





Abstract
Nitroglycerin (GTN) acts through release of a nitric oxide (NO)-related activator of soluble guanylate cyclase in vascular smooth muscle. Besides enzymatic GTN bioactivation catalysed by aldehyde dehydrogenase, non-enzymatic reaction of GTN with ascorbate also results in the formation of a bioactive product. Using an established guinea pig model of ascorbate deficiency, we investigated whether endogenous ascorbate contributes to GTN-induced vasodilation.
Guinea pigs were fed either standard or ascorbate-free diet for 2 or 4 weeks prior to measuring the GTN response of aortic rings and isolated hearts. The effects of ascorbate on GTN metabolism were studied with purified mitochondrial aldehyde dehydrogenase (ALDH2) and isolated mitochondria. Ascorbate deprivation led to severe scorbutic symptoms and loss of body weight, but had no (2 weeks) or only slight (4 weeks) effects on aortic relaxations to a direct NO donor. The EC50 of GTN was increased from 0.058 ± 0.018 to 0.46 ± 0.066 and 5.5 ± 0.9 µM after 2 and 4 weeks of ascorbate-free diet, respectively. Similarly, coronary vasodilation to GTN was severely impaired in ascorbate deficiency. The potency of GTN was reduced to a similar extent by ALDH inhibitors in control and ascorbate-deficient blood vessels. Up to 10 mM ascorbate had no effect on GTN metabolism catalysed by purified ALDH2 or liver mitochondria isolated from ascorbate-deficient guinea pigs.
Our results indicate that prolonged ascorbate deficiency causes tolerance to GTN without affecting NO/cyclic GMP-mediated vasorelaxation.
1. Introduction
There is general agreement that the anti-anginal drug nitroglycerin (GTN) acts through release of nitric oxide (NO) or a NO-related species that activates soluble guanylate cyclase (sGC) in vascular smooth muscle, resulting in cyclic GMP (cGMP)-mediated vasorelaxation.1–3 Several enzymes have been suggested to catalyse bioactivation of GTN,3 but current evidence suggests that aldehyde dehydrogenase-2 (ALDH2), localized in the mitochondrial matrix, is essential for GTN-triggered vasodilation in rodents and humans.4–7 The initial product of the ALDH2 reaction is thought to be nitrite, which becomes reduced to bioactive NO by an, as yet, elusive nitrite reductase activity in vascular smooth muscle.8 As GTN causes mechanism-based inactivation of ALDH2 through thiol oxidation, a reductant is required for reactivation of the enzyme and sustained GTN action. Recently, it has been suggested that dihydrolipoic acid serves as physiological reductant of ALDH2 in blood vessels.9 ALDH2 reactivation may become impaired upon prolonged GTN application, resulting in the well-known phenomenon of nitrate tolerance.10
In addition to enzymatic pathways, non-enzymatic reactions of GTN bioactivation have been described. Besides the well-established reaction with cysteine,2,11,12 GTN reacts with ascorbate to yield a product with NO-like biological activity.13 Although GTN caused significant activation of purified sGC in the presence of physiological concentrations of ascorbate, it has remained questionable whether this reaction contributes to the haemodynamic effects of GTN. In a preliminary study, we observed no effect of ascorbate supplementation on GTN-induced reduction of blood pressure of mice (see Supplementary material, Figure S1). However, we had to consider that normal ascorbate levels may be sufficient for optimal GTN conversion or that the ascorbate reaction is relevant only as a backup mechanism of GTN bioactivation in tolerant blood vessels. To address this issue, we studied the cardiovascular effects of GTN on aortic rings and the coronary circulation of isolated hearts from ascorbate-deficient and/or nitrate-tolerant guinea pigs and found that ascorbate deficiency resulted in markedly impaired macro- and microvascular relaxation to GTN. As the response to a direct NO donor was only marginally affected after 4 weeks of feeding ascorbate-free diet, ascorbate deficiency appears to specifically interfere with the bioactivation of GTN without considerably affecting NO/cGMP-mediated vasorelaxation.
2. Methods
2.1 Animals and experimental groups
Studies were performed on randomly selected normotensive Dunkin Hartley guinea pigs of either sex (initial body weight 300–400 g). Control groups were fed pelleted commercial guinea pig food (Altromin 3023 containing 1 g/kg ascorbate; Altromin, Lage, Germany). Ascorbate deficiency was induced by feeding ascorbate-free diet (Altromin C3015, Altromin) for 2 or 4 weeks. This protocol was reported to result in >99% depletion of tissue ascorbate levels.14 The effect of ascorbate deficiency was also studied in nitrate-tolerant guinea pigs either fed regular or ascorbate-free diet for 4 weeks. Nitrate tolerance was induced by subcutaneous injection of 50 mg/kg GTN (G. Pohl–Boskamp, Hohenlockstedt, Germany) four times a day for the last 3 days.15 After a feeding period of 2 or 4 weeks, hearts and thoracic aortas were removed, placed in chilled buffer, and immediately used for functional studies. All animals received care in accordance with the Austrian law on experimentation with laboratory animals (last amendment, 2004), which is based on the US National Institutes of Health guidelines. DEA/NO was from Alexis Corp. (Lausen, Switzerland). All chemicals were obtained from Sigma-Aldrich (Vienna, Austria).
