





Abstract
We investigated the mechanism by which proinflammatory stimulation induces microvascular endothelial barrier dysfunction. Since protein phosphatase type 2A (PP2A) can mediate paracellular leak and can be inactivated by tyrosine phosphorylation in its catalytic subunit (PP2Ac), we hypothesized that microvascular endothelial cells exposed to proinflammatory stimulation produce peroxynitrite that nitrates PP2Ac, and this nitration inhibits tyrosine phosphorylation of PP2Ac and thereby increases PP2A activity to mediate endothelial barrier dysfunction.
Exposure of mouse skeletal muscle microvascular endothelial cell monolayers to a proinflammatory stimulus [lipopolysaccharide (LPS) + interferon (IFN)γ] increased permeability to albumin, and this barrier dysfunction was attenuated by PP2A inhibitor okadaic acid or by siRNA (small interfering ribonucleic acid) against PP2Ac. LPS + IFNγ increased synthesis of peroxynitrite precursors nitric oxide (NO) and superoxide by inducible NO synthase (iNOS) and NADPH oxidase, respectively. PP2Ac immunoprecipitates isolated from LPS + IFNγ- or peroxynitrite-treated cells showed increased tyrosine nitration, decreased tyrosine phosphorylation and increased phosphatase activity. 3-Nitrotyrosine immunoprecipitates from LPS + IFNγ-stimulated cells also exhibited increased PP2A activity. Further, iNOS inhibitor 1400W, iNOS deficiency, NADPH oxidase inhibitor apocynin, or p47phox deficiency prevented the increase in PP2A activity and preserved barrier function.
LPS + IFNγ stimulates endothelial cells to produce iNOS-derived NO and NADPH oxidase-derived superoxide, which form peroxynitrite that nitrates tyrosine residues in PP2Ac and inhibits their phosphorylation. This nitration in PP2Ac is correlated with PP2A activation that mediates endothelial barrier dysfunction.
1. Introduction
Proinflammatory stimuli [e.g. lipopolysaccharide (LPS) and the cytokines tumour necrosis factor (TNF)α, interleukin (IL)-1β and interferon (IFN)γ] trigger tissue injury in inflammatory diseases such as sepsis and rheumatoid arthritis.1,2 These stimuli induce endothelial barrier dysfunction that permits extravasation of plasma and leukocytes and consequently leads to oedema and tissue injury.3,4 In support of this view, proinflammatory cytokines have been shown to increase endothelial cell monolayer permeability in the absence of leukocytes, through direct actions on endothelial cells.4–7 Moreover, the cytokines induce a delayed permeability increase that develops over hours to days in endothelial monolayers.4–7 However, the mechanisms causing the permeability increase are not clear.
Endothelial cells respond to proinflammatory stimuli by producing nitric oxide (NO), superoxide and peroxynitrite.2,8 NO generation by endothelial cells is necessary for the maintenance of endothelial barrier function.2,9,10 However, the protective effect of NO is diminished under inflammatory conditions due to the simultaneous production of superoxide, which reacts with NO to form peroxynitrite.11,12 Peroxynitrite has been implicated as a causative factor for endothelial barrier dysfunction that is not attributable to cell death.13,14 Inhibition of the production of superoxide or NO with, respectively, superoxide dismutase or NO synthase inhibitors, preserves the permeability barrier in endothelial cell monolayers challenged with LPS and cytokines.10,15
Peroxynitrite oxidizes cysteine residues and nitrates tyrosine residues, leading to modification of the functional activity of target proteins.11,12 This modification could lead to endothelial barrier disruption. For example, nitration by endogenous peroxynitrite of the cytoskeletal protein actin may account for TNFα-induced permeability increase in endothelial cell monolayers.16 Other peroxynitrite-mediated mechanisms also may be involved in this permeability increase, since the increase in permeability persists after nitrated actin returns to the control level.16
Protein phosphatase type 2A (PP2A) mediates dephosphorylation and redistribution of tight junction proteins that can cause paracellular leak in epithelial cell monolayers.17,18 Inhibition of PP2A with calyculin A prevents the redistribution of tight junction proteins and the increase in epithelial permeability caused by enteropathogenic Escherichia coli (E. coli) infection.19 PP2A activity can be modulated by the tyrosine phosphorylation state of its catalytic subunit (PP2Ac), because phosphorylation of tyrosine residues in PP2Ac decreases phosphatase activity.20–22 We hypothesized that microvascular endothelial cells exposed to proinflammatory stimulation produce peroxynitrite that nitrates PP2Ac and this nitration inhibits tyrosine phosphorylation of PP2Ac and thereby augments PP2A activity to mediate endothelial barrier dysfunction.
2. Methods
2.1 Endothelial cell culture
The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996). Microvascular endothelial cells were isolated from the hindlimb skeletal muscles of male wild-type, iNOS(inducible NO synthase)-deficient and p47phox-deficient mice with C57BL/6 background (obtained from Jackson Laboratory, Bar Harbor, ME, USA) and were cultured by methods we have published.23 Endothelial cell identification was carried out by methods we also have described.24 For experimental use, the cells were cultured in Dulbecco's modified Eagle medium (DMEM) containing 2% heat-inactivated foetal bovine serum.
