





Abstract
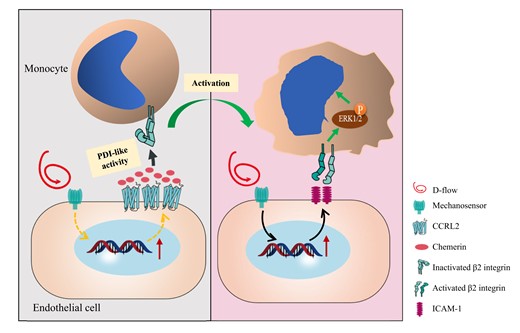
Chemoattractants and their cognate receptors are essential for leucocyte recruitment during atherogenesis, and atherosclerotic plaques preferentially occur at predilection sites of the arterial wall with disturbed flow (d-flow). In profiling the endothelial expression of atypical chemoattractant receptors (ACKRs), we found that Ackr5 (CCRL2) was up-regulated in an endothelial subpopulation by atherosclerotic stimulation. We therefore investigated the role of CCRL2 and its ligand chemerin in atherosclerosis and the underlying mechanism.
By analysing scRNA-seq data of the left carotid artery under d-flow and scRNA-seq datasets GSE131776 of ApoE−/− mice from the Gene Expression Omnibus database, we found that CCRL2 was up-regulated in one subpopulation of endothelial cells in response to d-flow stimulation and atherosclerosis. Using CCRL2−/−ApoE−/− mice, we showed that CCRL2 deficiency protected against plaque formation primarily in the d-flow areas of the aortic arch in ApoE−/− mice fed high-fat diet. Disturbed flow induced the expression of vascular endothelial CCRL2, recruiting chemerin, which caused leucocyte adhesion to the endothelium. Surprisingly, instead of binding to monocytic CMKLR1, chemerin was found to activate β2 integrin, enhancing ERK1/2 phosphorylation and monocyte adhesion. Moreover, chemerin was found to have protein disulfide isomerase-like enzymatic activity, which was responsible for the interaction of chemerin with β2 integrin, as identified by a Di-E-GSSG assay and a proximity ligation assay. For clinical relevance, relatively high serum levels of chemerin were found in patients with acute atherothrombotic stroke compared to healthy individuals.
Our findings indicate that d-flow-induced CCRL2 promotes atherosclerotic plaque formation via a novel CCRL2-chemerin-β2 integrin axis, providing potential targets for the prevention or therapeutic intervention of atherosclerosis.
Time of primary review: 18 days
Modulation of endothelial functional molecules that respond to d-flow in the blood vessel is believed to be a reasonable approach to inhibit proatherogenic mechanisms. Our findings that vascular CCRL2 expression is up-regulated by d-flow and the novel CCRL2-chemerin-β2 integrin axis contributes to atherosclerosis provide potential targets for the prevention or therapeutic intervention of atherosclerosis. Furthermore, the identification of chemerin as an independent risk factor for acute atherothrombotic stroke suggests its clinical application as a potential biomarker.
1. Introduction
Atherosclerosis is a chronic inflammatory disease of the arterial wall elicited by the accumulation of LDL and leucocytes in the subendothelium.1–4 It preferentially develops at predilection sites with disturbed blood flow (d-flow), where the dysfunctional endothelial cell phenotype mediates leucocyte rolling, adhesion, and transmigration, initiating plaque development.5–8 Normally, endothelial cells (ECs) that cover the vessel lumen protect vascular integrity and homeostasis by sensing and responding to physical, chemical, and biological stimuli.9–12 When exposed to d-flow, these cells undergo major phenotypic changes, which lead to increased endothelial permeability, cytokine release, and leucocyte adhesion.1,13–15 Monocytes, the major cells that infiltrate into the subendothelium, differentiate into macrophages that are transformed into foam cells by taking up cholesterol deposits, forming fatty streaks16–19 that eventually become complex vulnerable plaques liable to rupture, which may lead to myocardial infarction and stroke.20
Chemoattractants and their receptors are critical players in atherosclerosis, as they not only direct atherogenic recruitment of leucocytes but also tune their functional capabilities.21,22 For example, CCL2 mediates the recruitment and retention of CCR2+ monocytes to atherosclerotic plaques, while dendritic cell-derived CCL17 perturbs regulatory T-cell homeostasis and promotes atherogenesis.23 Upon ligand binding, most chemoattractant receptors signal via activation of heterotrimeric G proteins. Unlike these classical chemoattractant receptors, atypical chemoattractant receptors are G protein-independent and mainly function as scavenger receptors for chemoattractants.24 The ACKR family mainly comprises ACKR1/DARC (Duffy Antigen Receptor for Chemokines), ACKR2/D6, ACKR3/CXCR7, ACKR4/CCRL1 (CC-Chemokine Receptors like 1), ACKR5/CCRL2, and ACKR6 (Pitpnm3). ACKR5/CCRL2, an atypical chemoattractant receptor chemokine (C-C motif) receptor-like 2, is expressed by mast cells, activated macrophages, and vascular ECs,25,26 while its ligand chemerin is a multifunctional leucocyte chemoattractant protein secreted predominantly by adipose tissue, skin, liver, and vascular endothelium. It is known primarily for its chemotactic and adipokine properties27–29 and was reported to regulate thermogenesis30 and adipogenesis.31 In contrast to typical chemokine receptors, chemerin does not trigger CCRL2 internalization, chemotaxis, or calcium mobilization. Rather, CCRL2 acts to focus and facilitate chemerin activity by increasing local chemerin concentrations and presenting bound chemerin to its signalling receptor, chemokine-like receptor 1 (CMKLR1), on adjacent cells.25,26 CCRL2 is expressed in a tissue- and activation-dependent manner, regulating local chemerin levels and bioactivity and enhancing chemerin- and CMKLR1-dependent leucocyte/EC adhesion in vitro and recruitment to inflamed airways in vivo.26
By analysing scRNA-seq data of the left carotid arteries (LCAs) under d-flow and scRNA-seq datasets GSE131776 of ApoE−/− mice from the Gene Expression Omnibus database, we found that endothelial CCRL2 was up-regulated under d-flow and expressed in atherosclerosis plaques. We thus investigated the role of vascular CCRL2 in the development of atherosclerosis in mice and showed that deletion of CCRL2 significantly reduced macrophage accumulation and plaque formation in the aorta of ApoE−/− mice. Notably, d-flow-induced endothelial expression of CCRL2 increased chemerin accumulation, which contributes to monocyte adhesion by activating β2 integrin via the protein disulfide isomerase (PDI)-like activity of chemerin rather than through CMKLR1. Moreover, CCRL2 and chemerin were detectable in human carotid plaques, and patients with acute atherothrombotic stroke (AAS), a prevalent cerebral vascular atherosclerotic disease, showed higher serum levels of the CCRL2 ligand chemerin.