2.2 Aortic ring experiments
The aorta was cleaned and cut into small rings of approximately 3 mm length. Rings were suspended in 5 mL organ baths for isometric tension measurement containing oxygenated Krebs–Henseleit buffer (composition in millimolars: NaCl 118.4, NaHCO3 25, KCl 4.7, KH2PO4 1.2, CaCl2 2.5, MgCl2 1.2, d-Glucose 10.1; pH 7.4) as described.16 After equilibration for 90 min at 2 g, rings were contracted with the thromboxane A2 receptor agonist 9,11-dideoxy-11α, 9α-epoxymethanoprostaglandin F2α (U-46619; 50 nM) to obtain a total tone of approximately 5 g, corresponding to approximately 90% of maximal contractile activity (determined with 100 mM K+; see Supplementary material, Figure S2). When the contraction had reached a stable plateau, cumulative dose–response curves were performed using GTN (1 nM–100 µM) or 2,2-diethyl-1-nitroso-oxyhydrazine (DEA/NO; 1 nM–10 µM) in separate tissues. The contractile force corresponding to each agonist concentration was recorded and expressed as percent of precontraction (=baseline). Where indicated, rings were incubated for 45 min with chloral hydrate (5 mM) or daidzin (0.3 mM) before addition of DEA/NO or GTN. To test for the effect of polyethylene glycol-conjugated superoxide dismutase (PEG-SOD; Sigma) and polyethylene glycol-conjugated catalase (PEG-catalase; Sigma), rings were incubated with a combination of 100 U/mL PEG-SOD and 500 U/mL PEG-catalase 60 min before addition of GTN or DEA/NO. This protocol was shown previously to effectively reduce the generation of superoxide17 and hydrogen peroxide.18
The role of the endothelium in GTN-induced vasorelaxation was tested in aortic rings of standard diet-fed guinea pigs. The endothelium was removed by rubbing the intimal surface with a wooden stick19 and dose–response curves to acetylcholine (Ach; 1 nM–10 µM) and GTN were recorded in separate rings. Two minutes after addition of the highest dose of agonist, the tissues were freeze-clamped in liquid nitrogen and stored at −70°C pending cGMP analysis by radioimmunoassay (RIA).
2.3 Determination of cyclic GMP levels in aortic tissue
Freeze-clamped aortic rings were homogenized in 5% trichloroacetic acid and the acid was removed by diethyl ether extraction. The aqueous extracts were appropriately diluted and analysed for cGMP by RIA as described.20
2.4 Isolated heart experiments
After excision, hearts were immediately mounted on a perfusion apparatus (Hugo Sachs Elektronik/Harvard Instruments, March-Hugstetten, Germany) and retrogradely perfused with oxygenated Krebs–Henseleit buffer at 55 mmHg (constant pressure perfusion) as previously described.21 A fluid-filled balloon was inserted into the left ventricle and connected to a pressure transducer. The following cardiac parameters were monitored in unpaced hearts: coronary flow (an index of coronary arterial function), left ventricular developed pressure (LVDevP), and heart rate (electronically derived from the pressure signal). The latter two parameters were recorded to test for myocardial and atrial effects of GTN and DEA/NO.
After equilibration for 30 min (baseline), coronary relaxation was tested with GTN given as bolus injections through a sideline in non-cumulative manner, resulting in concentrations of approximately 1 nM to 100 µM (5 min per dose). After the last dose, GTN was washed out for 30 min and baseline re-established. Thereafter, a dose–response curve to DEA/NO (1 nM–10 µM) was performed (total duration of the experiment was 120 min). In both cases, LVDevP and heart rate were also recorded.
2.5 Nitroglycerin metabolism
The rates of 1,2- and 1,3-GDN formation by purified recombinant human liver ALDH2 or isolated liver mitochondria were determined by radio thin-layer chromatography as described previously.16
2.6 Determination of ascorbate levels in guinea pig plasma
Plasma ascorbate levels were determined by high-performance liquid chromatography (HPLC) and ultraviolet detection at 264 nm according to an established protocol.22 Whole blood was centrifuged at 2000 g for 10 min at 4°C. Plasma was immediately acidified with an equal volume of 10% (v/v) meta-phosphoric acid (MPA), and centrifuged again at 3000 g for 15 min at 4°C. The clear supernatant was stored at −70°C until analysis. Aliquots (10 µL) were injected onto a LiChrospher 100 RP-18 HPLC column with 5 µm particle size (Merck, Darmstadt, Germany) and eluted at a flow rate of 1 mL/min with water containing acetonitrile (2%; v/v), NaH2PO4 (2.5 mM), dodecyltrimethyl ammonium chloride (2.5 mM) and diethylene triamine pentaacetic acid (tetra sodium salt, 1.25 mM). The method was calibrated daily with ascorbate standard solutions (10–300 µM).
2.7 Statistical analysis
Data are presented as mean values ± SEM of experiments. Individual dose–response curves were fitted to a Hill-type model giving estimates of agonist potency (EC50) and efficacy (Emax). Analysis of variance with post hoc Bonferroni–Dunn test was used for comparison between groups using StatView® (version 5.0). The level of significance was set to P < 0.05.
3. Results
3.1 Scorbutic symptoms and ascorbate levels
Guinea pigs fed ascorbate-free diet developed typical symptoms of scurvy. They lost approximately 20% of their body weight during week 3 and 4 (Figure 1) and exhibited general weakness in combination with paralysis of hind legs and signs of haemorrhage in knee joints. The slight decrease in body weight after 2 weeks of ascorbate-free diet was not significant (P = 0.092). Loss of body weight was not accompanied by a significant reduction of heart weight, which was 2.5 ± 0.1 g in controls and 2.2 ± 0.1 g in ascorbate-deficient animals (n = 6 each). Induction of nitrate tolerance did not affect body or heart weights.