2.2 Endothelial monolayer permeability assay
Diffusion of Evans blue-coupled albumin through endothelial monolayers was determined as previously described with minor modifications.25 Cells were plated on gelatin-coated culture inserts (3 µm pore size) in 12-well plates (BD Biosciences, San Jose, CA, USA) and grown to confluence in the growth medium. The cells were treated in phenol red-free DMEM containing 2% serum, as described in the figure legends. Subsequently, Evans blue-coupled albumin was added to the upper chamber and uncoupled albumin was added to the lower chamber, so that no oncotic pressure gradient existed. After incubation of the monolayers with Evans blue-coupled albumin for 1 h, medium was collected from the lower chamber and the absorbance of Evans blue was measured at 595 nm. Absorbance values were referenced to a standard curve and the flux of albumin was expressed as μg/h.
2.3 Cell viability assay
Endothelial cell viability was measured using dimethylthiazolyldiphenyltetrazolium bromide (MTT) assay. Cells were cultured in 12-well plates in DMEM containing 2% serum and treated as indicated in the figure legends. One hour prior to end-point, the culture medium was changed to phenol red-free DMEM containing 2% serum and 0.25 mg/mL MTT. After 1 h incubation at 37°C, the medium was removed and 1 mL of dimethyl sulphoxide was added to each well, followed by trituration to dissolve the blue formazan. Two hundred microlitres from each well were transferred, in triplicate aliquots, to a 96-well plate and absorbance at 550 nm was read in a spectrophotometer.
2.4 Transfection of small interfering ribonucleic acid
Cells were seeded in 12-well plates and allowed to grow for 24 h in antibiotic-free DMEM containing 10% serum. Next, each well was incubated for 24 h with 80 pmol of PP2Ac siRNA (small interfering ribonucleic acid) or scrambled RNA and 3.15 µL of Oligofectamine® reagent in 1 mL of antibiotic-free DMEM containing 2% serum. The transfected cells were treated with vehicle or LPS + IFNγ for another 24 h and then analysed for monolayer permeability and PP2A activity.
2.5 Western blot analysis
Western blot analysis of immunoprecipitates or cells harvested from each well of the 12-well plates was carried out with the procedures we have described.23
2.6 Measurement of PP2A activity
PP2A activity was determined as okadaic acid-inhibitable phosphatase activity by the method described previously.26 Cells harvested from each well of 12-well plates were lysed by sonication in 100 µL assay buffer [50 mM Tris–HCl, pH 7.0, 0.1 mM EDTA and 1 mM phenylmethylsulphonylfluoride (PMSF)]. The cell extract was combined with 100 µL assay buffer containing 10 mM p-nitrophenyl phosphate (p-NPP) (final concentration = 5 mM) and 6 mM MnCl2 (final concentration = 3 mM), with or without 50 nM okadaic acid, and was incubated for 30 min at 30°C. The hydrolysis of p-NPP was measured at 405 nm and PP2A activity was calculated as total phosphatase activity minus okadaic acid-insensitive activity.
PP2A activity was also measured in immunoprecipitates prepared with anti-PP2Ac or anti-3-nitrotyrosine antibodies. For phosphatase assay, the immunoprecipitate was washed with assay buffer and resuspended in 200 µL assay buffer containing 5 mM p-NPP and 3 mM MnCl2, with or without 50 nM okadaic acid, and incubated for 30 min at 30°C. The hydrolysis of p-NPP was measured at 405 nm.
2.7 Superoxide production by lucigenin assay
Production of superoxide in intact cells was measured by lucigenin chemiluminescence assay as we have described previously.23
2.8 Measurement of nitrate and nitrite
The content of NO metabolites in the culture medium was measured to estimate the total NO production of cultured cells by methods we have published.27
2.9 Immunoprecipitation
Cells in 10 cm plates were washed with ice-cold phosphate-buffered saline and incubated on ice for 5 min with 500 µL lysis buffer (20 mM Tris–HCl, pH 7.0, 1% NP-40, 137 mM NaCl, 1 mM PMSF, 1 µM pepstatin, and 1 µM aprotinin). The cells were scrape-harvested and then sonicated on ice four times for 5 s each. Nuclear and cellular debris were removed by centrifugation for 10 min at 14 000g at 4°C. The supernatants were incubated with primary polyclonal antibodies for 18 h at 4°C, followed by addition of 20 µL protein A-agarose beads for 3 h at 4°C. The immunoprecipitates were washed four times with ice-cold lysis buffer by centrifugation for 3 min at 1000g at 4°C. They were then analysed for PP2A activity by the method described earlier or else were heated with Laemmli’s sample buffer for electrophoresis and western blot analysis.
2.10 Peroxynitrite treatment of cells
Cells were exposed to 500 µM authentic peroxynitrite, 500 µM degraded peroxynitrite or vehicle (0.5% of stock solution of 0.3 M NaOH + 0.3 M NaCl) for 2 h in DMEM containing 2% serum. This vehicle was used because 0.3 M NaOH + 0.3 M NaCl was employed by the supplier to preserve peroxynitrite. The peroxynitrite concentration and incubation time were based on the publication by Knepler et al.,13 which showed that no cytotoxic effect occurred with this protocol. After peroxynitrite treatment, cells were subjected to immunoprecipitation of PP2Ac and determination of monolayer permeability to Evans blue-coupled albumin by the methods described earlier.
