless and sickly = 4. The weight loss scoring rule was established as follows: no change = 0; <5% = 1; 6%–10% = 2; 11%–20% = 3; > 20% = 4. The severity of diarrhea scoring rule was established as follows: normal = 0; pasty and semiformal stools = 2; liquid stools = 4. The rectal bleeding scoring rule was established as follows: no blood = 0; visible blood in the rectum = 2; visible blood on the fur = 4. On Day 9, blood was collected from the mouse orbital sinus and the mice were sacrificed by cervical dislocation. Furthermore, the main organs of the heart, liver, spleen, lungs, kidneys, and whole colon tissues were isolated for further analysis. Colon length was measured as an indirect marker of inflammation.
Amlexanox treatment
The mice in ALX group were administered amlexanox (50 mg/kg) by gavage every other day from Days 0 to 6, and the mice in DSS group were administered an equal volume of solvent (10% DMSO + 40% PEG300 + 5% Tween-80 + 45% saline).
Survival analyses
For survival analyses, the mice in each group were fed 3% DSS for 7 days followed by dH2O. Drug treatment was extended to Day 8. Deaths were recorded daily to plot the survival curve. The survival observation period lasted for 60 days.
Cell isolation and culture
Splenocytes were isolated from C57BL/6 mice, and cultured in RPMI-1640 complete medium containing 10% fetal bovine serum. The splenocytes was stimulated with 20 μM ALX or not in 24-well plates (2 × 106 cells/well) for 24 h. For macrophages, cells can be harvested directly after ALX stimulation for flow cytometry analysis. For Th17 cells, ALX stimulation was followed by further stimulation with 2 μl/well leukocyte activation cocktail and BD GolgiPlugTM (containing Brefeldin A; BD Pharmingen, CA, USA) for 6 h, after which flow cytometry analysis was performed.
Histopathology
Heart, liver, spleen, lung, kidney, and colon tissues were washed with saline, fixed overnight with 4% paraformaldehyde, embedded in paraffin, sectioned at 4 μm, and stained with hematoxylin and eosin (H&E). The histopathological score of the colon tissue sections was determined, including severity of inflammation, depth of mucosal infiltration, degree of crypt breakage, and extent of the lesion [38, 39]. The severity of inflammation scoring rule was as follows: none = 0; mild = 1; moderate = 2; severe = 3. The depth of mucosal infiltration scoring rule was as follows: none = 0; pericryptal infiltration = 1; infiltration of the muscularis mucosa = 2; generalized infiltration of the muscularis mucosa with thickening of the mucosa = 3; and infiltration of the submucosal layer = 4. The degree of crypt disruption scoring rule was as follows: none = 0; damage to < 1/3 of crypt glands = 1; damage to < 2/3 of crypt glands = 2; complete loss of crypt glands = 3; destruction of the crypt and mucosal epithelium with marked inflammatory cell infiltration = 4. The extent of the lesion scoring rule was as follows: none = 0; 1%–25% = 1; 26%–50% = 2; 51%–75% = 3; 76%–100% = 4.
Enzyme-linked immunosorbent assay (ELISA)
The levels of IL-1β, IL-6, IL-10, TNF-α, TGF-β, and IFN-α in the mouse colon tissues, TBK1 levels in mouse serum, and IgA levels in mouse feces were measured using ELISA kits (Ruixin Biotech, Quanzhou, China) based on the manufacturer’s instructions. Briefly, mouse colon tissues were homogenized in RIPA buffer containing a protease/phosphatase inhibitor mixture (Roche, Basel, Switzerland) using a tissue homogenizer. The protein concentration in the supernatant was quantified using a BCA protein quantification kit. The cytokine content of the intestinal extracts is expressed as the amount per gram of tissue. Mouse feces were collected, washed three times with PBS (final feces mass: PBS volume = 1:10), and pulverized by ultrasonication. The supernatant was extracted after centrifugation and detected. Orbital blood was placed at room temperature for 10 min, and then centrifuged at 3000 rpm for 10 min to take serum for ELISA experiment.