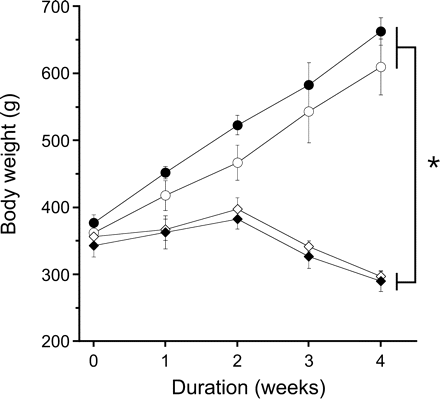
Effect of ascorbate deprivation on body weight. Open circles denote control; closed circles denote nitrate-tolerant; open diamonds denote ascorbate-deficient; closed diamonds denote ascorbate-deficient and nitrate-tolerant. Data are mean values±SEM of three guinea pigs per group. *P < 0.05 compared with corresponding standard diet-fed animals. Nitrate tolerance had no significant effect on body weight.
Ascorbate plasma levels of standard diet-fed animals were 129.4 ± 9.6 µM (n = 14). This value agrees well with the plasma levels reported previously for rats23 and ascorbate-fed guinea pigs.24 After 2 and 4 weeks of ascorbate deprivation, plasma levels were decreased to 12.5 ± 1.1 (n = 9) and 8.9 ± 2.5 µM (n = 6), respectively. The small difference between mean plasma levels measured after 2 and 4 weeks of ascorbate deprivation was not significant (P = 0.17). In GTN-tolerant animals, ascorbate plasma levels were significantly decreased to 54.8 ± 7.2 µM (n = 6; P < 0.001).
3.2 Effect of ascorbate deprivation on vascular relaxation to nitroglycerin and 2,2-diethyl-1-nitroso-oxyhydrazine (DEA/NO)
Incubation of aortic rings with U46619 (50 nM) increased aortic tone in all experimental groups to the same extent (5.0 ± 0.1 g). Figure 2A shows relaxation of aortic rings in response to GTN. As expected, induction of nitrate tolerance in standard diet-fed animals resulted in a rightward shift of the GTN dose–response curve (EC50: 0.076 ± 0.012 µM vs. 2.7 ± 1.1 µM, P < 0.05) and a slight reduction of maximal relaxation at 0.1 mM GTN (−96 ± 1% vs. −88 ± 1%, respectively; P < 0.05). In aortas of ascorbate-deficient animals, GTN was two orders of magnitude less potent than in control vessels (EC50: 5.5 ± 0.9 µM; P <0.05 vs. control). The response to the organic nitrate was further impaired in nitrate-tolerant aortas of ascorbate-deficient animals (EC50: 10.7 ± 3.1 µM; Emax: −44 ± 4% at 0.1 mM GTN; P < 0.05 vs. ascorbate-deficient, non-tolerant).

Relaxation and cyclic GMP levels of control and nitrate-tolerant aortic rings after 4 weeks of ascorbate deprivation. (A) Relaxation to nitroglycerin (GTN); (B) relaxation to 2,2-diethyl-1-nitroso-oxyhydrazine (DEA/NO). Open circles denote control; closed circles denote nitrate-tolerant; open diamonds denote ascorbate-deficient; closed diamonds denote ascorbate-deficient and nitrate-tolerant. C and D show cyclic GMP levels in response to maximally effective concentrations of GTN (C) and DEA/NO (D). Open columns: control; hatched columns: ascorbate-deficient. Data are mean values±SEM of 18 (GTN) or six (DEA/NO) rings derived from three animals. *P < 0.05 compared with corresponding standard diet-fed animals. #P < 0.05 compared with corresponding non-tolerant animals.
To see whether the reduced response to GTN in ascorbate deficiency was because of vascular dysfunction, we measured relaxation to the direct NO donor DEA/NO in separate rings. As shown in Figure 2B, there was a slight but statistically significant rightward shift of the DEA/NO concentration–response curve in ascorbate-deficient blood vessels (EC50: 44 ± 7 nM vs. 122 ± 5 nM; n = 6; P < 0.05). Nitrate tolerance had no effect on the response to DEA/NO, neither in standard diet nor in ascorbate-free diet-fed animals.
Impaired vasorelaxation to GTN in conditions of ascorbate deficiency and/or nitrate tolerance was accompanied by decreased aortic cGMP levels. As shown in Figure 2C, cGMP levels were reduced from 1178 ± 157 fmol/mg to 640 ± 82 and 443 ± 37 fmol/mg in nitrate-tolerant and ascorbate-deficient blood vessels, respectively. Induction of nitrate tolerance in ascorbate-deficient guinea pigs further decreased vascular cGMP levels (354 ± 18 fmol/mg; P < 0.05 vs. ascorbate-deficient, non-tolerant). Accumulation of cGMP in response to the direct NO donor DEA/NO was not significantly different in the four experimental groups (Figure 2D).
As ascorbate deficiency may cause endothelial dysfunction, we considered that vasorelaxation to GTN could be partially endothelium-dependent in guinea pig aorta. However, as shown in Supplementary material, (Figure S3), disruption of the endothelium did not affect vasodilation to GTN (Panel A), while the effect of the endothelium-dependent vasodilator acetylcholine was virtually abolished (Panel B).
In another set of experiments, we determined the GTN response of aortic rings obtained from guinea pigs that were fed ascorbate-free diet for a shorter period of time (2 weeks). As shown in Figure 3A, the potency of GTN was reduced significantly (EC50: 0.46 ± 0.066 µM), but the effect was less pronounced than after 4 weeks of ascorbate deficiency. The response to DEA/NO was not affected at all by 2 weeks of ascorbate deficiency (Figure 3B). To examine the possibility that ascorbate deprivation caused nitrate tolerance through oxidative stress and superoxide production, we studied the effect of PEG-SOD in combination with PEG-catalase. As shown in Figure 3C, PEG-SOD/PEG-catalase had no effect on GTN-induced relaxation of control and ascorbate-deficient guinea pig aortas. Figure 3D shows that relaxation to DEA/NO was neither affected.