2.11 Chemicals and reagents
LPS (from E. coli 055:B5) and mouse IFNγ (recombinant) were obtained from Sigma/Aldrich Chemical Co. (St Louis, MO, USA). Apocynin and N-(3-aminomethyl)benzylacetamidine (1400W) were obtained from VWR (West Chester, PA, USA). Peroxynitrite and degraded peroxynitrite were obtained from Millipore Corporation (Temecula, CA, USA). Anti-phosphotyrosine and anti-nitrotyrosine antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA). Anti-PP2Ac and anti-β-actin antibodies were obtained from BD Biosciences (San Jose, CA, USA) and EMD Chemicals (San Diego, CA, USA), respectively. siRNA against mouse PP2Ac and scrambled siRNA were purchased from Dharmacon (Lafayette, CO, USA).
2.12 Statistical analysis
Data are presented as mean ± SD values and were analysed with the Prism statistical program. Comparisons between treatment groups were performed with one-way ANOVA followed by the Tukey multiple comparison test. P < 0.05 was considered significant. The n values represent the number of independent experiments.
3. Results
3.1 Lipopolysaccharide + interferon gamma increases PP2A activity that mediates endothelial monolayer leak
Mouse skeletal muscle microvascular endothelial cell monolayers leaked a low level of Evans blue-labelled albumin under basal conditions (Figure 1A). Stimulation of the cell monolayers with LPS (25 ng/mL) + IFNγ (100 U/mL) markedly increased the permeability to albumin (Figure 1A). This permeability increase was attenuated by okadaic acid at concentrations of 0.1 and 1.0 nM (Figure 1A). Okadaic acid is a selective PP2A inhibitor with IC50 values of 0.1–2 nM.28,29 Treatment of the cells with LPS + IFNγ or okadaic acid at concentrations of 0.1 and 1.0 nM did not influence cell viability (Figure 1B).
![Inhibition of protein phosphatase type 2A (PP2A) activity prevents lipopolysaccharide (LPS) + interferon (IFN)γ-induced endothelial monolayer leak. Wild-type mouse skeletal muscle microvascular endothelial cells were incubated with Control (phosphate-buffered saline) or LPS (25 ng/mL) + IFNγ (100 U/mL) in the presence of vehicle (0.1% dimethyl sulphoxide) or PP2A inhibitor [okadaic acid (OA) at concentrations of 0.1 and 1.0 nM] for 24 h. (A) Shows the permeability of cell monolayers to Evans blue-labelled albumin (n = 4; *P < 0.05 compared with Control. #P < 0.05 compared with LPS + IFNγ). (B) Shows cell viability determined by dimethylthiazolyldiphenyltetrazolium bromide assay (n = 3; expressed as percentages of the values for vehicle-treated Control group).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/81/1/10.1093/cvr/cvn246/2/m_cvn24601.gif?Expires=1747909792&Signature=BmjLHTRmGqcm4c5dXAlxmcvQrtqtEY6ne9b3P--0e9-M3pyskv7MwiPsDtdy-ntePr6hz4JeDWQ4P6pbHuq-aruOxJjMYJCGQCm79awvU4Hu~M6pjQZPfSUqLiHzn8L9ya0qLOjabHUdSmAmiIgA5eV0cvxVXR3LXlK7iahv~n1JC-pD7sjPtREFVysEe8Lmnqf3CyiudzU6447LrCzgTEMuUKKWpoVGrybJhImZJ6ODANkEZ~iv7aglU1Tb0hw49jr2OrDjPNYulpxqL8xA3nabK-os83Dorfd1zmmlsgtPyKrH76Mn45qluigQx9C4MNcx3SvXjXDs0CDM5VTRGg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Inhibition of protein phosphatase type 2A (PP2A) activity prevents lipopolysaccharide (LPS) + interferon (IFN)γ-induced endothelial monolayer leak. Wild-type mouse skeletal muscle microvascular endothelial cells were incubated with Control (phosphate-buffered saline) or LPS (25 ng/mL) + IFNγ (100 U/mL) in the presence of vehicle (0.1% dimethyl sulphoxide) or PP2A inhibitor [okadaic acid (OA) at concentrations of 0.1 and 1.0 nM] for 24 h. (A) Shows the permeability of cell monolayers to Evans blue-labelled albumin (n = 4; *P < 0.05 compared with Control. #P < 0.05 compared with LPS + IFNγ). (B) Shows cell viability determined by dimethylthiazolyldiphenyltetrazolium bromide assay (n = 3; expressed as percentages of the values for vehicle-treated Control group).