Western blot analysis
After homogenizing the colon tissue, the supernatant was centrifuged, and the protein concentration was quantified using the BCA protein quantification kit. The proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane. After blocking, a primary antibody was added overnight at 4°C and the membrane was subsequently incubated with horseradish peroxidase-conjugated secondary antibody (1:5000). The following antibodies were used: TBK1 (Abcam, Cambridge, UK), IRF3, P65 (all 1:1000 dilution), and actin (1:5000 dilution). The gray value of the bands was analyzed using ImageJ software and the results were normalized. Relative expression was calculated.
Quantitative real-time polymerase chain reaction
Total RNA from the colon tissue was extracted using the mirVanaTM RNA isolation kit (Invitrogen, CA, USA) and cDNA was synthesized using a commercial qPCR kit (TransgenBiotech, Beijing, China). The primer sequences were as follows: TBK1, sense 5ʹ-TGGCTCCTGTCTGATATCC-3ʹ, antisense 5ʹ-CTTTTGACAG CATAGAGATCACC-3ʹ; Actin, sense 5ʹ-GGCTCCTAGCACCATGAAGA-3ʹ, antisense 5ʹ-AGCTCAGTAACAGTCCGCC-3ʹ. Reactions were performed on a LightCycler 480 II Real-time PCR instrument (Roche, Basel, Switzerland) using PerfectStartTM Green qPCR SuperMix (TransgenBiotech, Beijing, China). The values (relative expression level) were normalized to the level of the reference gene actin using the 2−ΔΔCT method.
Flow cytometry
After the mice were executed, spleens were crushed and splenocytes were isolated using 70 μm cell strainers (BD Biosciences, CA, USA). The erythrocytes were lysed, centrifuged to remove the supernatant, and resuspended in stain buffer (BD Biosciences, CA, USA) to obtain single-cell suspensions. For cell surface staining, single-cell suspensions were stained with fluorescent conjugated antibodies and incubated at 4°C for 30 min. For intracellular staining of IL-17A, the cells were stimulated with leukocyte activation cocktail and BD GolgiPlugTM (containing Brefeldin A; BD Pharmingen, CA, USA) for 6 h. Intracellular Foxp3 and IL-17A staining was performed using TF Fix/Perm buffer (BD Pharmingen, CA, USA), and intracellular CD206 staining was done using the fixation/permeabilization solution (BD Pharmingen, CA, USA) based on the manufacturer’s instructions. Anti-mouse antibodies (BD Biosciences, CA, USA) used for flow cytometry included anti-CD3-FITC, anti-CD4-BV605, anti-CD44-APC, anti-CD62L-PE-CY7, anti-CD25-PE, anti-IL17A-BV421, anti-Foxp3-AF647, anti-CD11b-PE, anti-CD11c-Cy5, anti-CD11b-PE, anti-CD11c-PE, PerCP-Cy5.5, anti-F4/80-BV421, and anti-CD206-FITC. Cells were detected using a BD FACSC Canto flow cytometer and the data were analyzed using FlowJo-v10.8.1 software. Harvested cells in vitro experiments stained in the same way as in vivo experiments.
Statistical analysis
All data were calculated based on three independent experiments and expressed as the mean ± SEM. Differences between two groups were compared by a two-tailed Student’s t-test, and P < 0.05 indicated that the data were significantly different.
Results
The expression level of TBK1 was correlated with the severity of colitis in mice
The study used the classical DSS-induced colitis model to determine whether TBK1 is associated with IBD (Fig. 1A). First, TBK1 levels were measured in the colonic tissues and serum of colitis and healthy mice. The results indicated that TBK1 levels were significantly higher in the DSS group compared with that in the control group (Fig. 1B and C). Next, we determined whether the severity of colitis, including weight loss and colon shortening, correlated with serum TBK1 levels. Interestingly, serum TBK1 levels were negatively correlated with colon length in the mice, whereas they were positively correlated with percent body weight reduction (Fig. 1D). The higher expression level of TBK1, the more severe symptoms of colitis. These data suggest that serum TBK1 levels are significantly associated with the severity of colitis. Western blot analysis revealed that TBK1 overexpression in mouse colon tissues activated the NF-κB and IRF3 signaling pathways (Fig. 1E). The activation of the TBK1-IRF3 signaling pathway can promote T-cell proliferation and lead to tissue damage, whereas the activation of the TBK1-NF-κB signaling pathway can promote the expression of inflammatory cytokines, and lead to inflammation. Thus, activation of pro-inflammatory signaling pathways by TBK1 exacerbates the inflammatory response in IBD. Taken together, these results suggest that TBK1 is involved in the development of DSS-induced colitis and its expression levels are associated with disease severity. Therefore, targeted inhibition of TBK1 may be effective in colitis treatment.