2. Methods
2.1 Human samples, enzyme-linked immunosorbent assay, and staining
A total of 111 patients with AAS and 111 age- and gender-matched control subjects were randomly drawn from the China Antihypertensive Trial in Acute Ischemic Stroke (CATIS), a randomized controlled clinical trial performed from August 2009 through May 2013 in 26 hospitals from China. The detail of CATIS was described elsewhere.32 Informed consent was obtained from each patient/family before collection of samples. Clinical data were collected from the electronic patient database from each hospital. Patients with cardiac disease, lung disease, infection, autoimmune disease, neoplasia, and under anti-inflammatory or anti-coagulant drugs were excluded. Those with cardioembolic stroke were also excluded from the current study. Forearm venous blood on admission was collected in EDTA anti-coagulated vacuum tubes and stored at −80°C for measurements of chemerin using enzyme-linked immunosorbent assay (ELISA). Sample preparation, analysis, and calculation were performed according to the manufacturer’s instructions (SEA945Hu, Cloud-Clone, Houston, TX). The study protocols are in strict accordance with the Declaration of Helsinki.33 The characteristics of the included subjects can be found in the Supplementary material online, Table S2. The association between serum chemerin and AAS was analysed using multivariate logistic regression. Areas under the receiver operator curve were calculated to evaluate the concordance of the logistic model. The AUC before and after adding chemerin to the conventional AAS risk factor was compared using Delong’s test. All anonymous human tissues were from a biobank at the Department of Pathology of Soochow University. The immunostaining details of human carotid plaques can be found in the Supplementary material.
2.2 Animals
All animals were housed in a specific pathogen-free facility, and all experiments were approved by the University Committee on Animal Care of Soochow University. CCRL2−/− mice on the C57BL/6J background were purchased from Jackson Laboratories (Bar Harbor, ME, USA). Wild-type (WT), ApoE−/− (C57BL/6J background), and green fluorescence protein (GFP)-transgenic mice (C57BL/6J background) were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China). CCRL2−/−ApoE−/− mice were generated by crossing CCRL2−/− mice with ApoE−/− mice. Littermate male ApoE−/− mice were fed a normal chow diet for 8 weeks and then switched to a high-fat diet (HFD, 0.15% cholesterol, and 21% fat without added cholate, Harlan Teklad diet No. 88137, Madison, WI, USA). Before the experiment, mice were anaesthetized by intraperitoneal injection of 1% pentobarbital (7 μL/g). Mouse partial carotid ligation (PCL) surgery was performed under isoflurane anaesthesia (3%). The anaesthesia degree of mice was examined by the response of pinching toes. At the end of the experiments, mice were euthanized by CO2 inhalation followed by cervical dislocation. More information about the mice is provided in the Supplementary material. All procedures followed the guidelines from Directive 2010/63/EU of the European Parliament.
2.3 Single-cell sequencing analysis
Single-cell suspension preparation, single-cell RNA sequencing, and single-cell data pre-processing were performed as previously described.34 In brief, the diced vascular tissue was collected and incubated in a solution containing dissociation enzyme for single-cell suspension. Then, the suspensions were loaded onto a Chromium Single Cell Controller (10x Genomics, Pleasanton, CA). Dimensionality reduction, visualization, and analysis of scRNA-sequencing data were performed with the R package Seurat (version 3.1.2). scRNA-seq libraries were prepared using the Single Cell 3′ Library and Gel Bead Kit V3 following the manufacturer’s instructions. Sequencing was performed on an Illumina (San Diego, CA) NovaSeq 6000 sequencer with a paired-end 150 bp (PE150) reading strategy (performed by CapitalBio Technology, Beijing). scRNA-sequencing data were analysed with the R package Seurat (version 4.1.0).
2.4 Analysis of atherosclerotic lesions
Atherosclerotic plaques were quantified in en face preparations stained with Sudan IV as described previously.35 Aortic images encompassing the entire aorta (arch, thorax, and abdomen) were captured with an Olympus SZX16 Telescope (Olympus, Japan) and analysed by Olympus cellSens Standard software (Japan). For the aortic roots, the hearts were cut at 50 µm increments until the valves appeared. Then, the sections were cut at 8 µm and stained with Oil Red O (NJJC, D027) followed by counterstaining with haematoxylin (NJJC, D006).
2.5 Immunofluorescence staining
Monocytes/macrophages were detected with rat anti-mouse MoMa-2 antibody (ab33451, Abcam, USA). Endothelial cells were detected with rat anti-mouse CD31 (553370, BD Pharmingen, USA). For CCRL2 and chemerin, we used rat anti-mouse CCRL2 antibody (498321, Thermo Fisher, USA) or mouse anti-mouse CCRL2 (ab88632, Abcam, USA), rabbit-anti-human/mouse chemerin antibody (ab103531, Abcam, USA) or rabbit anti-mouse chemerin antibody (bs-1501R, Bioss, USA), and IgG controls of corresponding species. The cytoskeleton of THP-1 cells was detected with rhodamine-labelled phalloidin (R415, Thermo Fisher, USA). Morphological adhesion of THP-1 cells with protruded pseudopodia was defined as activated THP-1 cells. The secondary antibodies were Alexa Fluor 488-conjugated donkey anti-mouse IgG, Alexa Fluor 488-conjugated donkey anti-rabbit IgG, Alexa Fluor 555-conjugated donkey anti-goat IgG, Alexa Fluor 568-conjugated donkey anti-mouse IgG, Alexa Fluor 568-conjugated donkey anti-hamster IgG, and Alexa Fluor 647-conjugated donkey anti-rat IgG (all from Abcam). Tissues were counterstained with DAPI before being mounted and examined by confocal microscopy (Olympus, Japan).
2.6 Induction of disturbed flow in vivo by partial carotid ligation
Partial carotid ligation was performed as described previously.7,36 Briefly, we ligated the external carotid artery, internal carotid artery, and occipital artery of the LCA and left the superior thyroid artery untouched. Detailed methods can be found in the Supplementary material.
2.7 Immunoblotting
Immunoblotting was performed as described previously.37 Primary antibodies included mouse anti-mouse CCRL2 (ab88632, Abcam, USA), rabbit anti-human CCRL2 (ab88624, Abcam, USA), mouse anti-mammalian β-actin (AA128, Beyotime, China), mouse anti-mammalian β-tubulin (70-ab009-040, Multi Sciences, China), rabbit anti-mammalian p-ERK (9102S, Cell Signaling Technology, USA), rabbit anti-mammalian ERK (4376S, Cell Signaling Technology, USA), and rabbit anti-human/mouse/rat CMKLR1 (PA5-50932, Thermo Fisher Scientific, USA). The following secondary antibodies were used: goat anti-rabbit IRDye 800CW, goat anti-mouse IRDye 800CW, and Licor Odyssey.
2.8 Bone marrow transplantation
Male CCRL2+/+ or CCRL2−/− mice (8 weeks old) were irradiated with a dose of 10 Gy and reconstituted with 1 × 107 bone marrow cells from GFP+ transgenic mice. Bone marrow cells were obtained by flushing the femurs and tibias of donor mice with IMDM. The donor mice were euthanized, and the collected bone marrow cells were transplanted into recipient mice by orbit posterior venous plexus vein injection. After bone marrow reconstitution, mice were maintained on a chow diet for 6–8 weeks and then used for leucocyte adhesion experiments.