Microvascular endothelial cells that were transfected with scrambled siRNA for Control expressed constitutively the PP2Ac at levels that were not changed by LPS + IFNγ (Figure 2A and B). Despite robust expression of PP2Ac protein, cells that were not incubated with LPS + IFNγ showed a low level of PP2A activity (Figure 2C). LPS + IFNγ markedly increased PP2A activity (Figure 2C). Further, the increased PP2A activity was accompanied by augmented monolayer permeability (Figure 2C and D). Transfection of the cells with siRNA against PP2Ac remarkably decreased the expression of PP2Ac protein in both unstimulated and LPS + IFNγ-stimulated endothelial cells (Figure 2A and B). PP2Ac siRNA did not affect PP2A activity or monolayer permeability in unstimulated cells, but it prevented increases in these parameters by LPS + IFNγ (Figure 2C and D). Together, these results indicate that the elevated PP2A activity mediates the permeability increase in LPS + IFNγ-stimulated endothelial cell monolayers.
![Inhibition of protein phosphatase type 2A (PP2A) expression attenuates lipopolysaccharide (LPS) + interferon (IFN)γ-induced PP2A activity and endothelial monolayer leak. Wild-type endothelial cells were transfected with siRNA (small interfering ribonucleic acid) against mouse PP2Ac [si-PP2Ac (catalytic subunit of PP2A)] or with scrambled siRNA (Scram-si). The transfected cells were then incubated with Control (phosphate buffered saline) or LPS + IFNγ for 24 h. (A) Shows representative western blots for PP2Ac and β-actin from three independent experiments. (B) Summary of PP2Ac protein band intensity (n = 3; *P < 0.05 compared with scrambled siRNA Control group; #P < 0.05 compared with scrambled siRNA LPS + IFNγ group). (C) Summary of PP2A activity (expressed as percentages of the values for scrambled siRNA Control group; n = 3; *P < 0.05 compared with scrambled siRNA Control group; #P < 0.05 compared with scrambled siRNA LPS + IFNγ group). (D) Shows the summary of the permeability of cell monolayers to Evans blue-labelled albumin (n = 4; *P < 0.05 compared with scrambled siRNA Control group; #P < 0.05 compared with scrambled siRNA LPS + IFNγ group).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/81/1/10.1093/cvr/cvn246/2/m_cvn24602.gif?Expires=1747909792&Signature=ogKjVR6~m-pinuPD1fTob1WZoGeqI7Wf88F~044cJ1mh9I1qKj8dVAH~9EbvfZyTlBOFjGSbRI1PFlO4rqJo2JYUqpPi8saQ2~OsSjtTzzgNHy0Uc9mC3M9oIUE50biVYY3V5~oLJz1zPhc1j9fIcMFTLh0M8Wk21U4dVIakzU5nRmqI9wZptz7CmNIAaAzoFoENYAEGbKOh9dKxorj8EGiLnJ6jDPFq502n3Id9dW~AEJX~3Yd~UN-amwhstnvwgof3BsWZrbkbi7INicfm4RMlEZ0gAOK-ERYL9G~s90AeP2Z4z2j2Nt1RltR83IrF42ZGa7j3LJ62Kf2ua01kLA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Inhibition of protein phosphatase type 2A (PP2A) expression attenuates lipopolysaccharide (LPS) + interferon (IFN)γ-induced PP2A activity and endothelial monolayer leak. Wild-type endothelial cells were transfected with siRNA (small interfering ribonucleic acid) against mouse PP2Ac [si-PP2Ac (catalytic subunit of PP2A)] or with scrambled siRNA (Scram-si). The transfected cells were then incubated with Control (phosphate buffered saline) or LPS + IFNγ for 24 h. (A) Shows representative western blots for PP2Ac and β-actin from three independent experiments. (B) Summary of PP2Ac protein band intensity (n = 3; *P < 0.05 compared with scrambled siRNA Control group; #P < 0.05 compared with scrambled siRNA LPS + IFNγ group). (C) Summary of PP2A activity (expressed as percentages of the values for scrambled siRNA Control group; n = 3; *P < 0.05 compared with scrambled siRNA Control group; #P < 0.05 compared with scrambled siRNA LPS + IFNγ group). (D) Shows the summary of the permeability of cell monolayers to Evans blue-labelled albumin (n = 4; *P < 0.05 compared with scrambled siRNA Control group; #P < 0.05 compared with scrambled siRNA LPS + IFNγ group).
3.2 Lipopolysaccharide + interferon gamma induces PP2Ac tyrosine nitration, decreases PP2Ac tyrosine phosphorylation and increases PP2A activity
Microvascular endothelial cells under basal conditions produced superoxide at a rate that was partially inhibited by the NADPH oxidase inhibitor apocynin (Figure 3A). NADPH oxidase deficiency (i.e. knockout of the enzyme subunit, p47phox) did not change the basal level of superoxide production (Figure 3A). Stimulation of the cells with LPS + IFNγ markedly increased superoxide production (Figure 3A). This increase was attenuated by apocynin or NADPH oxidase deficiency (Figure 3A). On the other hand, unstimulated endothelial cells with or without iNOS gene knockout produced very low levels of NO metabolites (nitrite and nitrate) (Figure 3B). Stimulation of the cells with LPS + IFNγ remarkably increased the levels of NO metabolites and this increase was completely inhibited by iNOS inhibitor 1400W or iNOS deficiency (Figure 3B). These observations indicate that NADPH oxidase and iNOS are the principal sources of superoxide and NO, respectively, in LPS + IFNγ-stimulated endothelial cells.