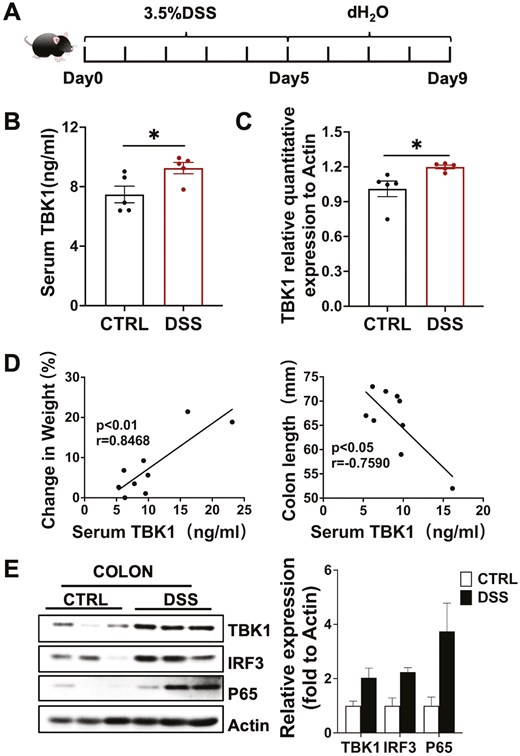
TBK1 levels are correlated with the disease severity of colitis. (A) Establishment of DSS-induced colitis model. (B) Serum TBK1 protein was detected by ELISA in both DSS and Control groups (n = 5/group). (C) Colon TBK1 mRNA was detected by real-time quantitative PCR (n = 5/group). P values determined by a two-tailed Student’s t-test. (D) The correlation between serum TBK1 levels and the severity of colitis, including colon length and percent body weight loss (n = 9/group). Statistically significant correlations were determined using linear regression. (E) The expression of TBK1 and the activation of TBK1-IRF3/NF-κB signaling in colon tissue was detected by western blot (n = 3/group). Data are presented as the mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. ns, not statistically significant
Amlexanox-targeted inhibition of TBK1 exacerbates DSS-induced colitis in mice
To determine the effect of the ALX-targeted inhibition of TBK1 on DSS-induced colitis, we performed a survival analysis of mice in the DSS and ALX groups. The results indicated that colitis was more severe in the ALX group compared with that in the DSS group, and 90% of the mice in the DSS group survived to Day 60, whereas only 50% of the ALX group survived under the same conditions (Fig. 2A and B). Next, the severity of colitis in each group of mice was assessed. The ALX group exhibited more pronounced weight loss, shortened colon lengths, higher DAI scores, and significantly worse symptoms of colitis compared with the DSS group (Fig. 2A, C–E). Histological analysis indicated that mice in the DSS and ALX groups exhibited colonic mucosal defects and destruction or disappearance of crypt glands, accompanied by a large infiltration of lymphocytes in the mucosa, submucosa, and even the muscularis propria. Simultaneously, inflammation and tissue damage of the distal colons of the mice in the ALX group was much more severe, and the histopathological scores of the colon were significantly higher in mice compared with those in the DSS group (Fig. 2F and G). These data suggest that ALX exacerbates the onset and progression of DSS-induced colitis, which was not consistent with our expectations.