2.9 Ex vivo leucocyte adhesion on the endothelium of the aorta
The ex vivo model was modified from an existing method.7 In brief, mouse aortic arches and thoracic aortas were isolated from WT and CCRL2−/− mice (8 weeks old) and immediately placed into DMEM + 1% heat-inactivated FBS. The aortas were opened longitudinally and pinned onto sterile polydimethylsiloxane. Mouse serum was collected the day before and stored at −80°C. Isolated mouse leucocytes were obtained from WT mice. Leucocytes were labelled with Calcein-AM Molecular Probes (65-0855-39, Thermo Fisher, Waltham, MA) according to the manufacturer’s instructions. Aortas were incubated for 60 min with 1 × 106 fluorescence-labelled mouse leucocytes. After incubation, unbound monocytes were rinsed away, and the number of monocytes firmly bound to the aorta was counted using fluorescent microscopy. Mouse serum or chemerin blocking antibody (AF2325, R&D, Minneapolis, MN) was incubated, and PBS was used as a control.
2.10 Cell culture
As previously described,7 human umbilical venous endothelial cells (HUVECs) (cc-2519, Lonza, Switzerland) were maintained in M199 (MT10060CV, Coring, USA) base culture medium containing 10% FBS (SH30071, HyClone, USA), 1% penicillin/streptomycin/fungizone (15240-062, Gibco, USA), 10% GlutaMAX (25-005-cl, Coring, USA), 10% endothelial cell growth serum, and 0.4% heparin (404867, McKesson Medical, USA). THP-1 cells were cultured in RPMI-1640 medium (SH30809.01, HyClone, USA) containing 10% FBS and 1% penicillin/streptomycin/fungizone (15240-062, Gibco, USA).
2.11 Serum chemerin assays
Mouse serum was frozen at −80°C and then thawed and centrifuged prior to measurement. Chemerin levels were determined using murine Quantikine ELISA kits (MCHM00, R&D, USA) according to the manufacturer’s instructions.
2.12 Real-time quantitative polymerase chain reaction
Total RNA was obtained from aorta by an RNA simple Total RNA Kit (DP419, TIANGEN, China). Total RNA was reverse transcribed into cDNA using PrimeScript RT Master Mix (TAKARA, Japan). Real-time quantitative polymerase chain reaction (RT–qPCR) for specific genes was performed using SYBR Green PCR master mix (A46109, Thermo Fisher, Waltham, MA) with custom-designed primers on the Roche LightCycler480 Real-Time PCR System. Results were normalized to GAPDH RNA, and the fold change was determined using the 2−ΔΔCT method.38 Primer sequences for each gene are given in the Supplementary material online, Table S4.
2.13 CCRL2 expression in HUVECs and the aortic arch
HUVECs were cultured in a 10 cm petri dish. A modified cone-and-plate shear stress device was used to generate oscillatory shear stress (OS) and laminar shear stress (LS) in vitro.39 HUVECs (70% confluent) were stimulated with OS (10 ± 5 dyn/cm2) for 24 h, and HUVECs were treated with LS (30 dyn/cm2) for 24 h as a control. The cells were measured for CCRL2 mRNA and protein expression. In the mRNA assay, HUVECs were collected by an RNA simple Total RNA Kit (DP419, TIANGEN, China), and CCRL2 was detected by RT–qPCR. For the protein assay, HUVECs treated with or without OS were lysed with protein lysis buffer, and CCRL2 was detected by Western blotting.
CCRL2 expression in the regions of greater curvature (GC) and lesser curvature (LC) of the aortic arch was examined by en face staining. To define the GC and LC regions, we opened the aortic arch along the GC and spread it on a surface. The middle portion is the LC region with the GC area on each side. We then performed en face staining for CCRL2 (magenta) and CD31 (green).
2.14 Flow cytometric detection of activated β2 integrin
After 1 × 106 cells were stimulated with chemerin157S (Chem157S) for 5 min, mouse-anti-human CD11 + CD18 antibody (Mab24, ab13219, Abcam, USA) was incubated for 30 min at RT in PBS and 0.1% FBS and then washed in the same buffer prior to incubation with Alexa Fluor 488-conjugated donkey anti-mouse IgG under the same conditions. After final washes, cell fluorescence was assessed using a FACS counting 1 × 104 cells. Flow cytometry data were analysed with FlowJo 10.0 (BD Bioscience, San Jose, CA).
2.15 Assay of enzyme-dependent disulfide reduction
The activities of Chem157S and ERp57 disulfide reductase against Di-E-GSSG were monitored in PDI assay buffer (0.1 M potassium phosphate buffer, 2 mM EDTA, pH 7.0) by adding 200 nM Chem157S and 20 nM ERp57 to Di-E-GSSG (150 nM) in the presence of 5 μM DTT. The increase in fluorescence was monitored at 545 nm, with excitation at 525 nm, as described previously.37 The degree of inhibition was relative to the total amount of EGSH formed over the time period of the assay or by the initial velocity of the reaction (nM EGSH formed per minute).
2.16 THP-1 cell adhesion to HUVECs
Monolayer HUVECs were cultured in 24-well plates. THP-1 cells were stained with Dill (C1036, Beyotime, China) for 10 min before co-incubation with the HUVECs for 30 min. For some samples, the THP-1 were pre-treated with blocking antibodies against β2 integrin or inhibitor for ERK1/2 (MCE, HY-12031A, USA). After washing with PBS, the cells were fixed with 4% PFA for 10 min and stained with DAPI for the nuclei. Adherent THP-1 cells were observed and counted with a microscope (Leica DM2000, Germany) and the ImageJ software (NIH).
2.17 Proximity ligation assay
A proximity ligation assay (PLA) was used to detect in situ protein–protein interactions. Fixed and permeabilized THP-1 cells were blocked before incubation with primary antibodies, anti-CD18 antibody (10554-1-AP, Proteintech, China), and anti-chemerin antibody (MAA945Hu22, Cloud-Clone Corp, China) for immunofluorescence staining. Then, the cells were processed for PLA (DUO92101, Sigma, Burlington, MA) according to the manufacturer’s instructions. PLA signals were detected as discrete punctate foci with an Olympus FV3000 confocal microscope. The fluorescence intensity of the intracellular localization of the protein–protein complex was later quantified by ImageJ.
2.18 Statistics
Statistical analysis was performed with GraphPad Prism 6.0 (GraphPad Software, San Diego, CA) and SPSS 22.0 (IBM) software. All data are expressed as the mean ± standard error of the mean (mean ± SEM). Comparisons of data from different experimental groups were conducted using unpaired Student’s t-tests (two-tailed) and one-way or two-way ANOVA tests with post hoc analysis.
3. Results
3.1 Single-cell transcriptional profiling reveals subpopulational expression of vascular endothelial CCRL2 by proatherosclerosis stimulation
To screen the endothelial expression of atypical chemoattractant receptors that respond to d-flow stimulation, we performed scRNA-seq for the LCA with PCL surgery for 1 d or 2 d with untied LCA (non-PCL) as a control. Uniform Manifold Approximation and Projection revealed five distinct clusters of ECs (Figure 1A). Strikingly, Ackr5 (CCRL2) was up-regulated in the d-flow-stimulated EC4 cluster (Figure 1B). Although Ackr3 (CXCR7) was expressed mainly in three EC clusters, its expression showed a slight change in response to d-flow stimulation (see Supplementary material online, Figure S1C). In addition, Ackr4 (CCRL1) was weakly expressed in several EC clusters (see Supplementary material online, Figure S1D), while Ackr1 (Darc), Ackr2 (D6), and Ackr6 (Pitpnm3) were undetectable in all EC clusters (see Supplementary material online, Figure S1A, B, and E). To examine the endothelial expression of Ackrs in atherosclerosis, single-cell datasets GSE13177640 of ApoE−/− mice from the Gene Expression Omnibus database were analysed. Results showed that CCRL2 was predominantly expressed in the EC1 subpopulation in the aortic root of ApoE−/− mice fed HFD for 16 weeks (Figure 1C and D). However, as illustrated in the Supplementary material online, Figure S2, Ackr3 was expressed in all EC subpopulations except EC2 in the aortic root atherosclerotic plaques of ApoE−/− mice fed HFD for 16 weeks (see Supplementary material online, Figure S2C), while Ackr1 and Ackr6 were almost undetectable in all EC clusters (see Supplementary material online, Figure S2A and E). Additionally, Ackr2 and Ackr4 were weakly expressed in the EC3 subpopulation in the aortic root atherosclerotic plaques of ApoE−/− mice fed HFD for 16 weeks (see Supplementary material online, Figure S2B and D). Together, CCRL2 appears to be up-regulated in an EC subcluster by both d-flow and HFD, proatherosclerosis stimulations, leading us to investigate its role in atherosclerosis.
