





Abstract
A reduction in the level of thioredoxin 1 (Trx1) has been proposed as a possible mechanism for the tumor-specific growth arrest caused by inhibition of histone deacetylases (HDACs). In this study, we investigated the effect of trichostatin A (TSA), a potent HDAC inhibitor, on the proliferation and migration of vascular smooth muscle cells (VSMCs), and we examined the role of reduced Trx1 levels in this effect.
TSA treatment time-dependently decreased Trx1 expression in rat VSMCs at both the mRNA and protein levels. It also enhanced platelet-derived growth factor (PDGF)-induced proliferation and migration of the VSMCs. By potentiating Akt phosphorylation, the siRNA-induced downregulation of Trx1 also enhanced VSMC proliferation and migration in response to PDGF or serum treatment. Consistent with these results, TSA administration increased neointimal thickening in a murine model of post-angioplastic restenosis.
These data demonstrate that TSA enhances vascular proliferative activity by downregulating Trx1, thus activating an Akt-dependent pathway. Our results indicate that, in addition to its apoptotic effects in tumour cells, the downregulation of Trx1 has a proliferative role in primary VSMCs.
1. Introduction
Superoxide anions, hydrogen peroxide, and other reactive oxygen species (ROS) are generated inside cells in response to physiological stimulation by growth factors or cytokines. ROS affect signal transduction pathways by directly or indirectly modifying signal molecules such as protein kinases, transcription factors, phospholipases, phosphatases, ion channels, and G proteins.1 Dysregulated ROS generation has been implicated in the pathophysiology of various human diseases, including cancer and cardiovascular disease.1
By modulating gene expression, the regulation of histone acetylation by histone acetyl transferase and histone deacetylase (HDAC) plays an important role in cell growth and differentiation. Preclinical studies have demonstrated that HDAC inhibitors, such as trichostatin A (TSA), suberoylanilide hydroxamic acid, and NSC3852,2 exert significant anti-proliferative effects on various cancers by inducing apoptosis, growth arrest, and differentiation.3–6 A reduced level of thioredoxin 1 (Trx1) leading to generation of ROS and ultimately to mitochondrial dysfunction is alleged to be responsible for the HDAC inhibitor-mediated cytotoxicity observed in transformed tumour cells.2,7
By acting as a hydrogen donor for many protein targets and scavengers of ROS, such as peroxiredoxin (Prx), Trx plays a key role in the redox regulation of signal transduction. Physiologically, Trx is thought to exert a cytoprotective effect against oxidative stress by scavenging ROS, either by itself or in conjunction with Prx.8 The exact function of Trx1 in normal tissues remains unclear, however; Trx1-deficient embryos exhibit early lethality, suggesting that Trx1 is essential for normal development.9
The abnormal proliferation and migration seen in vascular smooth muscle cells (VSMCs) are major pathophysiological processes seen in various cardiovascular diseases, including atherosclerosis, post-angioplastic vascular remodelling, and transplant vasculopathy.10 Accordingly, the aim of this study was to examine the therapeutic potential of TSA as an anti-proliferative drug for vascular disease. Unexpectedly, we found that TSA has a paradoxical pro-atherogenic effect on VSMCs via the reduction of Trx1.
2. Methods
2.1 Cell culture
Primary cultures of rat aortic smooth muscle cells were prepared from the thoracic and abdominal aortas of 5-week-old male Sprague-Dawley rats (150–200 g). Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and were used between passages 5 and 10. For the experiments, a confluent monolayer of primary cultured VSMCs was subcultured every 5–7 days (1:5 ratio) up to 10 passages. For the standard experiments, primary VSMCs were maintained at 80% of confluence. The purity of the cultures was analysed using FACStar (Becton Dickinson, Mountain View, CA, USA) after staining with fluorescein isothiocyanate (FITC)-conjugated antibody against α-smooth muscle actin (SMA). Cells were used for experiments when more than 97% of the cells were positive for SMA.