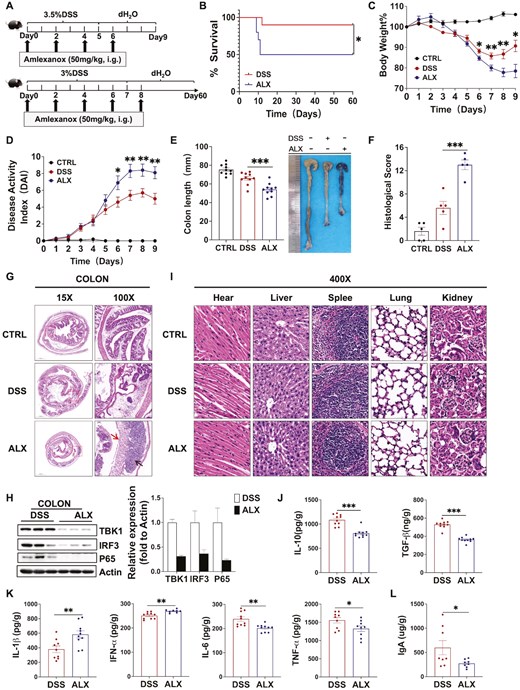
ALX-targeted inhibition of TBK1 exacerbates the occurrence and development of DSS-induced colitis. (A) Establishment of DSS-induced colitis model and amlexanox treatment. (B) Survival observation to Day 60. (C) Changes in body weight (%), (D) disease activity index, (E) colon length, (F) histology scores, and (G) representative H&E-stained colon sections of mice in the Control, DSS, and ALX groups. Severe submucosal inflammatory infiltration (arrow below), disappearance of crypt structures, and reduction of thrush cells (arrow above) were observed in the ALX group (H&E, 100×). (H) Western blot analysis showing that ALX inhibits the activation of the TBK1-IRF3/NF-κB signaling pathway in mouse colon tissues. (I) Major organs, including heart, liver, spleen, lung and kidney, were collected for histopathologic analysis (H&E, 400×). (J) ELISA measuring the concentration of anti-inflammatory cytokines IL-10 and TGF-βin colon. (K) ELISA measuring the concentration of pro-inflammatory cytokines IL-1β, IFN-α, IL-6, and TNF-αin colon. (L) ELISA results showing that the protein concentration of secretory IgA (sIgA) in the feces of mice in the ALX group is significantly lower compared with the DSS group. These results demonstrate that ALX-targeted inhibition of TBK1 exacerbates the occurrence and development of DSS-induced colitis and that the difference is independent of drug toxicity. Summary data are presented as the mean ± SEM, with P values determined by Kaplan–Meier analyses with the log-rank Mantel–Cox test (B) or a two-tailed Student’s t-test (C–K). *P < 0.05; **P < 0.01; ***P < 0.001. ns, not statistically significant
The assay of TBK1-regulated pro-inflammatory signaling pathway revealed the opposite result. Western blot analysis revealed that ALX inhibited the TBK1-NF-κB/IRF3 pro-inflammatory signaling pathway in mouse colon tissues, which was consistent with our expectation (Fig. 2H). Based on the difference between the clinical manifestation of colitis and the results of proinflammatory signaling, we further determined whether the exacerbation of colitis in the ALX group was associated with drug toxicity. The in vivo toxicity of ALX was assessed by a histopathological analysis of the heart, liver, spleen, lungs and kidneys of mice. At the experimental dose of ALX, the mice showed no infiltration or degenerative lesions in the normal structures of all organs (Fig. 2I). ALX was absorbed primarily through the gastrointestinal tract and excreted via the kidneys without accumulation in the organs, which also indicates that ALX was not toxic to the mice. These results suggest that ALX exacerbates the occurrence and development of DSS-induced colitis, but it is independent of drug toxicity.