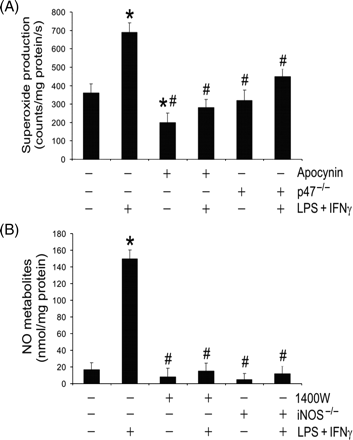
Lipopolysaccharide (LPS) + interferon (IFN)γ stimulates production of superoxide and nitric oxide (NO). Wild-type, p47phox-deficient (p47−/−), or iNOS(inducible NO synthase)-deficient (iNOS−/−) microvascular endothelial cells were treated with vehicle (0.1% dimethyl sulphoxide), apocynin (250 µM) or 1400W (50 µM) for 1 h. Subsequently they were incubated with the same drugs plus LPS + IFNγ for another 24 h. (A) Shows cellular superoxide production rate (n = 3; *P < 0.05 compared with wild-type Control group; #P < 0.05 compared with wild-type LPS + IFNγ group). (B) Shows NO metabolite (nitrite and nitrate) concentrations in the culture media (n = 3; *P < 0.05 compared with wild-type Control group; #P < 0.05 compared with wild-type LPS + IFNγ group).
Superoxide and NO react to form peroxynitrite. This reactive nitrogen species nitrates the tyrosine residues of target proteins, leading to formation of 3-nitrotyrosine. We investigated if PP2Ac is a target of endogenous peroxynitrite in LPS + INFγ-stimulated endothelial cells. PP2Ac isolated from LPS + INFγ-stimulated wild-type endothelial cells, but not vehicle-treated wild-type endothelial cells, contained 3-nitrotyrosine (Figure 4A and B). Further, 3-nitrotyrosine was undetectable in PP2Ac from LPS + IFNγ-stimulated endothelial cells that were deficient in p47phox or iNOS (Figure 4A and B). We then investigated if PP2Ac tyrosine nitration by endogenous peroxynitrite inhibits the tyrosine phosphorylation. Phosphotyrosine was detectable in PP2Ac isolated from endothelial cells. Incubation of wild-type cells with LPS + IFNγ diminished tyrosine phosphorylation levels in PP2Ac (Figure 4A and C). However, tyrosine phosphorylation in PP2Ac was not altered by LPS + IFNγ if the cells were deficient in p47phox or iNOS (Figure 4A and C). Notably, only the immunoprecipitates of PP2Ac that contained 3-nitrotyrosine exhibited an increased phosphatase activity (Figure 4D). Moreover, 3-nitrotyrosine immunoprecipitates isolated from LPS + INFγ-stimulated endothelial cells also showed a moderately increased PP2A activity (Figure 4D). These results indicate that nitration inhibits tyrosine phosphorylation in PP2Ac and is associated with PP2A activation.
![Lipopolysaccharide (LPS) + interferon (IFN)γ causes PP2Ac (catalytic subunit of protein phosphatase type 2A) tyrosine nitration, decreases PP2Ac tyrosine phosphorylation and increases PP2A activity. Wild-type, p47phox-deficient (p47−/−) or iNOS(inducible nitric oxide synthase)-deficient (iNOS−/−) endothelial cells were incubated with Control (phosphate-buffered saline) or LPS + IFNγ for 24 h, and then they were harvested for immunoprecipitation of PP2Ac (PP2Ac-IP) or 3-nitrotyrosine (3-NT-IP). (A) Shows representative western blots (n = 3) for 3-nitrotyrosine (3-NT), phosphotyrosine (p-Tyr) and PP2Ac. (B) Summary of 3-NT band intensity (n = 3; *P < 0.05 compared with wild-type Control group; #P < 0.05 compared with wild-type LPS + IFNγ group). (C) Summary of p-Tyr band intensity (n = 3; *P < 0.05 compared with wild-type Control group). (D) Summary of PP2A activity [expressed as percentages of the values for that in PP2Ac-IP (or in 3-NT-IP) from wild-type Control group; n = 3; *P < 0.05 compared with PP2Ac-IP wild-type Control group; #P < 0.05 compared with PP2Ac-IP wild-type LPS + IFNγ group; †P < 0.05 compared with 3-NT-IP wild-type Control group].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/81/1/10.1093/cvr/cvn246/2/m_cvn24604.gif?Expires=1747909792&Signature=TdM20kWwwA2AdWrG8NyzB4iNAHfBTy~UL6Vakd0dfCvj5756S0OjESSBepV~R3JjthI-kfIo93-aqJvkQtyeXFPX9J9CGkPkuFS8pNfo~PlgYJbcTYYCyQeOdeBQtTP09a1fVTrD11KNDyodGJlj9EVBfPMG-koJrApFgZh3dFZqq-N3nHyfgMmDWF0X69bLq~lKMauzJG5f3E9XM2EA0FJxXn0Ob7gjQccWYBh-WBoM9x2rZawId7MyFWAIhhiDKs5A7A6jQ1q8qFv2LYRqBagcHGiPZOFGWT1usqXO9HerF3mCFFPC1qGofXQJ2HkEX5W4rmKuczZ5kw9UI-0sNg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Lipopolysaccharide (LPS) + interferon (IFN)γ causes PP2Ac (catalytic subunit of protein phosphatase type 2A) tyrosine nitration, decreases PP2Ac tyrosine phosphorylation and increases PP2A activity. Wild-type, p47phox-deficient (p47−/−) or iNOS(inducible nitric oxide synthase)-deficient (iNOS−/−) endothelial cells were incubated with Control (phosphate-buffered saline) or LPS + IFNγ for 24 h, and then they were