2.2 Reagents
Trichostatin A and platelet-derived growth factor (PDGF)-BB were obtained from Sigma-Aldrich (St Louis, MO, USA). LY294001 and U0126 were purchased from Calbiochem (La Jolla, CA, USA). HeLa cells were grown in DMEM supplemented with 10% FBS. 3-(4,5-Dimethylthiaziazol-2-yl)2,5-diphenyl tetrazolium bromide (MTT) and dimethylsulfoxide (DMSO) were purchased from Sigma-Aldrich. 2',7'-dichlorofluorescein diacetate (H2DCF-DA) and TRIzol reagent were from Invitrogen (Carlsbad, CA, USA). Antibodies against Trx1, Trx2, Prx1, and Prx2 (Ab Frontier, Suwon, Korea) were used to detect antioxidants. Antibodies against p-Akt, Akt, p-ERK, ERK, and β-actin were obtained from Cell Signaling Technology (Beverly, MA, USA). LipofectAMINE (Invitrogen) was used for the knockdown experiment, and small interfering RNA (siRNA) against rat Trx 1 and Prx 2 was obtained (Bioneer, Daejeon, Korea), and a scramble siRNA was used as a control.
2.3 MTT assay
Cell growth and viability were analysed using the MTT method. Briefly, cells were incubated with 0.5 g/L MTT for 2 h. After the formation of formazan crystals, the culture supernatant was removed, and the formazan crystals were dissolved in DMSO. The absorbance of the solution at 570 nm was measured using a microplate reader (Bio-Rad, Richmond, CA, USA).
2.4 Measurement of intracellular ROS levels
To analyse intracellular ROS levels, the oxidation-sensitive fluorescence probe H2DCF-DA was used as previously described.11 H2DCF-DA was pre-diluted to a final concentration of 2.5 µmol/L. Cellular fluorescence was measured using an inverted epifluorescence microscope (Zeiss, Göttingen, Germany).
2.5 Western blot analysis
Soluble cell extracts were prepared, and aliquots containing 20 µg of total protein were separated on 12 or 13.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS–PAGE) gels and transferred to nitrocellulose membranes. The membranes were probed with specific antibodies against the corresponding proteins.
2.6 Reverse transcriptase–polymerase chain reaction
Total RNA was extracted using TRIzol reagent. First-strand cDNA was synthesized from 1 µg of total RNA under RNase-free conditions and amplified by PCR using primers for human and rat Trx1, rat Prx 2, and various components of NADPH oxidase complexes. The primers used for PCR are given in Table 1. PCR reactions were performed using an initial denaturation step at 95°C (5 min) followed by 20–25 cycles of 94°C (20 s), 60°C (30 s), and 72°C (30 s).
Nucleotide sequences of primer used for RT–PCR
| Forward (5′–3′) | Reverse (3′–5′) | |
|---|---|---|
| Trx 1 | TCAGGAGGCCCTGGCCGCTG | TAGCACCAGAGAACTCCCCAACC |
| Prx 2 | CCGAAAGCTAGGCTGCGAGG | AAGGCCTGGACGAGGCGGAG |
| NOX 1 | GTGGCTTTGGTTCTCATGGT | TGAGGACTCCTGCAACTCCT |
| NOX 2 | ACCCTTTCACCCTGACCTCT | TCCCAGCTCCCACTAACATC |
| NOX 4 | GGGCCTAGGATTGTGTTTGA | CTGAGAAGTTCAGGGCGTTC |
| p22 phox | TTGTTGCAGGAGTGCTCATC | CTGCCAGCAGGTAGATCACA |
| β-Actin | ATGCCCCGAGGCTCTCTTCC | TAGGAGCCAGGGCAGTAATCT |
| Forward (5′–3′) | Reverse (3′–5′) | |
|---|---|---|
| Trx 1 | TCAGGAGGCCCTGGCCGCTG | TAGCACCAGAGAACTCCCCAACC |
| Prx 2 | CCGAAAGCTAGGCTGCGAGG | AAGGCCTGGACGAGGCGGAG |
| NOX 1 | GTGGCTTTGGTTCTCATGGT | TGAGGACTCCTGCAACTCCT |
| NOX 2 | ACCCTTTCACCCTGACCTCT | TCCCAGCTCCCACTAACATC |
| NOX 4 | GGGCCTAGGATTGTGTTTGA | CTGAGAAGTTCAGGGCGTTC |
| p22 phox | TTGTTGCAGGAGTGCTCATC | CTGCCAGCAGGTAGATCACA |
| β-Actin | ATGCCCCGAGGCTCTCTTCC | TAGGAGCCAGGGCAGTAATCT |
Nucleotide sequences of primer used for RT–PCR