Amlexanox bidirectionally regulates cytokines secretion and negatively modulates intestinal mucosal immunity
After excluding the cause of drug toxicity, we measured the concentration of cytokines in the colon tissues, including IL-1β, IL-6, IL-10, TNF-α, TGF-β, and IFN-α. ELISA revealed that the anti-inflammatory cytokines IL-10 and TGF-β were significantly reduced, whereas the pro-inflammatory cytokines IL-1β and IFN-α were significantly increased; however, IL-6 and TNF-α were significantly decreased in the ALX group compared with the DSS group (Fig. 2J and K). These data suggest that ALX has a bidirectional role in regulating cytokines secretion in mice with colitis. ELISA detection of fecal secretory immunoglobulin A (sIgA) levels revealed a significantly lower sIgA concentration in the ALX group compared with that in the DSS group (Fig. 2L). sIgA is an important intestinal immunoglobulin that maintains intestinal immune balance by neutralizing and clearing pathogens in the intestine. Therefore, the inhibition of sIgA secretion by ALX reduces the intestinal immune defense capability of mice with colitis. These data indicate that the ALX-targeted inhibition of TBK1 bidirectionally regulates cytokines secretion and negatively modulates intestinal mucosal immunity, and therefore disrupts the balance of the immune system and the direction of inflammation regulation in mice with colitis.
Amlexanox promotes T-cell proliferation, activation, and differentiation in mice with colitis
As a target of ALX, TBK1 is not only involved in immunity and inflammation but also a key mediator of cell proliferation and growth. Additionally, the secretion of cytokines influenced by a variety of immune cells. Therefore, the anti-inflammatory activity of ALX may be affected by the regulatory effect of TBK1 on immune cell function. To determine the effect of ALX on the regulation of different immune cells in IBD, flow cytometry was used to detect the phenotype and the number of different immune cells in the spleen of mice with DSS induced colitis. Flow cytometry revealed that compared with the DSS group, the proportion of CD4+ T cells in the spleen of mice in the ALX group was significantly higher and the absolute number was significantly increased (Fig. 3A). Among the CD4+ T-cell subsets in the ALX group, the proportion of naive T cells (CD44-CD62L+) was significantly decreased, whereas the absolute number of effector T cells (CD44+CD62L−) was significantly increased. Additionally, both the absolute number and the proportion of memory T cells (CD44+CD62L+) were significantly lower (Fig. 3B). This suggests that ALX promotes CD4+ T lymphocyte proliferation and activation. Next, we performed flow cytometry analysis of T helper type 17 cells (CD4+ IL−17A+, Th17) and regulatory T cells (CD4+CD25+Foxp3+, Treg), which are subsets differentiated from CD4+ initial T lymphocytes, in the spleens of mice of the DSS and ALX group. The flow cytometry results indicate that compared with the DSS group, Treg cells were slightly reduced in the ALX group, but the difference was not statistically significant, whereas the proportion of Th17 cells was significantly higher in the ALX group, and the absolute number was also significantly higher (Fig. 3C and D). These results suggest that ALX-targeted inhibition of TBK1 promotes the proliferation, activation, and differentiation of T lymphocytes, resulting in increased differentiation of CD4+ T cells into Th17 cells, and ultimately aggravates the inflammatory response in mice with colitis.