harvested for immunoprecipitation of PP2Ac (PP2Ac-IP) or 3-nitrotyrosine (3-NT-IP). (A) Shows representative western blots (n = 3) for 3-nitrotyrosine (3-NT), phosphotyrosine (p-Tyr) and PP2Ac. (B) Summary of 3-NT band intensity (n = 3; *P < 0.05 compared with wild-type Control group; #P < 0.05 compared with wild-type LPS + IFNγ group). (C) Summary of p-Tyr band intensity (n = 3; *P < 0.05 compared with wild-type Control group). (D) Summary of PP2A activity [expressed as percentages of the values for that in PP2Ac-IP (or in 3-NT-IP) from wild-type Control group; n = 3; *P < 0.05 compared with PP2Ac-IP wild-type Control group; #P < 0.05 compared with PP2Ac-IP wild-type LPS + IFNγ group; †P < 0.05 compared with 3-NT-IP wild-type Control group].
3.3 Treatment of cells with authentic peroxynitrite causes PP2Ac tyrosine nitration, decreases PP2Ac tyrosine phosphorylation, and increases PP2A activity and endothelial monolayer leak
To test further if peroxynitrite alters PP2A activity, we performed additional experiments in which authentic peroxynitrite was added directly to microvascular endothelial cell cultures. Controls were treated with vehicle or degraded peroxynitrite. The results showed that PP2Ac isolated from peroxynitrite-treated endothelial cells, but not from vehicle- or degraded peroxynitrite-treated cells, contained 3-nitrotyrosine (Figure 5A and B). In contrast, phosphotyrosine was detectable in PP2Ac from vehicle- or degraded peroxynitrite-treated cells but not peroxynitrite-treated cells (Figure 5A and C). Furthermore, treatment of cells with peroxynitrite, but not with degraded peroxynitrite, increased PP2A activity and endothelial monolayer permeability (Figure 5D and E). These results indicate that peroxynitrite-dependent tyrosine nitration and inhibition of tyrosine phosphorylation in PP2Ac are associated with increases in PP2A activity and endothelial monolayer leak.
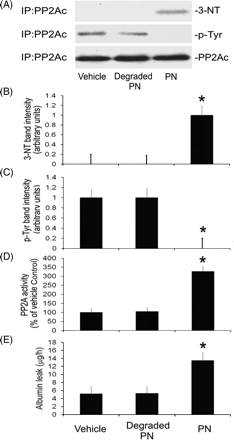
Authentic peroxynitrite causes PP2Ac (catalytic subunit of protein phosphatase type 2A) tyrosine nitration, decreases PP2Ac tyrosine phosphorylation, and increases PP2A activity and monolayer permeability to albumin. Wild-type endothelial cells were incubated with vehicle (0.5% 0.3 M NaOH + 0.3 M NaCl), degraded peroxynitrite (PN) or authentic PN for 2 h, and then they were harvested for immunoprecipitation of PP2Ac or were analysed for monolayer permeability to Evans blue-coupled albumin. (A) Shows representative western blot analysis of PP2Ac immunoprecipitates (n = 3) for 3-nitrotyrosine (3-NT), phosphotyrosine (p-Tyr) and PP2Ac. (B) Summary of 3-NT band intensity (n = 3; *P < 0.05 compared with vehicle-treated group). (C) Summary of p-Tyr band intensity (n = 3; *P < 0.05 compared with vehicle-treated group). (D) Summary of PP2A activity (expressed as percentages of the values for vehicle-treated group; n = 3; *P < 0.05 compared with vehicle-treated group). (E) Shows the summary of the permeability of cell monolayers to Evans blue-labelled albumin (n = 3; *P < 0.05 compared with vehicle-treated group).
3.4 Inhibition of inducible nitric oxide synthase or NADPH oxidase attenuates lipopolysaccharide + interferon gamma-induced increases in PP2A activity and endothelial monolayer leak
Finally, we investigated the roles of endogenous superoxide and NO in PP2A activity and barrier function in cell monolayers. Our results showed that stimulation of the cells with LPS + IFNγ markedly increased PP2A activity and the monolayer permeability to albumin (Figure 6A and B). The increase in PP2A activity induced by LPS + IFNγ was inhibited partially, but significantly, by apocynin or 1400W (Figure 6A). Whereas an effect of LPS + IFNγ on PP2A activity was still detectable in cells pretreated with apocynin or 1400W, the effect was abolished by p47phox deficiency or iNOS deficiency (Figure 6A). Moreover, the increase in endothelial monolayer permeability induced by LPS + IFNγ was blocked by apocynin, 1400W, p47phox deficiency or iNOS deficiency (Figure 6B). Apocynin, p47phox deficiency, 1400W or iNOS deficiency did not affect cell survival since similar cell viabilities were observed after these treatments (Figure 6C). These results indicate that inhibition of either NADPH oxidase or iNOS can attenuate LPS + IFNγ-induced increases in PP2A activity and endothelial monolayer leak.