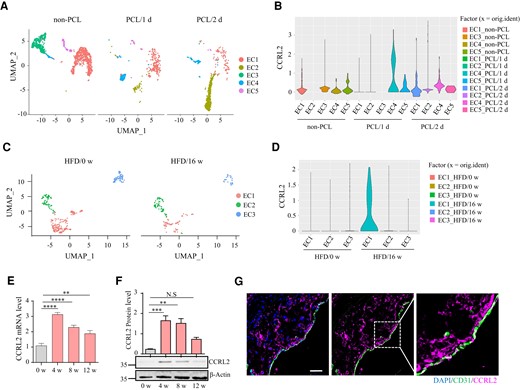
Single-cell transcriptional profiling reveals subpopulational expression of vascular endothelial CCRL2 by proatherosclerosis stimulation. (A) Uniform Manifold Approximation and Projection (UMAP) plot illustrating cell clusters identified in the full single-cell dataset from the left carotid artery of wild-type mice 1 or 2 days after partial carotid ligation (PCL) surgery or without PCL (non-PCL). (B) Violin plots of CCRL2 expression in five EC clusters. (C) UMAP projection of all endothelial cells identified in the full single-cell dataset from the aortic root of ApoE−/− mice fed a high fat diet (HFD) for 0 or 16 weeks. (D) Violin plots of CCRL2 expression in three EC clusters. (E) Male ApoE−/− mice were fed HFD for 0, 4, 8, or 12 weeks. Aortic CCRL2 mRNA expression was determined by real-time quantitative PCR (RT–qPCR), normalized to GAPDH and displayed as fold-change induction relative to baseline (HFD for 0 week) (n = 5 mice per group). **P < 0.01, ****P < 0.0001 by one-way ANOVA with Dunnett’s test for multiple comparisons. (F) Aortic CCRL2 protein expression detected by Western blotting was normalized to β-actin and displayed as fold-changes relative to baseline (n = 3 mice per group). Full blots can be found in the Supplementary material online, Figure S8A. **P < 0.01, ***P < 0.001 by one-way ANOVA with Dunnett’s test for multiple comparisons. (G) Aortic sections from ApoE−/− mice fed HFD for 4 weeks were stained for CCRL2 and CD31. Bar = 20 μm.
We next examined the changes in aortic CCRL2 expression during vascular atherogenic transformation in ApoE−/− male mice fed HFD. Compared with baseline (HFD for 0 week), CCRL2 mRNA expression in the aortic wall reached a peak induction level on HFD for 4 weeks (3.0-fold) and then remained significantly elevated on HFD for 8 weeks (2.0-fold) and 12 weeks (1.7-fold) (Figure 1E). Consistently, CCRL2 protein expression in the aortic wall reached peak induction on HFD for 4 weeks (6.9-fold) and then remained significantly elevated on HFD for 8 weeks (5.3-fold) but returned to normal levels on HFD for 12 weeks compared to baseline (Figure 1F and Supplementary material online, Figure S8A). Furthermore, immunofluorescence staining of ApoE−/− mice fed HFD for 4 weeks showed that CCRL2 was expressed both in the plaque and on the endothelium of atherosclerotic vessels, which was confirmed by co-staining with the endothelial marker CD31 (Figure 1G). Together, the increased local vascular expression of CCRL2 is associated with flow disturbance, hyperlipidaemia, and plaque formation, implying a role for CCRL2 in atherogenesis.
3.2 CCRL2 deficiency reduces lipid deposition and macrophage accumulation in the aorta of ApoE−/− mice
To investigate the potential role of CCRL2 in atherogenesis, CCRL2−/− mice were crossed with ApoE−/− mice to generate CCRL2−/−ApoE−/− mice. After 4 or 16 weeks on HFD, mice were sacrificed, and the plaque size was analysed by en face preparation following Sudan IV staining. As illustrated in Figure 2A, compared with CCRL2+/+ApoE−/− mice, the plaque size in the whole aorta of CCRL2−/−ApoE−/− mice was significantly reduced by 32.6% on HFD for 4 weeks and 27.2% on HFD for 16 weeks. The protective effect of CCRL2 deficiency was localized primarily to the aortic arch, as atherosclerotic lesion size in the aortic arch in CCRL2−/−ApoE−/− mice was significantly reduced by 38.8% on HFD for 4 weeks and 37.8% on HFD for 16 weeks compared with CCRL2+/+ApoE−/− mice, while there was no genotype-dependent difference in the descending aorta after 4 or 16 weeks of HFD (Figure 2A). Significant protection conferred by CCRL2 deletion against plaque formation in mice fed HFD for 16 weeks was also confirmed by Oil Red O staining of aortic root cross-sections, with a 32.9% reduction in lesion size in CCRL2−/−ApoE−/− mice compared with CCRL2+/+ApoE−/− mice (0.53 ± 0.05 mm2 vs. 0.80 ± 0.05 mm2, P < 0.01) (Figure 2B). Therefore, CCRL2 deficiency protects against plaque formation primarily on the aortic arch in ApoE−/− mice.
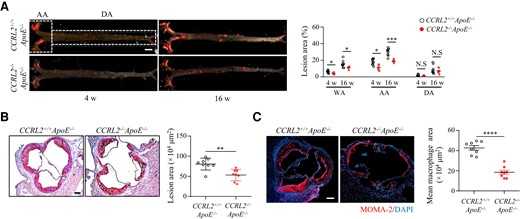
CCRL2 deficiency reduces lipid deposition and macrophage accumulation in the aorta of ApoE−/− mice. (A) CCRL2+/+ApoE−/− and CCRL2−/−ApoE−/− mice were fed HFD for 4 or 16 weeks and analysed for plaque size. Lipid deposition in representative images of en face Sudan IV stained whole aorta is shown. Bar = 2 mm. n = 8 mice per group. The lesion area was quantified and displayed as the percentages of lipid deposition area of the whole aorta (WA), aortic arch (AA) or descending aorta (DA) (n = 8 mice per group). *P < 0.05, **P < 0.01, ***P < 0.001 by two-way ANOVA with Sidak test for multiple comparisons. (B) Representative images of frozen aortic root cross-sections from CCRL2−/−ApoE−/− and CCRL2+/+ApoE−/− mice fed HFD for 16 weeks were stained with Oil red O (n = 8 mice per group). Bar = 200 µm. The lesion area was quantified. **P < 0.01 by unpaired Student’s t-test. (C) Aortic root sections from CCRL2−/−ApoE−/− and CCRL2+/+ApoE−/− mice fed HFD for 16 weeks were stained and quantified for macrophages (n = 8 mice per group). Bar = 200 µm. ****P < 0.0001 by unpaired Student’s t-test.