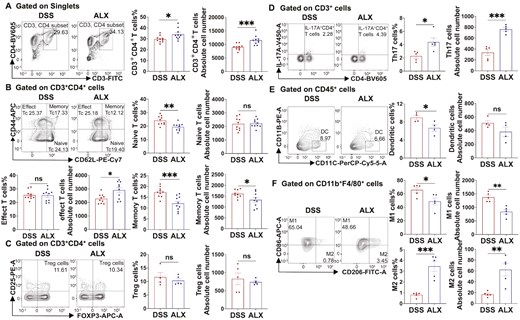
ALX regulates the function of T cells, dendritic cells, and macrophages in colitis mice. (A, B) CD4+ T lymphocytes and their subsets of naive T cells (CD44−CD62L+), memory T cells (CD44+CD62L+), and effector T cells (CD44+CD62L−) in spleen of mice in both DSS and ALX groups (n = 8–10/group) were analysis by flow cytometry. (C, D) Th17 cells (CD4+ IL-17A+) and regulatory T cells (Treg) (CD4+ CD25+ Foxp3+) in the spleen were analysis by flow cytometry (n = 4–5/group). (E, F) DCs (CD45+CD11B−CD11C+) and macrophages in spleen were analysis by flow cytometry (n = 4–5/group). According to the expression of CD206, macrophages were divided into M1 macrophages (CD86+ CD206-) and M2 macrophages (CD86+CD206+). Data are expressed as the mean ± SEM. P values were tested using a two-tailed Student’s t-test. * P < 0.05; ** P < 0.01; *** P < 0.001. ns, not statistically significant
Amlexanox suppresses DC generation and M1 macrophage polarization in mice with colitis
In addition to the assay of T cells, we further investigated the effect of ALX on dendritic cells (DC) and macrophages by flow cytometry. The results indicated that compared with the DSS group, the proportion of DC (CD45+CD11B−CD11C+) and M1 macrophages (CD86+CD206−) in the ALX group were significantly lower, whereas the proportion and absolute number of M2 macrophages (CD86−CD206+) was significantly higher (Fig. 3E and F). This suggests that the ALX suppresses DC generation and inflammatory macrophage polarization. Dendritic cells are known to be antigen presenting cells that are involved in the cross-talk between innate and adaptive immunity, and are responsible for T-cell activation and induction of the adaptive immune response. Macrophages are also involved in the pathogenesis of IBD, among which M1 macrophages have pro-inflammatory effects and participate in the secretion of inflammatory cytokines, whereas M2 macrophages exhibit anti-inflammatory effects and are involved in tissue repair following injury. These data show that ALX-targeted inhibition of TBK1 suppresses DC generation and M1 macrophage polarization, thus exerting an attenuating effect on inflammation.
Amlexanox promotes Th17 cells production and inhibits M1 macrophage polarization in vitro
We next sought to determine whether ALX could affect these immune cells in vitro. Isolated splenocytes from healthy mice were cultured in the presence or absence of ALX (20 μM) for 24 h and then stimulated with leukocyte activation cocktail for 6 h. Flow cytometry was performed on the harvested cells to detect Treg cells, Th17 cells, DC, and macrophages in different groups. The results showed a significantly higher proportion of Th17 cells and M2 macrophages and a significant decrease in M1 macrophages in the ALX group compared with the control group (Fig. 4B and D). Meanwhile, there was no significant difference in Treg cells between the two groups (Fig. 4A). These results are consistent with the effects of ALX on these immune cells in vivo. However, inconsistent with the performance of the in vivo experiments, ALX did not affect DC production in vitro (Fig. 4C). Thus, in vitro experiments confirmed that ALX targeted-inhibited TBK1 promoted Th17 production and inhibited M1 macrophage polarization.
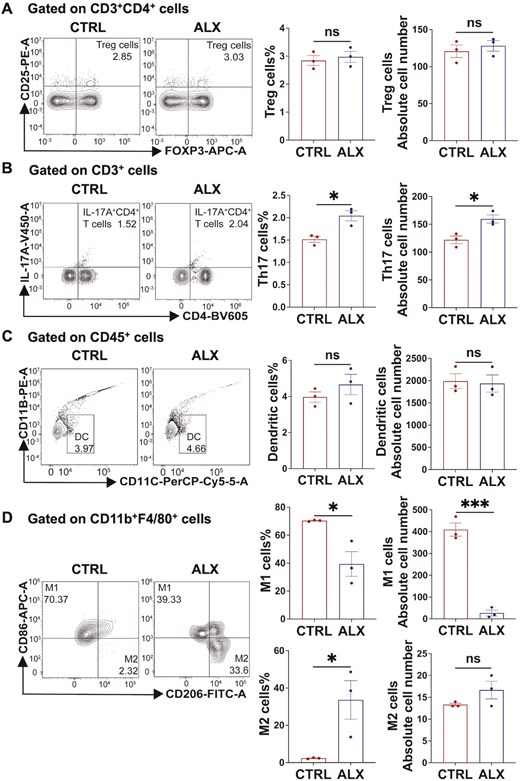
Amlexanox promotes Th17 cells production and inhibits M1 macrophage polarization in vitro. (A, B) Flow cytometry of Th17 cells and Treg cells from the ALX and control groups in vitro (n = 3/group). (C, D) Flow cytometry of DCs and macrophages from the ALX and control groups in vitro (n = 3/group). These data indicated that compared with the CTRL group, the ALX group had significantly higher proportions of Th17 cells and M2 macrophages, and significantly lower M1 macrophages. Treg and DC were not significantly different between the two groups. Data are expressed as the mean ± SEM. P values were determined by a two-tailed Student’s t-test. *P < 0.05; **P < 0.01; ***P < 0.001. ns, not statistically significant.