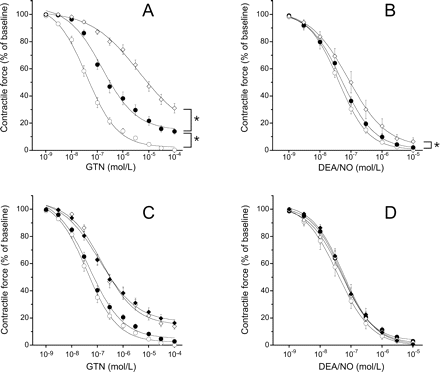
Relaxation of aortic rings after 2 weeks of ascorbate deprivation. Data obtained after 4 weeks of ascorbate deficiency taken from Figures 2A and B are shown for comparison. (A) Relaxation to nitroglycerin (GTN); (B) relaxation to 2,2-diethyl-1-nitroso-oxyhydrazine (DEA/NO). Open circles denote control; closed circles denote 2 weeks of ascorbate deprivation; open diamonds denote 4 weeks of ascorbate deprivation. (C) Relaxation to GTN (C) or DEA/No (D) in the absence or presence of PEG polyethylene glycol-conjugated superoxide dismutase (PEG-SOD) (100 U/mL) in combination with polyethylene glycol-conjugated catalase (500 U/mL). Open circles denote control; closed circles denote control in the presence of PEG-SOD/PEG-catalase, open diamonds denote ascorbate-deficient; closed diamonds denote ascorbate-deficient in the presence of PEG-SOD/PEG-catalase. Data are mean values±SEM of 18 (GTN) or six (DEA/NO) rings derived from three animals.
3.3 Effects of aldehyde dehydrogenase inhibitors on nitroglycerin-induced vasodilation
To test whether vascular tolerance to GTN in ascorbate deficiency is because of inactivation of ALDH2, we studied the effects of the selective ALDH2 inhibitor daidzin and the non-selective drug chloral hydrate on GTN-induced relaxation of control vessels and vessels obtained from guinea pigs fed with ascorbate-free diet for 2 weeks. We reasoned that, if ALDH2 inactivation were involved, the residual GTN response of ascorbate-deficient rings should be insensitive or at least less sensitive to the inhibitors than controls. However, as shown in Figure 4, daidzin (0.3 mM) and chloral hydrate (5 mM) significantly attenuated relaxations to GTN in both control (Panel A) and ascorbate-deficient (Panel B) blood vessels. In the presence of daidzin, the EC50 of GTN was increased from 0.059 ± 0.013 to 0.22 ± 0.090 µM and from 0.46 ± 0.068 to 2.3 ± 0.7 µM in control and ascorbate-deficient rings, respectively. In the presence of chloral hydrate, the EC50 values of GTN were 0.39 ± 0.074 µM (control) and 6 ± 1.6 µM (ascorbate-deficient). The inhibitors had no effect on relaxations induced by DEA/NO neither under control conditions (Figure 4C) nor in ascorbate deficiency (Figure 4D).
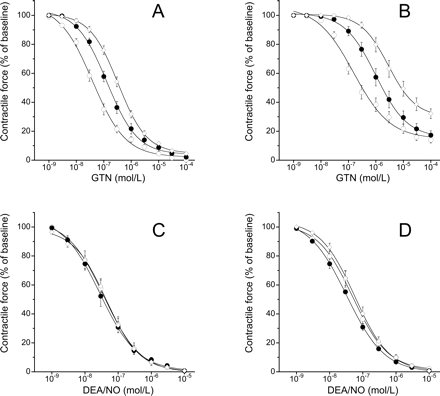
Effects of aldehyde dehydrogenase inhibitors on relaxation of aortic rings after 2 weeks of ascorbate deprivation. (A) Nitroglycerin (GTN), control; (B) GTN, ascorbate-deficient; (C) 2,2-diethyl-1-nitroso-oxyhydrazine (DEA/NO), control; (D) DEA/NO, ascorbate-deficient. Open circles denote vehicle; closed circles denote daidzin (0.3 mM); open diamonds denote chloral hydrate (5 mM). Data are mean values±SEM of 18 (GTN) or six (DEA/NO) rings derived from three animals.
3.4 Effects of ascorbate deficiency and/or nitrate tolerance on coronary flow, myocardial function, and heart rate
Relaxation of the microvasculature of the four experimental groups was tested by measuring coronary flow in isolated hearts perfused at constant pressure (n = 3 per group). In hearts obtained form standard diet-fed non-tolerant guinea pigs, coronary flow was increased by GTN in a dose-dependent manner from about 20 to 30 mL/min (EC50: 0.020 ± 0.005 µM; Figure 5A). As in aortic rings, the effect of GTN was markedly attenuated in hearts of nitrate-tolerant and ascorbate-deficient (4 weeks) animals (EC50: 3.2 ± 2.5 µM and 1.5 ± 0.7 µM, respectively; P < 0.05 vs. control); the most pronounced impairment of vasorelaxation was observed in hearts obtained from animals that were ascorbate-deficient and nitrate-tolerant (EC50: 9.5 ± 3.9 µM).

Effects of nitroglycerin (GTN) (A) or 2,2-diethyl-1-nitroso-oxyhydrazine (DEA/NO) (B) on coronary flow of isolated perfused hearts. Open circles denote control; closed circles denote nitrate-tolerant; open diamonds denote ascorbate-deficient; closed diamonds denote ascorbate-deficient and nitrate-tolerant. Data are mean values ± SEM of three hearts per group. *P < 0.05 compared with corresponding standard diet-fed animals. #P < 0.05 compared with corresponding non-tolerant animals.
The response of the microvasculature to DEA/NO was identical in all four treatment groups (Figure 5B), indicating that nitrate tolerance and/or ascorbate deficiency did not attenuate NO/cGMP-mediated relaxation of the coronary microvasculature. The respective EC50 values were 25 ± 10 nM (control), 27 ± 13 nM (nitrate-tolerant), 21 ± 16 nM (ascorbate-deficient), and 17 ± 9 nM (ascorbate-deficient and nitrate-tolerant).