| Forward (5′–3′) | Reverse (3′–5′) | |
|---|---|---|
| Trx 1 | TCAGGAGGCCCTGGCCGCTG | TAGCACCAGAGAACTCCCCAACC |
| Prx 2 | CCGAAAGCTAGGCTGCGAGG | AAGGCCTGGACGAGGCGGAG |
| NOX 1 | GTGGCTTTGGTTCTCATGGT | TGAGGACTCCTGCAACTCCT |
| NOX 2 | ACCCTTTCACCCTGACCTCT | TCCCAGCTCCCACTAACATC |
| NOX 4 | GGGCCTAGGATTGTGTTTGA | CTGAGAAGTTCAGGGCGTTC |
| p22 phox | TTGTTGCAGGAGTGCTCATC | CTGCCAGCAGGTAGATCACA |
| β-Actin | ATGCCCCGAGGCTCTCTTCC | TAGGAGCCAGGGCAGTAATCT |
| Forward (5′–3′) | Reverse (3′–5′) | |
|---|---|---|
| Trx 1 | TCAGGAGGCCCTGGCCGCTG | TAGCACCAGAGAACTCCCCAACC |
| Prx 2 | CCGAAAGCTAGGCTGCGAGG | AAGGCCTGGACGAGGCGGAG |
| NOX 1 | GTGGCTTTGGTTCTCATGGT | TGAGGACTCCTGCAACTCCT |
| NOX 2 | ACCCTTTCACCCTGACCTCT | TCCCAGCTCCCACTAACATC |
| NOX 4 | GGGCCTAGGATTGTGTTTGA | CTGAGAAGTTCAGGGCGTTC |
| p22 phox | TTGTTGCAGGAGTGCTCATC | CTGCCAGCAGGTAGATCACA |
| β-Actin | ATGCCCCGAGGCTCTCTTCC | TAGGAGCCAGGGCAGTAATCT |
2.7 Transfection of small interfering RNA
Cells were transfected with small interfering RNA (siRNA) using LipofectAMINE 2000 according to the manufacturer's protocol. The siRNAs targeting human and rat Trx1 were generated based on nucleotides 246–264 and 268–286, respectively, of the Trx1 cDNAs from these organisms and were each used at 40 nmol/L. The siRNAs targeting rat Prx2 were generated based on nucleotides 789–807 of the Prx2 cDNAs. A scrambled siRNA was used as a negative control.
2.8 Wound-healing assay
Adherent cells (2 × 105) were scraped off the bottom of a culture plate using a pipette tip to create a cell-free (wounded) area. The cell culture was washed with phosphate-buffered saline to remove cell debris and then incubated with PDGF for 48 h. Migration distance was determined using i-Solution (iMTechnology, Korea), and the shortest distance between cells that had moved into the wounded region and their respective starting points was determined.
2.9 Murine carotid ligation model
The carotid artery ligation model was described previously.12 Male C57/BL6 mice (20–25 g) were used for all experiments and were handled in compliance with the Guide for the Care and Use of Laboratory Animals of Ewha Women's University, Seoul, Korea. Mouse carotid artery remodelling was induced through ligation of the left common carotid proximal to its bifurcation. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
2.10 Statistical analysis
Data are presented as the mean ± significant deviation (SD). Levels of significance for comparisons between two independent samples were determined using the Student's t-test. Groups were compared using ANOVA with Tukey's ‘honestly significant difference’ post hoc test applied to significant main effects (SPSS 12.0K for Windows, SPSS, Chicago, IL, USA).
3. Results
3.1 TSA potentiates vascular proliferation in vitro and in vivo
To test the effect of TSA on vascular proliferation in vivo, we used a mouse carotid ligation model. Two weeks after ligation, quantitative analysis demonstrated that the neointimal area was 2.5 times larger in TSA-treated animals than in control animals (0.020 ± 0.014 vs. 0.050 ± 0.052 mm2; P = 0.007, Student t-test) (Figure 1). The neointimal/medial area ratio was as much as 2.2 times larger in TSA-treated animals than in non-treated animals (2.01 ± 1.67 vs. 0.57 ± 0.45, P = 0.013). Contrary to our initial expectation that an HDAC inhibitor might have an anti-proliferative effect, these results clearly indicate that TSA enhances VSMC proliferation after carotid ligation injury in vivo.