Discussion
In this study, we used DSS-induced colitis in mice to determine the correlation between TBK1 and colitis severity. We confirmed a positive correlation between serum TBK1 levels and the severity of colitis (Fig. 1A–D). The results of western blot analysis also demonstrated that over expression of TBK1 activates the pro-inflammatory signaling pathway associated with IBD (Fig. 1E). Using ALX-targeted inhibition of TBK1, we found that ALX exacerbates DSS-induced colitis. Compared with mice in the DSS group, the ALX group had reduced survival time and exhibited more severe symptoms of colitis, including greater weight loss, shortened colon length, higher disease activity, and more severe colorectal inflammatory infiltration (Fig. 2B–G); however, ALX inhibited the activation of the TBK1-NF-κB/IRF3 signaling pathway (Fig. 2H). We next examined the basis for the discrepancy between the clinical manifestations of colitis and the results of the pro-inflammatory signaling. After excluding the possibility that drug toxicity was responsible for the more severe symptoms of colitis, we measured the concentration of cytokines in colonic tissues (Fig. 2I–K). Interestingly, ALX regulated cytokines secretion in a bidirectional manner. Because cytokine secretion is influenced by a variety of immune cells, the inflammatory regulatory effects of ALX may be affected by the regulatory effects of TBK1 on different immune cell functions [20, 34, 40].
To examine the mechanism of this unexpected finding, we determined the effect of ALX on different immune cells in mice with DSS-induced colitis by flow cytometry. The results indicated that ALX promotes T-cell proliferation, activation, and differentiation, but has an inhibitory effect on the production of DC and polarization of M1 macrophages (Fig. 3A–F). Also, flow cytometry results from in vitro assays have confirmed that ALX promotes Th17 cell production and inhibits M1 macrophage polarization (Fig. 4A–D). T cells play an important role in the exacerbation of colonic inflammation and pathological damage in inflammatory bowel disease; however, the reduction in DC and M1 macrophages attenuates the severity of the disease. Consistent with our findings, in a DSS-induced mouse model of colitis, conditional knockout TBK1 in myeloid cells significantly increased serum concentrations of pro-inflammatory cytokines, mainly IL-1β, leading to exacerbation of colitis in mice [36]. Similarly, conditional knockout TBK1 in intestinal epithelial cell leads to increased secretion of IL-1β in intestinal macrophages, which in turn promotes T-cell proliferation and differentiation of pro-inflammatory Th17 cells [41]. These findings suggest that ALX has a regulatory effect on different immune cells, and different regulatory effects indirectly affect the inflammatory response of colitis mice. Thus, ALX in different immune cells regulates inflammation differently in colitis mice. Previous studies have also demonstrated that TBK1 knockdown in different cells exhibits varying effects on inflammatory diseases. In the same EAE mouse model, the specific knockdown of the TBK1 gene in DC and T cells results in completely different effects. Studies of conditional knockout of T cells TBK1 in mice have shown that TBK1 deficiency impairs T-cell migration, resulting in the retention of T cells in lymph node and a decrease in the number of T cells in the central nervous system, thereby inhibiting the development of EAE in mice [20]. However, conditional knockout of dendritic cells TBK1 results in increased expression of the costimulatory molecules CD80 and CD86 in mouse splenic DCs, which promotes DC-mediated immune stimulation of T cells, leading to earlier onset and increased severity of EAE [42]. These studies suggest that TBK1 knockdown in different immune cells has different effects on inflammatory disease, even in the same disease model. This confirms that the functional regulation of TBK1 on different immune cells affects its role in the regulation of inflammation. Furthermore, a study using ALX to treated mice with EAE showed that ALX inhibited DC maturation by attenuating TBK1/AKT and TBK1/IRF3 signaling, as well as suppressed the production of Th1 and Th17 cells, which attenuated the development of EAE [34]. This indicates a difference in the regulation of immune cell function between the application of ALX and TBK1-specific knockdown. Unlike TBK1-specific knockdown, ALX inhibits TBK1 in all cells in vivo. The regulatory effect of ALX on inflammation is the sum of the effects of various immune cells, and ultimately exhibit either anti-inflammatory or pro-inflammatory effects. This explains why the clinical manifestations of DSS-induced colitis are exacerbated despite the inhibition of the pro-inflammatory signaling pathway by ALX-targeted inhibition of TBK1.