LVDevP, a measure of left ventricular contractility, and heart rate of isolated hearts of the four experimental groups are shown in Table 1. Basal LVDevP and heart rate were not different between experimental groups. While GTN (0.1 mM) did not affect cardiac contractility, DEA/NO (10 µM) had a slight positive inotropic effect in hearts from all four treatment groups.
Haemodynamic parameters of isolated guinea pig hearts at baseline and after application of nitroglycerin (GTN) or 2,2-diethyl-1-nitroso-oxyhydrazine (DEA/NO)
| Baseline | GTN (100 µM) | DEA/NO (10 µM) | |
|---|---|---|---|
| (A) Control | |||
| LVDevP (mmHg) | 97 ± 4 | 100 ± 4 | 117 ± 6* |
| Heart rate (b.p.m.) | 188 ± 5 | 188 ± 4 | 192 ± 2 |
| (B) Nitrate-tolerant | |||
| LVDevP (mmHg) | 94 ± 5 | 100 ± 8 | 118 ± 8* |
| Heart rate (b.p.m.) | 203 ± 10 | 205 ± 8 | 201 ± 11 |
| (C) Ascorbate-deficient | |||
| LVDevP (mmHg) | 99 ± 5 | 105 ± 2 | 119 ± 2* |
| Heart rate (b.p.m.) | 201 ± 11 | 203 ± 8 | 206 ± 10 |
| (D) Nitrate-tolerant and ascorbate-deficient | |||
| LVDevP (mmHg) | 95 ± 1 | 97 ± 1 | 121 ± 2* |
| Heart rate (b.p.m.) | 201 ± 8 | 196 ± 7 | 201 ± 9 |
| Baseline | GTN (100 µM) | DEA/NO (10 µM) | |
|---|---|---|---|
| (A) Control | |||
| LVDevP (mmHg) | 97 ± 4 | 100 ± 4 | 117 ± 6* |
| Heart rate (b.p.m.) | 188 ± 5 | 188 ± 4 | 192 ± 2 |
| (B) Nitrate-tolerant | |||
| LVDevP (mmHg) | 94 ± 5 | 100 ± 8 | 118 ± 8* |
| Heart rate (b.p.m.) | 203 ± 10 | 205 ± 8 | 201 ± 11 |
| (C) Ascorbate-deficient | |||
| LVDevP (mmHg) | 99 ± 5 | 105 ± 2 | 119 ± 2* |
| Heart rate (b.p.m.) | 201 ± 11 | 203 ± 8 | 206 ± 10 |
| (D) Nitrate-tolerant and ascorbate-deficient | |||
| LVDevP (mmHg) | 95 ± 1 | 97 ± 1 | 121 ± 2* |
| Heart rate (b.p.m.) | 201 ± 8 | 196 ± 7 | 201 ± 9 |
LVDevP, left ventricular developed pressure. Data are mean values±SEM of three hearts per group.
*P < 0.05 vs. corresponding baseline.
Haemodynamic parameters of isolated guinea pig hearts at baseline and after application of nitroglycerin (GTN) or 2,2-diethyl-1-nitroso-oxyhydrazine (DEA/NO)
| Baseline | GTN (100 µM) | DEA/NO (10 µM) | |
|---|---|---|---|
| (A) Control | |||
| LVDevP (mmHg) | 97 ± 4 | 100 ± 4 | 117 ± 6* |
| Heart rate (b.p.m.) | 188 ± 5 | 188 ± 4 | 192 ± 2 |
| (B) Nitrate-tolerant | |||
| LVDevP (mmHg) | 94 ± 5 | 100 ± 8 | 118 ± 8* |
| Heart rate (b.p.m.) | 203 ± 10 | 205 ± 8 | 201 ± 11 |
| (C) Ascorbate-deficient | |||
| LVDevP (mmHg) | 99 ± 5 | 105 ± 2 | 119 ± 2* |
| Heart rate (b.p.m.) | 201 ± 11 | 203 ± 8 | 206 ± 10 |
| (D) Nitrate-tolerant and ascorbate-deficient | |||
| LVDevP (mmHg) | 95 ± 1 | 97 ± 1 | 121 ± 2* |
| Heart rate (b.p.m.) | 201 ± 8 | 196 ± 7 | 201 ± 9 |
| Baseline | GTN (100 µM) | DEA/NO (10 µM) | |
|---|---|---|---|
| (A) Control | |||
| LVDevP (mmHg) | 97 ± 4 | 100 ± 4 | 117 ± 6* |
| Heart rate (b.p.m.) | 188 ± 5 | 188 ± 4 | 192 ± 2 |
| (B) Nitrate-tolerant | |||
| LVDevP (mmHg) | 94 ± 5 | 100 ± 8 | 118 ± 8* |
| Heart rate (b.p.m.) | 203 ± 10 | 205 ± 8 | 201 ± 11 |
| (C) Ascorbate-deficient | |||
| LVDevP (mmHg) | 99 ± 5 | 105 ± 2 | 119 ± 2* |
| Heart rate (b.p.m.) | 201 ± 11 | 203 ± 8 | 206 ± 10 |
| (D) Nitrate-tolerant and ascorbate-deficient | |||
| LVDevP (mmHg) | 95 ± 1 | 97 ± 1 | 121 ± 2* |
| Heart rate (b.p.m.) | 201 ± 8 | 196 ± 7 | 201 ± 9 |
LVDevP, left ventricular developed pressure. Data are mean values±SEM of three hearts per group.
*P < 0.05 vs. corresponding baseline.