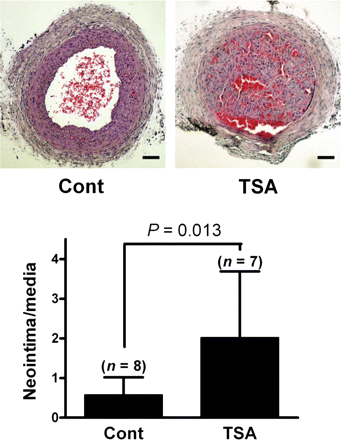
Prior exposure to an HDAC inhibitor potentiates neointima formation in balloon-injured arteries. Animals were injected with either vehicle (10 mL/kg, 2% DMSO in saline) (n = 8) or TSA (1 mg/kg; at 10 mL/kg, 2% DMSO in saline) (n = 7) intraperitoneally at the time of ligation injury, 1 day before injury, and 6 days after injury. The quantification of neointimal thickening was performed 2 weeks after ligation using the computer program i-Solution. Scale bar = 100 µm.
We also examined whether TSA exposure affected the proliferation of VSMCs in vitro. To this end, VSMCs were exposed to TSA before stimulating growth with PDGF. As expected, the growth rate of VSMCs treated with 1 µg/L PDGF for 24 h was up to three times greater than that of the untreated control VSMCs. This stimulatory effect of PDGF on VSMC growth was significantly increased by a 48 h pre-treatment of the cells with TSA (Figure 2A), indicating that exposure to TSA significantly increases VSMC proliferation.
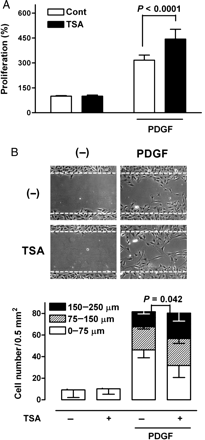
Effect of TSA on PDGF-induced growth and migration of VSMCs. (A) VSMCs were incubated in the absence or presence of 0.5 µmol/L TSA for 48 h and then treated with 1 µg/L PDGF for an additional 24 h. Growth rates were measured using the MTT test. (Tukey post hoc test applied to significant group effects in analysis of variance: F3,28 = 205.19, P < 0.001). (B) VSMCs were incubated in the absence or presence of TSA for 48 h and scraped with a pipette tip. Cells were then incubated in the absence or presence of 5 µg/L PDGF for 48 h. P-values are indicated for statistically significant differences (Student's t-test). The vertical axis represents the number of cells that migrated into the wounded region. Stacked bars display the portion of migratory cells classified by migration distance.
Next, we analysed the effect of TSA on PDGF-induced migration, another important phenotype of VSMCs, using a wound-healing assay. TSA had a minimal effect on the total number of cells induced to migrate by PDGF, but it did increase the proportion of cells that migrated farther: the proportion that moved more than 150 µm was higher than in the group treated with PDGF alone (Figure 2B). These results clearly indicate that the PDGF-induced growth and migration of VSMCs are augmented by TSA in vitro and in vivo.
3.2 TSA increases the level of intracellular ROS by downregulating Trx1 expression
In transformed tumour cells, HDAC inhibitors have been associated with increased intracellular oxidative stress resulting from the transcriptional perturbation of antioxidant proteins.13 Therefore, we examined the effect of TSA on intracellular ROS levels in VSMCs. We found that exposure of VSMCs to TSA led to a significant increase in the level of intracellular ROS (Figure 3A). When we subsequently examined whether TSA exposure affected the expression of antioxidant proteins in VSMCs, we found that TSA exposure led to a significant decrease in the level of Trx1 in a time-dependent manner; whereas the expression of Trx 2, another isotype of Trx was unaffected by the same treatment (Figure 3B). The expression of Prx1 and Prx 2 was also suppressed by TSA treatment. RT–PCR analysis showed that TSA treatment decreased the levels of Trx1 and Prx2 mRNA (Figure 3C). These results clearly indicate that TSA treatment reduced the expression of Trx1 and Prx2, at least at the mRNA level. We further tested the possibility that the TSA-induced increase in intracellular ROS is caused by altered expression of NADPH oxidase complex, which is the major enzyme responsible for intracellular ROS generation in response to various extracellular stimuli.14 As expected, TSA treatment had no significant effect on the mRNA expression levels of NADPH oxidase components including NOX proteins (Figure 3D).