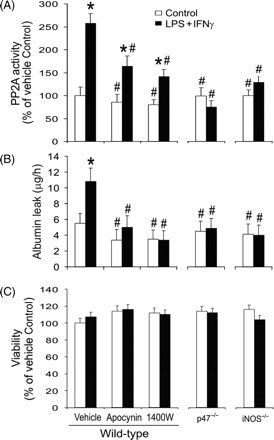
Inhibition of inducible nitric oxide synthase (iNOS) or NADPH oxidase prevents lipopolysaccharide (LPS) + interferon (IFN)γ-induced increases in protein phosphatase type 2A (PP2A) activity and endothelial monolayer leak. Wild-type, p47phox-deficient (p47−/−) or iNOS-deficient (iNOS−/−) endothelial cells were pretreated with vehicle, apocynin or 1400W for 1 h. Subsequently they were incubated with the same drugs plus LPS + IFNγ for another 24 h. (A) Summary of PP2A activity (expressed as percentages of the values for wild-type vehicle-treated Control group; n = 3; *P < 0.05 compared with wild-type vehicle-treated Control group; #P < 0.05 compared with wild-type vehicle-treated LPS + IFNγ group). (B) Shows the summary of monolayer permeability to Evans blue-labelled albumin (n = 4; *P < 0.05 compared with wild-type vehicle-treated Control group; #P < 0.05 compared with wild-type vehicle-treated LPS + IFNγ group). (C) Shows cell viability determined by MTT assay (n = 3; expressed as percentages of the values for wild-type vehicle-treated Control group).
4. Discussion
Proinflammatory stimuli induce barrier dysfunction in endothelial and epithelial cell monolayers through disruption of intercellular tight junctions.4,6,30 The present study discovered that this effect is likely mediated through activation of PP2A. Further, PP2A activation is associated with peroxynitrite-dependent tyrosine nitration and inhibition of tyrosine phosphorylation in PP2Ac.
LPS + IFNγ is a potent inducer of iNOS and NADPH oxidase in various cell types.23,27,31 Consistently, our results show that LPS + IFNγ stimulates the synthesis of NO and superoxide by iNOS and NADPH oxidase, respectively, in microvascular endothelial cells. iNOS-derived NO and NADPH oxidase-derived superoxide react to form peroxynitrite. This reactive nitrogen species could cause massive oxidation and potential destruction of cellular proteins, leading to the induction of cell death through both apoptosis and necrosis.11,12 However, our results further show that LPS + IFNγ-induced permeability increase is not caused by cell death, because similar levels of cell viability are observed in Control and LPS + IFNγ-stimulated endothelial cells. Similarly, other studies reported that cell death is not the cause of permeability increase in endothelial and epithelial cell monolayers after exposure to cytokines.7,30 Moreover, exogenous peroxynitrite that induces endothelial barrier dysfunction does not cause cell death.13
We observed that unstimulated endothelial cells express large amounts of PP2Ac protein. However, they have relatively low levels of PP2A activity and monolayer permeability to albumin. Transfection with PP2Ac siRNA decreases PP2Ac protein expression but does not affect PP2A activity in unstimulated endothelial cells. The explanation for this may be that most PP2Ac in unstimulated endothelial cells is inactive because of high levels of tyrosine phosphorylation in this catalytic subunit. In support of this explanation, we observed that incubating the cells with LPS + IFNγ did not change PP2Ac protein expression but increased PP2A activity and decreased PP2Ac tyrosine phosphorylation. Inhibition of PP2A activity with okadaic acid or inhibition of PP2Ac expression with siRNA against PP2Ac attenuates LPS + IFNγ-induced increases in PP2A activity and monolayer permeability to albumin. These results establish that LPS + IFNγ-induced increase in PP2A activity mediates barrier dysfunction in endothelial cell monolayers. The mechanism of action for PP2A to induce barrier dysfunction has been studied extensively in epithelial cells. PP2A is associated with the junction proteins ZO-1 and occludin and can directly modulate the phosphorylation state of these junction proteins.17,18,32,33 PP2A activity causes dephosphorylation and redistribution of the tight junction proteins, leading to epithelial barrier dysfunction.17,18
Our results show that PP2A activity is increased by LPS + IFNγ and this increase is not due to increased expression of PP2Ac protein. Previous studies found that PP2A activity and function can be regulated by post-translational modifications of the catalytic subunit (i.e. cysteine oxidation, tyrosine phosphorylation, or leucine methylation of PP2Ac).34,35 Peroxynitrite formed in the LPS + IFNγ-stimulated endothelial cells may oxidize the cysteine residues and nitrate the tyrosine residues in PP2Ac, leading to modulation of PP2A activity.11,12 In preliminary studies we found that the thiol-specific reagent dithiothreitol increases further the PP2A activity in PP2Ac immunoprecipitates isolated from LPS + IFNγ-stimulated cells (data not shown), suggesting that oxidation by endogenous peroxynitrite of the cysteine residues in PP2Ac inhibits the enzyme activity. Other studies also show that oxidants such as hydrogen peroxide inhibit the phosphatase activity of PP2A in cells or cell lysates.36,37 Therefore, oxidation of the cysteine residues in PP2Ac is unlikely to be the mechanism by which LPS + IFNγ increases PP2A activity.