3.5 Effects of ascorbate on nitroglycerin metabolism by purified aldehyde dehydrogenase 2 and isolated liver mitochondria
To extend and corroborate the organ bath experiments performed in the presence of ALDH2 inhibitors, we studied whether GTN metabolism by mitochondrial ALDH required ascorbate as a cofactor. In particular, we were interested to see whether ascorbate could substitute reduced thiols that are required for ALDH2 reactivation. As shown in Table 2, the rates of 1,2- and 1,3-GDN formation catalysed by purified ALDH2 in the absence and presence of thiols (2 mM GSH+2 mM dithiothreitol) were not significantly affected by up to 10 mM ascorbate. However, if ascorbate were involved in the regeneration of an endogenous ALDH2 cofactor, its effect would not become apparent in experiments with purified ALDH2. Therefore, we repeated the experiments with liver mitochondria isolated from ascorbate-deficient guinea pigs. The rates of GTN metabolism shown in Table 2 were similar to those reported previously for rat liver mitochondria.16 Again, up to 10 mM ascorbate had no effect on reaction rates determined in the absence and presence of thiols, further suggesting that ALDH2 inactivation did not contribute to nitrate tolerance in ascorbate deficiency. Formation of 1,2-GDN by purified ALDH2 and isolated mitochondria was completely inhibited by 10 mM chloral hydrate (data not shown).
Effects of ascorbate on nitroglycerin (GTN) metabolism by purified aldehyde dehydrogenase2 (ALDH2) and liver mitochondria isolated from ascorbate-deficient guinea pigs
| Purified ALDH2 | Isolated mitochondria | ||||
|---|---|---|---|---|---|
| 1,2-GDN (nmol × min−1 × mg−1) | 1,3-GDN (nmol × min−1 × mg−1) | 1,2-GDN (pmol × min−1 × mg−1) | 1,3-GDN (pmol × min−1 × mg−1) | ||
| + Thiols | Control | 9.38 ± 0.026 | 0.23 ± 0.009 | 95.48 ± 0.44 | 32.99 ± 0.42 |
| + Asc 0.1 mM | 9.33 ± 0.077 | 0.23 ± 0.003 | 93.06 ± 0.88 | 32.28 ± 0.15 | |
| 1 mM | 9.40 ± 0.042 | 0.21 ± 0.014 | 96.33 ± 0.43 | 31.85 ± 0.20 | |
| 10 mM | 9.27 ± 0.099 | 0.16 ± 0.001 | 97.26 ± 2.79 | 29.28 ± 3.27 | |
| − Thiols | Control | 1.71 ± 0.16 | 0.063 ± 0.036 | 23.83 ± 3.15 | 3.39 ± 0.78 |
| + Asc 0.1 mM | 1.98 ± 0.10 | 0.024 ± 0.008 | 27.06 ± 3.89 | 4.74 ± 0.89 | |
| 1 mM | 1.75 ± 0.27 | 0.031 ± 0.003 | 27.40 ± 1.90 | 4.00 ± 0.45 | |
| 10 mM | 1.90 ± 0.13 | 0.041 ± 0.011 | 22.94 ± 4.05 | 3.20 ± 0.36 | |
| Purified ALDH2 | Isolated mitochondria | ||||
|---|---|---|---|---|---|
| 1,2-GDN (nmol × min−1 × mg−1) | 1,3-GDN (nmol × min−1 × mg−1) | 1,2-GDN (pmol × min−1 × mg−1) | 1,3-GDN (pmol × min−1 × mg−1) | ||
| + Thiols | Control | 9.38 ± 0.026 | 0.23 ± 0.009 | 95.48 ± 0.44 | 32.99 ± 0.42 |
| + Asc 0.1 mM | 9.33 ± 0.077 | 0.23 ± 0.003 | 93.06 ± 0.88 | 32.28 ± 0.15 | |
| 1 mM | 9.40 ± 0.042 | 0.21 ± 0.014 | 96.33 ± 0.43 | 31.85 ± 0.20 | |
| 10 mM | 9.27 ± 0.099 | 0.16 ± 0.001 | 97.26 ± 2.79 | 29.28 ± 3.27 | |
| − Thiols | Control | 1.71 ± 0.16 | 0.063 ± 0.036 | 23.83 ± 3.15 | 3.39 ± 0.78 |
| + Asc 0.1 mM | 1.98 ± 0.10 | 0.024 ± 0.008 | 27.06 ± 3.89 | 4.74 ± 0.89 | |
| 1 mM | 1.75 ± 0.27 | 0.031 ± 0.003 | 27.40 ± 1.90 | 4.00 ± 0.45 | |
| 10 mM | 1.90 ± 0.13 | 0.041 ± 0.011 | 22.94 ± 4.05 | 3.20 ± 0.36 | |
The rates of 1,2- and 1,3-GDN formation from 2 µM 14C-labelled GTN were determined by radio thin-layer chromatography as described16 with or without GSH/dithiothreitol (2 mM each; ±Thiols) in the absence (Control) or presence of the indicated concentrations of ascorbate (Asc). Data are mean values ± SE from three independent experiments.