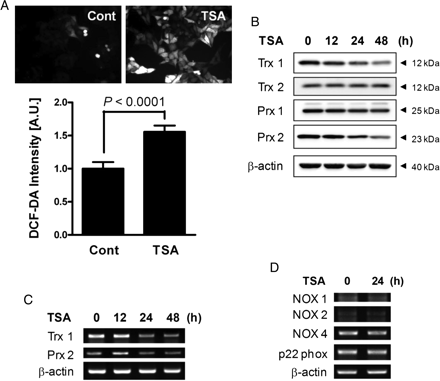
The effect of TSA on antioxidant expression and intracellular ROS levels. (A) VSMCs were incubated in the absence or presence of 0.5 µmol/L TSA for 48 h, and intracellular ROS levels were determined by staining with DCF-DA. P-values are indicated for statistically significant differences (Student's t-test). (B) VSMCs were incubated with TSA for various lengths of time, and soluble cell lysates were subjected to western blotting for Trx1/2 and Prx1/2. The results shown are representative of three independent experiments. (C) Trx1 and Prx2 mRNA levels were determined by RT–PCR. (D) VSMCs were incubated in the absence or presence of 0.5 µmol/L TSA for 24 h, and mRNA levels of various NADPH oxidase components were determined using RT–PCR.
A role for Trx1 downregulation in the observed increase in ROS levels was confirmed using Trx1-specific siRNA. Downregulation of the Trx1 protein level by the transfection of Trx1-specific siRNA (Figure 4A) significantly increased the DCF-DA fluorescence intensity (Figure 4B), indicating an increase in ROS accumulation. We observed similar results in HeLa cells (see Supplementary material online, Figure S1). These results suggest that TSA increases intracellular ROS levels in VSMCs and HeLa cells by suppressing the expression of antioxidant proteins such as Trx1 and Prx 2.
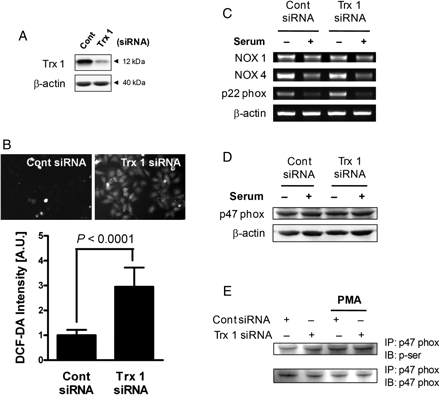
Effect of Trx 1 knockdown on the activation of the NADPH oxidase component. (A) VSMCs were transfected with Trx1-specific siRNA and cultured for 48 h, at which time the efficiency of transfection was determined by western blotting. (B) In Trx1 siRNA-transfected cells, intracellular ROS levels were determined by staining with DCF-DA. P-values are indicated for statistically significant differences (Student's t-test). (C) VSMCs transfected with Trx1-specific siRNA were incubated in the absence or presence of 5% serum for 4 h, and then the mRNA levels of NADPH oxidase components were determined by RT–PCR. (D) VSMCs transfected with Trx1-specific siRNA were incubated in the absence or presence of 5% serum for 4 h, and soluble cell lysates were subjected to immunoblot analysis for p47phox. (E) VSMCs were transfected with Trx1 siRNA for 48 h, and the lysates were then immunoprecipitated with anti-p47 phox. Phorbol myristate acetate (PMA)-treated cells were used as a positive control. The immunocomplex was subjected to immunoblotting with anti-phosphoserine or anti-p47 phox, as indicated on the right.
To further confirm the involvement of Trx 1 on the expression of NADPH oxidase components, we examined the effect of Trx 1 knockdown on the serum-induced expression of NADPH oxidase components. Serum addition suppressed the NOX 4 and p22 phox expression, and this suppressive effect did not differ between cont siRNA- and Trx 1 siRNA-transfected cells (Figure 4C). We also showed that the expression and phosphorylation of p47 phox, the cytosolic component of NADPH oxidase, were not changed by Trx 1 knockdown (Figure 4D and E). These results collectively indicate that Trx 1 knockdown does not affect the activation of membranous and cytosolic NADPH oxidase components.