We next examined the effect of CCRL2 deletion on the accumulation of macrophages in atherosclerotic plaques in ApoE−/− mice challenged with HFD for 16 weeks. Macrophage accumulation was significantly reduced by 58.1% in CCRL2−/−ApoE−/− mice compared with CCRL2+/+ApoE−/− mice, as quantified by MOMA2-positive surface area (0.43 ± 0.06 mm2 vs. 0.18 ± 0.06 mm2, P < 0.001) (Figure 2C). However, the accumulation of neutrophils, lymphocytes, and smooth muscle cells did not change (see Supplementary material online, Figure S3). In addition, CCRL2 deficiency did not alter the number of circulating neutrophils, lymphocytes, or monocytes (see Supplementary material online, Figure S4). Therefore, CCRL2 contributes to efficient macrophage recruitment to and/or retention within atherosclerotic lesions.
Since the CCRL2 ligand chemerin can function as an adipokine, to determine whether lipid metabolism accounts for the decrease in atherosclerotic plaque development in CCRL2−/−ApoE−/− mice, serum lipid levels were measured after a 12-h fasting period. There were no significant differences in serum levels of total cholesterol (TC), triglycerides (TG), HDL, and LDL between CCRL2+/+ApoE−/− and CCRL2−/−ApoE−/− mice fed HFD for 16 weeks (see Supplementary material online, Table S1). Additionally, CCRL2 deletion did not affect the body weights of atherosclerosis-prone mice during the development of atherosclerosis (see Supplementary material online, Table S1). These data suggest that the protective effects of CCRL2 deficiency in atherogenesis are likely due to alterations in local inflammatory responses.
3.3 Disturbed blood flow up-regulates vascular CCRL2 expression that promotes leucocyte adhesion and plaque formation
Atherosclerotic lesions occur predominantly at locations subjected to disturbed blood flow (d-flow) (e.g. aortic arch) compared with locations subjected to relatively stable blood flow (s-flow) (e.g. descending aorta).1 Since the attenuation of plaque formation by CCRL2 deficiency initially and primarily occurred in the aortic arch, we hypothesized that CCRL2 might be differentially expressed in aortic regions exposed to d-flow vs. s-flow. Indeed, CCRL2 mRNA expression assessed by real-time quantitative PCR was significantly higher in the aortic arch than in the descending aorta in WT mice (Figure 3A). CCRL2 protein was largely restricted to the endothelium of the aortic arch rather than the descending aorta, co-localizing with the endothelial marker CD31 by immunofluorescence staining (Figure 3B). In particular, CCRL2 was predominantly expressed in the LC of the aortic arch, which is highly exposed to d-flow (Figure 3C). Therefore, CCRL2 expression in the aorta correlates with domains exposed to d-flow.
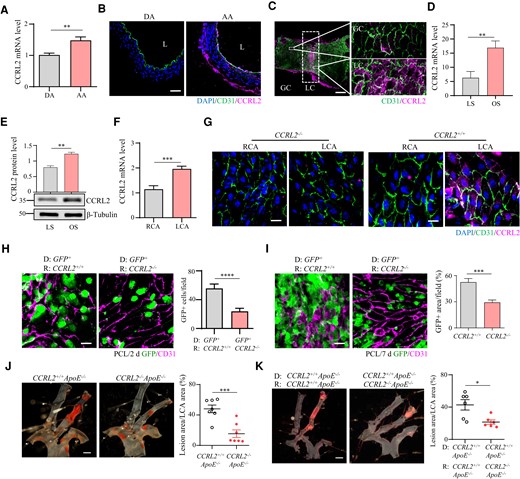
Disturbed blood flow up-regulates vascular CCRL2 expression that promotes leucocyte adhesion and plaque formation. (A) CCRL2 mRNA in the aortic arch (AA) and descending aorta (DA) of C57BL/6J mice was assessed by RT–qPCR (n = 5 per group). **P < 0.01 by unpaired Student’s t-test. (B) CCRL2 protein levels in the aortic arch and descending aorta of WT mice were measured by IF staining with anti-CCRL2 and anti-CD31 antibodies. Bar = 20 μm. (C) CCRL2 protein expression in the GC and LC of the aortic arch in WT mice was detected by en face staining. (red, CCRL2; green, CD31; blue, DAPI). Bar = 100 μm. (D and E) HUVECs were treated for 24 h by OS or LS with a cone-and-plate shear stress device. The expression of CCRL2 was examined by RT–qPCR (D) and Western blotting (E) (n = 3 per group). Full blots can be found in the Supplementary material online, Figure S8B. **P < 0.01 by unpaired Student’s t-test. (F) To induce d-flow, the left carotid artery (LCA) of WT or CCRL2−/− mice was partially ligated. Total carotid RNA before and 24 h after ligation was isolated and analysed for CCRL2 mRNA expression by RT–qPCR, normalized to GAPDH, and expressed as the ratio of ligated LCA to non-ligated (RCA) control (n = 5 per group). ***P < 0.001 by unpaired Student’s t-test. (G) Immunofluorescence staining was performed by en face staining of the carotid intima (red, CCRL2; green, CD31; blue, DAPI). Bar = 20 μm. (H and I) Chimeric mice were generated by transplanting CCRL2+/+ GFP+ bone marrow into irradiated CCRL2+/+ and CCRL2−/− recipients. After 6–8 weeks to allow engraftment and haematopoietic reconstitution, d-flow was induced in the mouse LCA by PCL. After 2 days or 7 days, leucocyte adhesion to the vascular endothelium of the LCA was assessed by en face staining. Green, GFP; red, CD31; Bar = 20 μm. The number of adherent GFP+ leucocytes (H) or the area of adherent GFP+ leucocytes (I) on the vessel wall was quantified in 10–17 fields from 3–4 mice. ***P < 0.001, ****P < 0.0001 by unpaired Student’s t-test. (J) CCRL2+/+ApoE−/− and CCRL2−/−ApoE−/− mice were subjected to PCL. After 2 weeks on HFD, lipid deposition (in red) on mouse LCA was analysed by en face Sudan IV staining. Bar = 1 mm. The lesion surface area was quantified and displayed as the percentage area of the LCA. **P < 0.01 by unpaired Student’s t-test. n = 7 mice per group. (K) Male CCRL2−/−ApoE−/− or CCRL2+/+ApoE−/− mice were lethally irradiated and transplanted with CCRL2+/+ApoE−/− mouse bone marrow cells. Plaque formation in chimeric mice induced by PCL and HFD. *P < 0.05 by unpaired Student’s t-test. n = 6–7 mice per group.