Of course, there are some limitations to our study, such as ALX is a dual inhibitor that targets TBK1 and IKKε, but this study only focused on its role in targeting TBK1. The main reason is that, unlike TBK1, which is widely expressed in all tissues, IKKε expression is restricted to specific tissues such as lymphoid tissues, peripheral blood lymphocytes, and pancreas [35]. Moreover, the high expression of IKKε in cells was mainly found in hepatic stellate cells, adipocytes, and adipose tissue macrophages [43]. Therefore, the evidence in the present studies does not support the relevance of IKKε to IBD, and IKKε is not a key target of interest in this study. Notably, for diseases with high IKKε expression such as nonalcoholic fatty liver disease (NAFLD), special attention should be paid to the dual targeting effect of ALX. This also suggests the urgent need to develop specific inhibitors for TBK1 in the future.
In this study, we demonstrated that ALX-targeted inhibition of TBK1 negatively regulates the development of IBD. Both in vitro and in vivo experiments confirmed that ALX aggravated the inflammatory response of colitis by promoting the production of Th17 and inhibiting the polarization of M1 macrophages; however, the specific molecular mechanism by which ALX regulates the function of T cells, dendritic cells and macrophages remains to be further studied. In conclusion, our study suggests that the inhibition of total TBK1 in all cells is ineffective for the treatment of colitis, and future studies can focus on exploring the anti-inflammatory mechanism of ALX targeting dendritic cells and macrophages, thereby providing new therapeutic strategies for the treatment of IBD.
Abbreviations:
- ALX
amlexanox
- DSS
dextran sodium sulfate
- IBD
inflammatory bowel disease
- UC
ulcerative colitis
- CD
Crohn’s disease
- 5-ASA
aminosalicylates
- TBK1
TANK-binding kinase 1
- EAE
experimental autoimmune encephalomyelitis
- dH2O
distilled water
- DAI
disease activity index
- H&E
hematoxylin and eosin
- ELISA
enzyme-linked immunosorbent assay
- sIgA
secretory Immunoglobulin A
- Th17
T helper type 17 cells
- Treg
regulatory T cells
- DCs
dendritic cells
- NAFLD
nonalcoholic fatty liver disease
Ethical statement
The animal study was reviewed and approved by Medical Ethics Committee of the Second West China Hospital of Sichuan University (No. 2,023,065).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the Sichuan Science and Technology Program (Grant No. 2023YFS0186 and 2023YFS0222) and the Clinical Research Foundation of West China Second University Hospital (Grant No. KL066 and KL076).
Data availability
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding authors.
Author contributions
Lu Hui and Ting Liu drafted the manuscript. Lu Hui, Meng-ke Huang, Qing-Kai Dai, Cheng-lin Miao, Yun-long Yang, Chen-Xi Liu, and Ting Liu generated the experimental results. YMJ and TL reviewed the manuscript for intellectual content. All authors approved the final version of the manuscript. All the authors have accepted responsibility for the entire content of this submitted manuscript and approved the submission.
References
Author notes
Lu Hui and Meng-ke Huang contributed equally to this work and share first authorship.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}