3.3 Downregulation of Trx1 increases proliferation and migration of VSMCs
To further investigate the involvement of Trx 1 in the growth of VSMCs, we examined the effect of Trx 1 downregulation on the proliferation of VSMCs. The results demonstrate that siRNA-induced inhibition of Trx1 expression significantly upregulated the growth rate of VSMCs in the presence of PDGF or serum (Figure 5A and B). This finding is clearly contradictory to previous reports that the inhibition of Trx1 by TSA treatment or siRNA significantly suppressed the growth of HeLa cells (see Supplementary material online, Figure S2).5,6
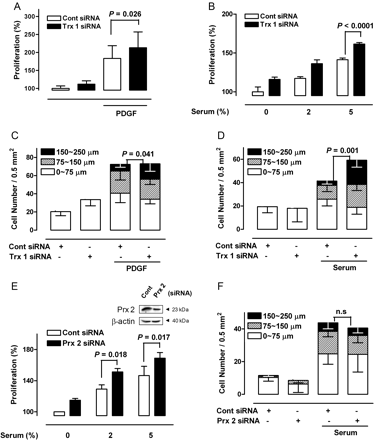
The effect of Trx 1 and Prx 2 inhibition on VSMC growth and migration. (A) VSMCs transfected with Trx1 siRNA were treated with PDGF for 24 h, and growth rates were determined using the MTT assay (Tukey post hoc test applied to significant group effects in analysis of variance, F3,61 = 57.38, P < 0.001). (B) After 48 h of transfection with Trx1-specific siRNA, VSMCs were incubated with various concentrations of serum for an additional 24 h, and growth rates were measured (Tukey post hoc test applied to significant group effects in analysis of variance, F5,18 = 136.96, P < 0.001). (C and D) VSMCs transfected with Trx1-specific siRNA were incubated in the absence or presence of PDGF or 5% serum for 48 h, and migration was determined using a migration assay. P-values are indicated for statistically significant differences (Student's t-test). The vertical axis represents the number of cells that migrated into the wounded region. Stacked bars display the portion of migratory cells classified by migration distance. (E and F) VSMCs transfected with Prx 2-specific siRNA were incubated with various concentrations of serum for an additional 24 h, and growth rates were measured (Tukey post hoc test applied to significant group effects in analysis of variance, F5,24 = 32.92, P < 0.001), and migration was determined using a migration assay. P-values are indicated for statistically significant differences (Student's t-test).
Migration in the presence of PDGF or serum was also enhanced by siRNA-induced inhibition of Trx1 (Figure 5C and D). Pre-incubation with various ROS scavengers such as N-acetylcysteine and vitamin E abrogated the enhancing effect of Trx1-specific siRNA on growth and migration by VSMCs (data not shown). These results clearly indicate that PDGF- and serum-induced growth and migration are enhanced by Trx1 inhibition in an ROS-dependent manner.
Because we observed reduced Prx2 expression with TSA treatment, we examined the effect of Prx2 inhibition on the growth and migration of VSMCs further by using Prx2-specific siRNA. In common with Trx 1 inhibition, reduced Prx 2 expression increased the proliferation of VSMCs (Figure 5E), which is consistent with our previous report that Prx 2 deficiency results in increased production of ROS and subsequent increased neointimal thickening in a murine restenosis model.15 However, Prx 2 knockdown did not induce any significant increase in PDGF- or serum-induced migration, in contrast to the results of Trx 1 knockdown (Figure 5F). These results suggest that the enhanced migration of VSMCs observed in our study was mediated by a decreased interaction of Trx1 with target proteins other than Prx 2.
3.4 Downregulation of Trx1 enhances serum-induced phosphorylation of Akt
Pre-treatment with LY294002, a pharmacological inhibitor of Akt, almost completely abrogated serum-induced proliferation and migration of VSMCs (Figure 6A and B). Treatment with serum induced rapid Akt phosphorylation, but this effect was dramatically suppressed by pre-treatment with various ROS scavengers (Figure 6C), clearly indicating that signal-dependent production of intracellular ROS is critical for the serum-dependent activation of the Akt signalling pathway. In addition, Trx1 inhibition and a subsequent rise in ROS enhanced the serum-dependent phosphorylation of Akt after 15 min of serum reconstitution and prolonged stimulation with serum (48 h) (Figure 6D).
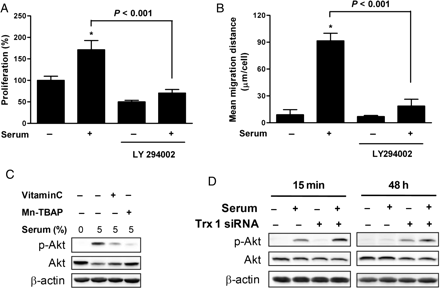
Effect of Trx1 inhibition on Akt activation. (A and B) VSMCs were pre-incubated in the absence or presence of LY294002 for 1 h and then treated with 5% serum for 24 h. Growth rates and migration activities were measured using MTT and migration assays (Tukey post hoc test applied to significant group effects in analysis of variance, F3,16 = 88.72, P < 0.001 in MTT assay and F3,16 = 179.31, P < 0.001 in migration assay, *P < 0.001). The soluble cell lysates were subjected to western blotting. Results shown are representative of three independent experiments. (C) VSMCs were pre-incubated in the absence or presence of 0.5 mmol/L Vitamin C or 25 µmol/L Mn-TBAP and then incubated with serum for 15 min. The soluble cell lysates were subjected to western blotting. (D) VSMCs were transfected with Trx1-specific siRNA and treated with 5% serum for 15 min or 48 h after overnight serum starvation. The soluble cell lysates were subjected to western blotting.