To directly evaluate the effect of d-flow on vascular CCRL2 expression, we first analysed the expression of CCRL2 in HUVECs after 24 h of stimulation with LS or oscillating shear stress (OS). The results showed that CCRL2 mRNA and protein levels were up-regulated in HUVECs under OS stimulation compared with LS-treated HUVECs (Figure 3D and E and Supplementary material online, Figure S8B). We then analysed CCRL2 expression by d-flow in WT or CCRL2−/− mice before or 24 h after PCL. Results showed that CCRL2 mRNA expression in the LCA, normalized to the right coronary artery (RCA), was significantly increased after partial ligation of the LCA for 24 h compared with that in the non-PCL RCA (Figure 3F). Consistent with mRNA expression, en face staining showed that CCRL2 protein was expressed on the vascular endothelium of the partially ligated LCA exposed to d-flow for 24 h but not on the non-PCL RCA exposed to s-flow or in the CCRL2-deficient carotid artery sections (Figure 3G). Taken together, our results indicate that d-flow induces the expression of vascular CCRL2.
We then asked whether vascular endothelial CCRL2 up-regulated by d-flow contributes to leucocyte adhesion in vivo. We generated chimeric mice by transplanting GFP+ bone marrow (BM) into irradiated CCRL2+/+ or CCRL2−/− recipients. These chimeric mice permitted the evaluation of recipient radio-resistant stromal CCRL2 expression without potential leucocyte expression confounding subsequent analyses. After allowing 6–8 weeks for engraftment and haematopoietic reconstitution (confirmed by a haematology analyser), d-flow was induced in the LCA of chimeric mice by PCL. The chimeric animals were euthanized, and leucocyte adhesion on the vascular endothelium of d-flow-exposed LCA was quantified by en face immunofluorescence staining. The number (PCL 2 d, Figure 3H) and area (PCL 7 d, Figure 3I) of adherent GFP+ leucocytes on the vessel wall (CD31+) in CCRL2−/− recipient mice were significantly lower than those in CCRL2+/+ recipients, indicating that radio-resistant stromal CCRL2 is required for maximal intravascular leucocyte adhesion induced by both acute and chronic models of experimental d-flow.
We then evaluated the effect of CCRL2 deletion on d-flow-induced plaque formation in vivo using an extended PCL model. In this model, CCRL2−/−ApoE−/− and CCRL2+/+ApoE−/− mice were challenged with PCL and fed HFD for 2 weeks, and their carotid arteries were examined for lipid deposition. The lesion size in CCRL2−/−ApoE−/− mice was reduced by 73.1% compared to that in CCRL2+/+ApoE−/− controls (lesion area as the percentage of total inner surface of the carotid artery) (Figure 3J), indicating that CCRL2 is required for maximal experimental d-flow-induced atherosclerotic plaque formation. BM transplantation experiments were performed to further elucidate the contribution of endothelial CCRL2 to atherogenesis. CCRL2−/−ApoE−/− or CCRL2+/+ApoE−/− mice were lethally irradiated and transplanted with CCRL2+/+ApoE−/− mouse bone marrow cells. After recovery for 6 weeks, mice underwent PCL surgery and were fed HFD for 2 weeks. We then examined atherosclerotic plaque formation on the left common carotid artery by Sudan IV staining. Results showed that the plaque size of the CCRL2−/−ApoE−/− recipients was smaller than that of the CCRL2+/+ApoE−/−recipients (Figure 3K). Similar results were obtained when CCRL2−/−ApoE−/− or CCRL2+/+ApoE−/− mice were transplanted with CCRL2−/−ApoE−/− mouse bone marrow cells (see Supplementary material online, Figure S5). Together, our data indicate that endothelial CCRL2 contributes to plaque formation.
3.4 Chemerin recruited by CCRL2 mediates leucocyte adhesion
To ask how vascular endothelial CCRL2 expression promotes leucocyte adhesion, we first examined the presence of chemerin in the aorta and in serum in atherosclerotic ApoE −/− mice on a HFD since chemerin is a ligand for CCRL2 and was reported to be recruited by CCRL2 to increase its local concentration.26 Similar to CCRL2 (Figure 1F), chemerin mRNA expression in the aortic artery reached its peak on HFD for 4 weeks (1.7-fold), remained significantly elevated (1.3-fold) on HFD for 8 weeks, and returned to normal levels on HFD for 12 weeks compared to baseline (Figure 4A). Serum chemerin in ApoE −/− mice was 2.5-fold higher than that in the control group (Figure 4B). We then examined the influence of CCRL2 deletion on chemerin retention in the aortic endothelium in WT or atherosclerotic ApoE−/− mice fed HFD. Results showed that CCRL2 deficiency reduced the carotid endothelial localization of chemerin induced by PCL (Figure 4C). Aortic section staining of the atherosclerotic mice on HFD showed that chemerin was enriched on the endothelium and in the plaques when CCRL2 was present but almost completely absent when CCRL2 was deleted (Figure 4D).
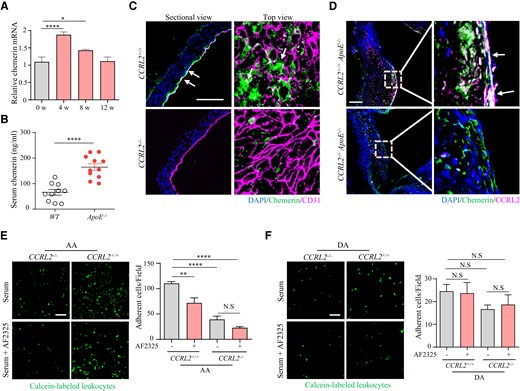
Chemerin recruited by CCRL2 mediates leucocyte adhesion. (A) ApoE−/− mice fed HFD for 0, 4, 8, or 12 weeks were euthanized. mRNA levels in the aorta were measured by RT–qPCR (n = 12). *P < 0.05, ****P < 0.0001 by unpaired Student’s t-test. (B) Serum from ApoE−/− mice fed HFD for 4 weeks was collected from the blood, and serum chemerin levels were measured by an ELISA kit (n = 10–11 mice per group). ****P < 0.0001 by unpaired Student’s t-test. (C) Representative immunofluorescence photomicrographs of aortic sections. En face preparations from CCRL2+/+ and CCRL2−/− mice 7 days after PCL were stained with antibodies against CD31, chemerin and DAPI. Bar = 50 μm. (D) Aortic root sections from CCRL2+/+ApoE−/− and CCRL2−/−ApoE−/− mice fed HFD for 4 weeks were stained for CCRL2 (red) and chemerin (green). Bar = 50 μm. (E and F) Calcein-labelled PBMCs from WT mice were incubated with mouse serum with or without 10 μg/mL anti-chemerin blocking antibody (AF2325) and then added to the luminal side of the aortic arch (E) or descending aorta (F) from CCRL2+/+ or CCRL2−/− mice. The number of adhered leucocytes on the vessel wall was quantified in 8–13 fields from 3 to 4 mice. Bar = 50 μm. **P < 0.01, ****P < 0.0001, N.S, not significant, by one-way ANOVA. EC, endothelial cells; Leu, leucocytes.
We next examined the role of chemerin in the regulation of leucocyte adhesion to the aortic endothelium using an in vitro cell adhesion system. Calcein-labelled peripheral blood leucocytes from WT mice were incubated with mouse serum (containing chemerin) with the addition of an anti-chemerin blocking antibody (AF2325) (100 ng/mL) as a negative control. Cells adherent to the aortic arch from CCRL2+/+ or CCRL2−/− mice were subsequently quantified by immunofluorescence microscopy. As illustrated in Figure 4E, in the aortic arch, chemerin neutralization by the blocking antibody reduced leucocyte adhesion to the endothelium by 34.8% (P < 0.01) in WT mice, but no significant reduction in leucocyte adhesion to the CCRL2−/− endothelium was observed. Moreover, deletion of CCRL2 reduced leucocyte adhesion to the endothelium by 64.2% (P < 0.0001), while chemerin neutralization reduced leucocyte adhesion to the aortic arch from CCRL2−/− mice by 80.7% (P < 0.0001). The differences observed in the aortic arch region were not seen when leucocytes were incubated with descending aorta (Figure 4F). Thus, these findings indicate that the CCRL2-chemerin axis plays a critical role in leucocyte adhesion in d-flow-associated regions of the aorta.