Effects of ascorbate on nitroglycerin (GTN) metabolism by purified aldehyde dehydrogenase2 (ALDH2) and liver mitochondria isolated from ascorbate-deficient guinea pigs
| Purified ALDH2 | Isolated mitochondria | ||||
|---|---|---|---|---|---|
| 1,2-GDN (nmol × min−1 × mg−1) | 1,3-GDN (nmol × min−1 × mg−1) | 1,2-GDN (pmol × min−1 × mg−1) | 1,3-GDN (pmol × min−1 × mg−1) | ||
| + Thiols | Control | 9.38 ± 0.026 | 0.23 ± 0.009 | 95.48 ± 0.44 | 32.99 ± 0.42 |
| + Asc 0.1 mM | 9.33 ± 0.077 | 0.23 ± 0.003 | 93.06 ± 0.88 | 32.28 ± 0.15 | |
| 1 mM | 9.40 ± 0.042 | 0.21 ± 0.014 | 96.33 ± 0.43 | 31.85 ± 0.20 | |
| 10 mM | 9.27 ± 0.099 | 0.16 ± 0.001 | 97.26 ± 2.79 | 29.28 ± 3.27 | |
| − Thiols | Control | 1.71 ± 0.16 | 0.063 ± 0.036 | 23.83 ± 3.15 | 3.39 ± 0.78 |
| + Asc 0.1 mM | 1.98 ± 0.10 | 0.024 ± 0.008 | 27.06 ± 3.89 | 4.74 ± 0.89 | |
| 1 mM | 1.75 ± 0.27 | 0.031 ± 0.003 | 27.40 ± 1.90 | 4.00 ± 0.45 | |
| 10 mM | 1.90 ± 0.13 | 0.041 ± 0.011 | 22.94 ± 4.05 | 3.20 ± 0.36 | |
| Purified ALDH2 | Isolated mitochondria | ||||
|---|---|---|---|---|---|
| 1,2-GDN (nmol × min−1 × mg−1) | 1,3-GDN (nmol × min−1 × mg−1) | 1,2-GDN (pmol × min−1 × mg−1) | 1,3-GDN (pmol × min−1 × mg−1) | ||
| + Thiols | Control | 9.38 ± 0.026 | 0.23 ± 0.009 | 95.48 ± 0.44 | 32.99 ± 0.42 |
| + Asc 0.1 mM | 9.33 ± 0.077 | 0.23 ± 0.003 | 93.06 ± 0.88 | 32.28 ± 0.15 | |
| 1 mM | 9.40 ± 0.042 | 0.21 ± 0.014 | 96.33 ± 0.43 | 31.85 ± 0.20 | |
| 10 mM | 9.27 ± 0.099 | 0.16 ± 0.001 | 97.26 ± 2.79 | 29.28 ± 3.27 | |
| − Thiols | Control | 1.71 ± 0.16 | 0.063 ± 0.036 | 23.83 ± 3.15 | 3.39 ± 0.78 |
| + Asc 0.1 mM | 1.98 ± 0.10 | 0.024 ± 0.008 | 27.06 ± 3.89 | 4.74 ± 0.89 | |
| 1 mM | 1.75 ± 0.27 | 0.031 ± 0.003 | 27.40 ± 1.90 | 4.00 ± 0.45 | |
| 10 mM | 1.90 ± 0.13 | 0.041 ± 0.011 | 22.94 ± 4.05 | 3.20 ± 0.36 | |
The rates of 1,2- and 1,3-GDN formation from 2 µM 14C-labelled GTN were determined by radio thin-layer chromatography as described16 with or without GSH/dithiothreitol (2 mM each; ±Thiols) in the absence (Control) or presence of the indicated concentrations of ascorbate (Asc). Data are mean values ± SE from three independent experiments.
4. Discussion
Our data demonstrate that ascorbate is essential for GTN-induced vasodilation in the macro- and microvasculature of guinea pigs. The potency of GTN was decreased by about 10- and 100-fold in both non-tolerant and nitrate-tolerant blood vessels after 2 and 4 weeks of ascorbate deprivation, respectively. Previous studies have shown that ascorbate supplementation prevents the development of nitrate tolerance in rodents,25 healthy humans,26 and patients with congestive heart failure.27 This effect is widely considered to result from reduction of oxidative stress in the vasculature,10 but two studies claimed that ascorbate prevents nitrate tolerance independently of its antioxidant function.28,29 It is unclear whether the earlier findings are related to our present observations. Ascorbate supplementation had no effect on GTN-induced reduction of blood pressure in mice (see Supplementary material, Figure S1), indicating that normal ascorbate levels are sufficient for GTN action in non-tolerant animals. However, chronic GTN administration could cause reduction of vascular ascorbate levels, leading to impaired GTN bioactivation that is restored by exogenous administration of ascorbate. Interestingly, the increase in the EC50 of GTN caused by nitrate tolerance was clearly less pronounced in ascorbate-deficient rings than in control vessels (cf. Figures 2A and 5A), suggesting that the development of nitrate tolerance may indeed compromise an ascorbate-dependent mechanism of GTN action.
Dozens of studies have documented the beneficial effects of ascorbate on endothelial dysfunction in a variety of pathological conditions, including atherosclerosis, type I and type II diabetes, coronary artery disease, and smoking.30 Interestingly, all of these pathologies are associated with decreased plasma ascorbate levels, suggesting that the beneficial effects of the vitamin in GTN tolerance could also be owing to correction of abnormally low plasma or tissue levels. Unfortunately, there is only little information available on the potential effects of long-term GTN application on tissue ascorbate levels. Milone et al.31 found no changes in ascorbate plasma levels after continuous therapy of human subjects with transdermal GTN. However, this result is not conclusive because—in contrast to most other studies—the authors did not observe improved haemodynamic effects of GTN by co-administration of ascorbate. McVeigh et al.32 also reported that ascorbate plasma levels remained unchanged after 3 days of transdermal GTN administration. To address this issue, we measured ascorbate plasma levels in control and GTN-tolerant guinea pigs and surprisingly found a decrease by about 50% in nitrate-tolerant animals. Although these data may support the hypothesis that ascorbate deficiency contributes to GTN tolerance, our functional data, indicating that tolerance develops only after prolonged periods of ascorbate deprivation, raise the question whether the decrease in ascorbate levels observed after 3 days of GTN application does indeed affect GTN vasoactivity. Further studies, in particular determination of ascorbate levels in blood vessels, are needed to clarify this issue.