Collectively, these results suggest that Trx1 inhibition increases serum-induced growth and migration of VSMCs by enhancing the activation of Akt, a key signalling protein responsible for cellular growth and migration.
4. Discussion
In our experiments, TSA treatment of VSMCs inhibited the expression of Trx1 and induced a subsequent accumulation of ROS. This TSA-induced downregulation of Trx1 was expected to inhibit cell growth. However, contrary to our expectations, Trx1 inhibition and the subsequent increase in intracellular ROS levels augmented PDGF-induced proliferation and migration in VSMCs. Although HDAC inhibitors have emerged as potential anticancer drugs, TSA has been shown to exacerbate neointimal proliferation in low-density lipoprotein receptor-deficient mice and to promote hypertrophy of cardiac myocytes in neonatal rats.16,17 Inhibition of Trx in the heart has also been reported to increase oxidative stress and cardiac hypertrophy.18 To our best knowledge, our findings are the first to demonstrate that the downregulation of Trx1 is associated with vascular proliferation in vitro and in vivo.
Trx serves a key redox function in regulating signal transduction; reduced Trx catalyses the reduction of disulfide bonds in many proteins, such as PTEN and ASK1.19,20 Trx-deficient mice are embryonic lethal, whereas Trx-overexpressing transgenic mice are more resistant than are control mice to various inflammatory responses.9,21 Physiologically, Trx is thought to exert a cytoprotective effect against oxidative stress by scavenging ROS, perhaps in conjunction with Prx.8 However, the exact function of Trx1 in normal tissues remains unclear because of the early lethality of the Trx1-deficient embryo. Because Trx functions as a hydrogen donor for many protein targets and scavengers of ROS, such as Prx, Trx downregulation might be anticipated to lead to ROS accumulation and the subsequent induction of vascular proliferation, as in the case in Prx deficiency.15 This possibility was supported by the finding that, in common with Trx 1 inhibition, Prx 2 knockdown increased the proliferation of VSMCs. However, we cannot exclude the possibility that the enhanced migration of VSMCs observed in our experiments was mediated by a decreased interaction of Trx1 with target proteins other than Prx2, as Prx 2 knockdown did not induce any significant increase in serum-induced migration.
Compounds that can influence ROS generation are currently under study for their potential use in the treatment of various diseases, including cancer. However, little has been reported regarding the ability of HDAC inhibitors to regulate the generation of ROS, especially in normal cells. In a study examining the effect of HDAC inhibitors on leukaemia and myeloid cells, they were shown to promote apoptosis through an ROS-regulated process.22,23 However, in the current study, we demonstrated a conflicting role for ROS in TSA-induced vascular proliferation; specifically, we found that exposure to TSA led to an increase in ROS accumulation and that treatment with ROS scavengers significantly inhibited activation of Akt, which is important for proliferation and migration of VSMCs. Given our finding that ROS regulate Akt activation, we speculate that, as a target of ROS, Akt is a molecular mediator for TSA-mediated vascular proliferation. This hypothesis is supported by the report of Nishida et al.,24 that Akt is an ROS-responsive serine/threonine kinase, and by our finding that downregulation of Trx1 increases ROS levels and augments serum or PDGF-mediated Akt activation in VSMCs.
In our experiment, we could not distinguish which Akt isoform made the major contribution to vascular proliferation on TSA treatment. A previous report showed that the Akt3 isoform is expressed in substantial amounts by aortic VSMCs and coronary artery VSMCs.25 That study also showed that exposure of VSMCs to PDGF led to a significant increase in Akt3 activity, which was much higher than the activity of Akt1 and Akt2.25 Consequently, we speculate that the Akt3 isoform plays an important role in the TSA-induced proliferation of VSMCs, although all three Akt isoforms are capable of increasing their respective activity on PDGF treatment.