3.5 Chemerin activates β2 integrin and downstream ERK1/2 in monocytes via its PDI-like activity
Endothelial CCRL2 was reported to bind chemerin and enhance CMKLR1+ cell adhesion and transendothelial migration under inflammatory stimulation.26,27 As expected, CCRL2 deficiency reduced F4/80+ monocyte adhesion to the endothelium of the mouse common carotid artery after PCL (Figure 5A, middle panel image). However, to our surprise, CMKLR1 deletion did not significantly reduce F4/80+ monocyte adhesion to the endothelium (Figure 5A, right panel image). Moreover, using the recombinant human bioactive chemerin variant chemerin157S (Chem157S) with the inactive chemerin isoform Chem155A41 (Figure 5B) as a negative control, we showed that over 60% of THP-1 cells (with undetectable CMKLR1 expression, see Supplementary material online, Figure S6) were activated (shape change and pseudopod formation) by Chem157S (Figure 5C), implying a role for CCRL2-recruited chemerin in cell adhesion independent of CMKLR1 under disturbed flow. We therefore asked how chemerin activates monocytes and promotes cell adhesion. Since the β2 integrin family plays a central role in the cell adhesion cascade in leucocytes,42 we examined whether chemerin activates β2 integrin in cultured THP-1 cells by flow cytometry using an antibody against activated β2 integrin. Results showed that Chem157S induced β2 integrin activation (Figure 5D) and triggered ERK1/2 phosphorylation (Figure 5E and Supplementary material online, Figure S8C) in cultured THP-1 cells, which critically regulates cytoskeletal changes in cell adhesion.43 Moreover, MEM148, a β2 integrin blocking antibody, significantly reduced ERK1/2 phosphorylation induced by Chem157S (Figure 5F and Supplementary material online, Figure S8D). Supportively, adhesion of THP-1 cells was significantly decreased when incubated with either β2 integrin blocking antibody MEM148 or ERK inhibitor U0126. Notably, when ERK signalling was blocked, the adhesion induced by Chem157 was eliminated compared with the β2 integrin blocking group (Figure 5G). These results further confirmed a β2 integrin-involved and ERK-dependent signalling pathway induced by Chem157 in THP-1 cells. Together, our results indicated that chemerin activates β2 integrin and downstream ERK1/2 in monocytes independent of CMKLR1 under disturbed flow, subsequently strengthening β2 integrin binding capacity to its ligands, including ICAM-1.
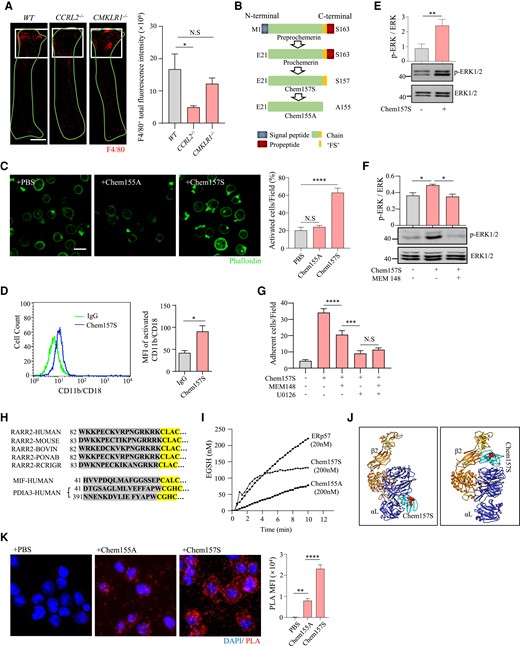
Chemerin activates β2 integrin and downstream ERK1/2 in monocytes via its PDI-like activity. (A) Whole mount staining of LCA from WT, CCRL2−/−, and CMKLR1−/− mice 7 days after PCL (n = 3 per group). The white square indicates the disturbed flow region. Red, F4/80. F4/80+ fluorescence intensity was quantified. Bar = 1 mm. *P < 0.05 by one-way ANOVA. (B) Schematic diagram of Chem157S (high bioactivity) and Chem155A (an inactive isoform). (C) Cultured THP-1 cells were stimulated with PBS, Chem155A or Chem157S and stained with phalloidin (n > 6 per group). Activated cells were morphologically defined by increased pseudopodia protrusions. *P < 0.05, ****P < 0.0001 by one-way ANOVA. (D) THP-1 cells were stimulated with Chem157S for 5 min and stained for β2 integrin (CD11b/CD18) with Mab24 and Alexa Fluor 488-conjugated donkey anti-mouse IgG (n = 5 per group). Fluorescence was assessed by flow cytometry. *P < 0.05 by one-way ANOVA. (E) THP-1 cells were treated with Chem157S (100 ng/mL) for 5 min, and ERK1/2 phosphorylation was detected by Western blotting (n = 3 per group). Full blots are shown in the Supplementary material online, Figure S8C. **P < 0.01 by unpaired Student’s t-test. (F) THP-1 cells were pre-incubated with the β2 integrin blocking antibody MEM148 and then stimulated with Chem157S for 5 min (n = 3 per group). The phosphorylation of ERK1/2 was quantified. Full blots can be found in the Supplementary material online, Figure S8D. *P < 0.05 by unpaired Student’s t-test. (G) Dil-labelled THP-1 cells were incubated with or without inhibitors (MEM148 and U0126), stimulated with Chem157S, and then cocultured with HUVECs. ***P < 0.001, ****P < 0.0001 by one-way ANOVA. (H) Partial amino acid sequence including the ‘CXXC’ motif of chemerin (RARR2) in different species, MIF, and Erp57 (PDIA3). (I) Di-E-GSSG assay for the enzymatic activities. A total of 150 nM Di-E-GSSG was incubated with or without ERp57, Chem155A or Chem157S. The relative fluorescence unit (RFU) of EGSH formed over time is shown. (J) A 3D ribbon display of the binding modes between human chemerin and integrin αL (left) or human chemerin and integrin β2 (right) was estimated using a rigid docking method. Disulfide bonds in integrins are marked in yellow. The putative enzymatic center of chemerin is labelled with red spheres. αL, integrin αL (blue). β2, integrin β2 (orange). (K) Proximity ligation assay (PLA) showing the interaction between Chem157S and β2 integrin in THP-1 cells (n = 9 per group). **P < 0.01, ****P < 0.0001 by one-way ANOVA.