It has to be considered that ascorbate deficiency may affect vascular morphology and function because of impaired synthesis of collagen.33,34In vitro angiogenesis assays using a rat aortic ring model showed that microvessels grown in the absence of ascorbate were large and dilated, whereas vessels grown in the presence of the vitamin resembled normal capillaries.35 The results of in vivo studies carried out either with ascorbate-deficient guinea pigs36 or with l-gulonolactone oxidase knockout mice37,38 are controversial. In our model, vessels of standard diet-fed animals and ascorbate-deficient animals showed identical contractions to U46619, and relaxation to DEA/NO was not affected by 2 weeks of ascorbate deficiency and only slightly reduced after 4 weeks. Moreover, even 4 weeks of ascorbate deprivation did not affect heart weight or relaxation of the coronary bed to DEA/NO (which would have shown as reduced coronary flow), indicating that morphological changes of blood vessels possibly caused by ascorbate deficiency must have been subtle, if at all present in our model. Thus, the effect of ascorbate depletion on GTN-induced relaxation appears to reflect impaired bioactivation of the nitrate rather than vascular dysfunction.
As expected, relaxation of aortic rings to both GTN and DEA/NO was accompanied by accumulation of cGMP. In agreement with the functional data, ascorbate deficiency caused a marked decrease in GTN-triggered cGMP accumulation in both non-tolerant and nitrate-tolerant vessels. The data obtained with DEA/NO are less clear. There was a tendency of reduced cGMP levels in non-tolerant ascorbate-deficient rings, but this effect was not significant. Thus, it remains unclear whether the slightly decreased vasodilatory potency of DEA/NO after 4 weeks of ascorbate deprivation reflects impaired sGC activation, e.g., because of decreased NO bioavailability, or vascular dysfunction downstream of cGMP formation.
At present we can only speculate about the molecular mechanism underlying the ascorbate dependence of GTN-induced vasodilation. Several not necessarily mutually exclusive explanations have to be considered: (i) ascorbate could trigger non-enzymatic bioactivation of GTN,13 (ii) it could act as a reducing cofactor supporting ALDH2-catalysed GTN metabolism,8,9 (iii) it could be essential for linking GTN metabolism to sGC activation, (iv) it could increase NO bioavailability by reducing oxidative stress in response to GTN,39 and (v) it could be required for sustained ALDH2 expression in vascular smooth muscle.
Although ascorbate depletion was virtually complete after 2 weeks, vascular GTN responsiveness was further decreased about 10-fold after 4 weeks of ascorbate deprivation. This delayed development of nitrate tolerance may be considered as evidence against an acute ascorbate dependence of GTN bioactivation, most likely excluding a role of ascorbate in non-enzymatic bioactivation or as a cofactor of the ALDH2 reaction. Our observations that ALDH2 inhibitors affected the GTN response in control and ascorbate-deficient blood vessels to a similar degree and that ascorbate had no effect on ALDH2-catalysed GTN metabolism by isolated mitochondria and purified ALDH2 provide further evidence against the involvement of ALDH2 inactivation. It is still unknown how ALDH-catalysed nitrite formation is linked to sGC activation. Conceivably, ascorbate could be essentially involved in this process simply by reduction of GTN-derived nitrite to NO. Considering the slow onset of GTN tolerance upon ascorbate deprivation, this would imply significantly delayed depletion of ascorbate in vascular smooth muscle. The same applies to a potential antioxidative function of ascorbate. There is evidence that long-term application of GTN triggers generation of superoxide that reduces NO bioavailability in nitrate tolerance,10,40 and prevention of vascular tolerance to GTN by co-administration of ascorbate has been attributed to superoxide scavenging.41 As our data revealed a marked ascorbate dependence of the GTN response of non-tolerant blood vessels, the oxidative stress hypothesis of ascorbate action would imply that NO availability is significantly attenuated by GTN-derived superoxide even upon acute administration of the nitrate. The lack of effect of PEG-SOD/PEG-catalase may argue against the idea that nitrate tolerance is caused by superoxide production, but the data do not exclude the involvement of mitochondrial superoxide not accessible to exogenously applied scavenging enzymes.
Finally, the slow development of GTN tolerance may result from transcriptional downregulation of an enzyme essentially involved in GTN bioactivation. Although the rates of GTN metabolism by guinea pig liver mitochondria were not reduced upon ascorbate depletion (data not shown), ascorbate may have a distinct effect on the expression of ALDH2 (or an auxiliary protein) in vascular tissue. As the sequence of guinea pig ALDH2 is unknown, we have no possibility at present to address this issue by reverse transcriptase-polymerase chain reaction or immunoblotting with sequence-directed antibodies. To circumvent this drawback of the guinea pig model, we are planning to establish l-gulonolactone oxidase-deficient mice in our laboratory as a more widely applicable animal model of ascorbate deficiency.37 This model would have the additional advantage of allowing non-invasive blood pressure measurements to judge the consequence of ascorbate deficiency on the GTN response in vivo.
In conclusion, the present study provides unequivocal evidence that prolonged ascorbate deficiency impairs vascular relaxation to GTN. Future studies addressing the potential molecular mechanisms underlying this effect may provide new insights into the complex processes involved in vascular GTN bioactivation. In case that ascorbate deficiency similarly affects the GTN response in vivo, our data may also have important clinical implications with respect to GTN therapy of patients suffering from various cardiovascular and non-cardiovascular diseases that are associated with oxidative stress and low ascorbate levels.
Supplementary material
Supplementary material is available at Cardiovascular Research Online.
Funding
Fonds zur Förderung der Wissenschaftlichen Forschung in Österreich (W901 DK Molecular Enzymology, P16690 and P20669 to B.M.).
Acknowledgements
We wish to thank Friedrich Brunner (1952–2007) for his essential contribution to this study and indispensable assistance during the preparation of an early version of the manuscript.
Conflict of interest: none declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}