Previous studies have suggested that HDAC inhibitors downregulate Trx and arrest cell growth in transformed fibroblast, VA-13, and prostate cancer cells.2,13 In concordance with these reports, we found that TSA treatment enhanced TRAIL-induced apoptosis in transformed HeLa cells via downregulation of Trx1 (see Supplementary material online, Figure S1A and B). We also found that Trx1 inhibition by siRNA led to increased levels of p38 and JNK phosphorylation in HeLa cells (data not shown), suggesting that the TSA-induced death of tumour cells is regulated through alterations in MAP kinase signalling that mediate cell death and are positively regulated by ROS. Our findings are supported by reports that antioxidants inhibit ROS-induced cell death and activation of p38 and JNK.26,27 The observation described here regarding tumour cells is consistent with previous reports that HDAC inhibitors act to reduce tumour cell viability.3,5 Although TSA treatment induced a reduction in Trx1 expression in both HeLa and VSMCs, the final outcomes were opposite: proliferative in VSMCs and pro-apoptotic in transformed HeLa cells. We believe that the observation of these divergent effects of TSA or Trx1 knockdown is unique to our study. However, the reasons as to why tumour and normal cells respond differently to the same TSA treatment remain unclear. It is tempting to speculate that tumour and normal cells have different ROS-regulated molecular targets. This hypothesis is supported by our finding that treatment with TSA or transfection with Trx1-specific siRNA increased the activation of different molecules in tumour cells (p38 and JNK) and normal cells (Akt). Taken together, these data suggest that although TSA exposure can increase ROS levels through the downregulation of Trx1 in both VSMCs and HeLa cells, the mechanisms of action in the two cell types may be distinct from one another.
In addition, we showed that the TSA-induced increase in ROS did not occur through altered expression of NADPH oxidase components. Of the NOX proteins, NOX 4 was expressed most highly in VSMCs. The expression of NOX 2 was also detected, but the expression level was significantly lower. As expected, TSA treatment did not affect the expression of NOX 1 or NOX 4 (Figure 3D). Although p22 phox was also highly expressed in VSMCs, its expression level was also not affected by TSA treatment (Figure 3D). To further confirm the involvement of Trx 1 on the expression of NADPH oxidase components, we examined the effect of Trx 1 knockdown on the serum-induced expression of NADPH oxidase components. Serum addition suppressed the NOX 4 and p22 phox expression, and this suppressive effect did not differ between cont siRNA- and Trx 1 siRNA-transfected cells (Figure 4C). This effect is almost consistent with a previous result that NOX 4 and p22 phox expression was affected by serum mitogen.28 The expression of p47 phox, the cytosolic component of NADPH oxidase, was not changed by Trx 1 knockdown (Figure 4D). These results suggest that Trx 1 knockdown does not change the expression of membranous and cytosolic NADPH oxidase components. We also showed that the phosphorylation of p47 phox was not affected by Trx 1 knockdown (Figure 4E). Given our findings, it is possible that the ROS accumulation on TSA treatment or Trx1 knockdown is not due to the activation of ROS-producing NADPH oxidase system, but to the downregulation of the ROS-scavenging antioxidant system including Trx1 and Prx 2.
The mechanism by which TSA exposure leads to decreased expression of antioxidant proteins remains to be elucidated. Recent evidence has suggested several regulatory mechanisms concerning the transcriptional or post-translational modification by HDAC inhibitors. HDACs generally act as components of large co-repressor complexes that prevent gene transcription. Alternatively, abundant evidence indicates that the reversible acetylation of transcriptional regulators, such as TFIIEβ, TFIIF, p53, NF-κB, GATA1, and ELK, affects their DNA binding properties.29 For example, deacetylation of a lysine residue in the IκB protein, an inhibitor of NF-κB, has been reported to promote its subsequent ubiquitination and proteasome-dependent degradation.30 Hyper-acetylation induced by HDAC inhibitors can therefore suppress the activation of NF-κB by stabilizing IκB. Because NF-κB has been reported to regulate Trx transcription,31 the inhibitory effect of TSA on Trx1 may be mediated through NF-κB inactivation.
In conclusion, TSA enhances vascular proliferative activity by downregulating Trx1 in an Akt-dependent manner. Because various HDAC inhibitors have been tested and put to practical use for cancer treatment, our results prompt the precaution that HDAC inhibitors might have unexpected paradoxical pro-atherogenic effects, especially in susceptible subjects with high risk factors.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: none declared.
Funding
This research was supported by a grant from the Korea Health 21 R&D Project (A060687) of the Ministry of Health and Welfare, Republic of Korea (to C.C.) and 21C Frontier Functional Proteomics Project (FPR08-B1-190) of Ministry of Education, Science & Technology (to S.W.K.). Funding to pay the Open Access publication charges for this article was provided by Chung Moon Soul Center for Bio-Information and Electronics.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}