Tyrosine phosphorylation of PP2Ac is another important mechanism for regulation of PP2A activity. PP2Ac can undergo tyrosine phosphorylation mediated by p60v-src, p56lck, epithelial growth factor receptors, and insulin receptors.20–22 The phosphorylation results in inactivation of the enzyme.21–22 Peroxynitrite nitrates tyrosine residues in proteins, leading to formation of 3-nitrotyrosine.11,12 This tyrosine nitration could inhibit tyrosine phosphorylation. It has been reported that nitration by authentic peroxynitrite of tyrosine residues in a cdc2(6–20) NH2 peptide inhibits completely phosphorylation of the tyrosine residues by p56lck tyrosine kinase.38 Moreover, studies in intact cells have shown that nitration by exogenous peroxynitrite of tyrosine residues in proteins inhibits their phosphorylation. For example, incubation of lymphocytes with authentic peroxynitrite causes protein tyrosine nitration and inhibits the tyrosine phosphorylation induced by anti-CD3 antibodies.39 Our data demonstrate that PP2Ac immunoprecipitates, prepared from LPS + IFNγ- or authentic peroxynitrite-treated endothelial cells, show 3-nitrotyrosine formation, decreased tyrosine phosphorylation, and increased PP2A activity. These changes in 3-nitrotyrosine, tyrosine phosphorylation and PP2A activity are not detectable in PP2Ac from iNOS-deficient or NADPH oxidase-deficient endothelial cells exposed to LPS + IFNγ. We infer that either endogenous peroxynitrite induced by LPS + IFNγ or authentic peroxynitrite can nitrate the tyrosine residues in PP2Ac and thereby inhibit tyrosine phosphorylation. Functionally this inhibition is associated with increased PP2A activity.
Our results indicate that iNOS-inhibitor 1400W, iNOS deficiency, NADPH oxidase-inhibitor apocynin, or p47phox deficiency prevents the increase in PP2A activity and preserves the permeability barrier in endothelial monolayers challenged with LPS + IFNγ. Moreover, exposure of endothelial cells to authentic peroxynitrite leads to augmented PP2A activity and increased monolayer leak. These results suggest that PP2A is a target of peroxynitrite that increases PP2A activity to mediate endothelial monolayer leak. This effect of peroxynitrite likely depends on tyrosine nitration of PP2Ac to inhibit tyrosine phosphorylation and activate this catalytic subunit. The observation of a moderate increase in PP2A activity in 3-nitrotyrine immunoprecipitates isolated from LPS + IFNγ-stimulated endothelial cells further supports an association between PP2Ac nitration and PP2A activity. Although nitration by exogenous peroxynitrite of the cytoskeletal protein actin has been reported to contribute to peroxynitrite-induced permeability increase in porcine pulmonary artery endothelial monolayers,13 our results indicate that PP2Ac is another target of peroxynitrite that mediates microvascular endothelial leak.
Stimulation of cells by growth factors or serum promotes phosphorylation of tyrosine residues in PP2Ac, thereby inactivating PP2A.20–22 Similarly, PP2A inactivation may occur in endothelial cells under the serum-supplemented cell culture conditions employed in the present study and may contribute to the barrier integrity of cell monolayers. Peroxynitrite produced by the cells in response to proinflammatory stimulation (i.e. LPS + IFNγ in the present study) nitrates the tyrosine residue in PP2Ac. This nitration prevents phosphorylation/inactivation of PP2A by the receptor tyrosine kinases of growth factors, such as epidermal growth factor and insulin, and consequently increases PP2A activity to mediate barrier dysfunction. It has been reported that enteropathogenic E. coli infection stimulates nitrative stress and serine/threonine phosphatase activity that mediates the increased permeability in epithelial monolayers.19,40 Further, it also has been reported that nitrative stress-induced epithelial barrier dysfunction can be prevented by treatment of cell monolayers with epidermal growth factor.41
Bacterial infection or tissue damage stimulates the innate immune response to produce proinflammatory cytokines such as IL-1β, TNFα and IFNγ. Stimulation by these cytokines causes progression of tissue injury in inflammatory diseases such as rheumatoid arthritis and sepsis.1,2 Vascular leak is an important pathological process of inflammatory diseases2,3 in which iNOS and peroxynitrite have been implicated11,12,42,43 The present study, for the first time, identifies peroxynitrite-dependent activation of PP2A as an important step in endothelial barrier dysfunction. This finding provides new pharmacological targets for therapy of inflammatory diseases.
Funding
This work was supported in part by National Institutes of Health grant AT003643-02 and the University at Buffalo Foundation.
Conflict of interest: none declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}