We next asked how chemerin activates monocyte β2 integrin. By comparing chemerin protein sequences in different species, we found a conserved ‘CLAC’ motif (Figure 5H). Several studies have reported that some proteins with ‘CXXC’ motifs have redox activity, such as PDI44 and macrophage migration inhibitor (MIF)45 (Figure 5H). Moreover, neutrophil extracellular PDI was reported to regulate αMβ2 integrin-mediated neutrophil recruitment during vascular inflammation,46 and we proposed that endothelial CCRL2-recruited chemerin activates monocyte β2 integrin via its ‘PDI-like activity’. To test this hypothesis, we examined the disulfide reductase activity of Chem157S using a Di-E-GSSG assay with ERp57 (Protein disulfide-isomerase A3, PIA3), a prominent member of the PDI family that exhibits PDI activity,47 as a positive control. Results showed that Chem157S, but not Chem155A, presented reductase activity in the GSSG assay comparable to that of ERp57, with an initial velocity of 49 nM EGSH/min (Figure 5I). Supportively, protein structural analysis by Chimera software suggested a potential binding site between Chem157S and the β2 integrin subunits (αL and β2) (Figure 5J). To confirm the interaction of chemerin with β2 integrin, we performed a PLA, which was designed to detect in situ protein–protein interactions. Fixed and permeabilized THP-1 cells were incubated with recombinant chemerin Chem157S or Chem155A followed by staining with primary PLA probe reagents for chemerin and β2 integrin. Results showed that a large amount of interaction signal of bioactive Chem157S with the β2 integrin subunits was detected compared to inactive Chem155A (Figure 5K). Together, these results indicate that chemerin has PDI-like activity that enables the activation of β2 integrin to promote leucocyte adhesion.
3.6 Serum levels of chemerin are elevated in patients with acute atherothrombotic stroke
In patients with cerebral or carotid atherosclerosis, rupture of vulnerable plaques may result in acute thrombus formation and block blood supply to the brain. This situation is known as AAS, one of the most prevalent and life-threatening cardiovascular disorders worldwide.48,49 Previous studies have suggested a positive link between serum chemerin levels and cardiovascular disease.50 To extend the clinical significance of CCRL2 in human atherosclerotic disease, we first examined the expression of CCRL2 and chemerin in human carotid plaques by immunohistochemistry, and both were detectable in the plaques (Figure 6A). We then measured the levels of serum chemerin in AAS patients and age- and sex-matched healthy controls. AAS patients had significantly increased serum chemerin levels compared with the control group [23.42 ng/mL (interquartile range, 19.90–31.60) vs. 18.27 ng/mL (interquartile range, 14.64–21.32), P < 0.001] (Figure 6B and Supplementary material online, Table S2). Compared to those with lower serum chemerin levels, individuals with higher chemerin levels had four times the odds of having AAS [odds ratio (OR), 4.72; 95% confidence interval (CI), 2.68–8.31; P < 0.0001]. After adjustment for known cardiovascular risk factors and baseline covariates, including body mass index, TC, TG, LDL cholesterol, HDL cholesterol, fasting glucose, smoking, and drinking status, higher serum chemerin levels remained a significant risk factor for AAS (OR, 6.00, 95% CI, 3.13–11.53; P < 0.0001). Furthermore, each SD increase in serum chemerin levels was correlated with a three-fold increase in the odds of AAS (see Supplementary material online, Table S3, unadjusted OR, 3.46; 95% CI, 2.21–5.40; P < 0.0001; adjusted OR, 3.95; 95% CI, 2.44–6.39; P < 0.0001). The addition of chemerin to the conventional risk model for AAS significantly improved the C-statistic from 0.726 to 0.827 (P < 0.01) (Figure 6C), suggesting serum chemerin as a potential biomarker for AAS.
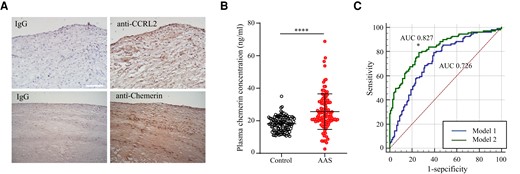
Serum levels of chemerin are elevated in patients with acute atherothrombotic stroke. (A) The expression of CCRL2 and chemerin in human carotid arteries was detected by immunohistochemistry. IgG was the isotype control. (B) Plasma samples were obtained from patients with acute ischemic atherothrombotic stroke (AAS) (n = 111) or age- and gender-matched healthy donors (control, n = 111). Plasma chemerin levels were measured by ELISA. The scatter plot shows individual chemerin concentrations, and horizontal lines represent the median and interquartile ranges. ****P < 0.0001 vs. control by Mann–Whitney U test. (C) Improved diagnostic power by adding chemerin to the conventional risk assessment model for AAS. Receiver operating characteristic (ROC) curves were constructed for a reference model with known AAS risk factors (Model 1) and an improved model combining known AAS risk factors and chemerin (Model 2). The area under the ROC curve (AUC) was calculated for each model. *P < 0.05 vs. Model 1.
4. Discussion
The migration of monocytes from the periphery to subintimal is a key step in the development of atherosclerosis and inflammation.17–19 Chemoattractants and their receptors direct atherogenic recruitment of leucocytes via activation of heterotrimeric G proteins. In this study, we investigated the role of the atypical chemoattractant receptor CCRL2 in the development of atherosclerosis. Global deletion of CCRL2 was beneficial in dampening essential aspects of disease pathogenesis, including reduced monocyte adhesion on activated vascular endothelium, diminished macrophage accumulation, and attenuated plaque formation in atherosclerotic-prone mice. Mechanistically, disturbed blood flow up-regulated CCRL2 in local vascular regions, which served to concentrate the chemoattractant chemerin on the cell surface, promoting the adhesion of circulating monocytes by activating β2 integrin via a novel PDI-like activity of chemerin. Moreover, serum levels of chemerin were elevated in patients with AAS.
In this study, CCRL2 appeared to be up-regulated in the EC subcluster by both d-flow and HFD by single-cell transcriptional profiling. However, the other members of the atypical chemoattractant receptor family in the dataset may also be worth pursuing. For example, Ackr3 was expressed in most EC subclusters, although there was a slight change in response to d-flow (see Supplementary material online, Figure S1C). Moreover, analysis on single-cell dataset from ApoE−/− mice on HFD showed that Ackr3 expression was undetectable only in EC2 subcluster in ApoE−/− mice fed HFD for 16 weeks (see Supplementary material online, Figure S2C). In fact, ACKR3 was reported to participate in the development of atherosclerosis, but its role in atherosclerosis is debated. Some reports have shown that ACKR3 plays an atheroprotective role via its scavenger function of chemokine CXCL12 and its anti-inflammatory function against macrophages,51 while other studies have indicated that endothelial ACKR3 fuels atherosclerosis by mediating endothelium-immune cell adhesion,52 influencing the secretion of proinflammatory factors, and regulating monocyte to macrophage differentiation.51 Potentially, one of the reasons for the complicated role of ACKR3 may be its wide expression (ECs, macrophages, and platelets) and multiple ligands (including CXCL12, CXCL11, PAMP-12, MIF, and opioid peptides).51 Another interesting finding in the dataset was the unique expression of Ackr2 in EC3 in ApoE−/− mice on HFD for 16 weeks (see Supplementary material online, Figure S2B), although there has been no report on its role in atherosclerosis. On the other hand, although Ackr1 was undetectable in our single-cell analysis, its deficiency was reported to be protective in an ApoE−/− mouse model of atherogenesis by reducing the total aorta content of T cells and limiting the inflammatory state of monocytes and macrophages.53
Although CCRL2 has been shown to be up-regulated in ECs by a variety of proinflammatory cytokines and TLR agonists,26 as well as d-flow in this study, no evidence indicates that CCRL2 alone has any effects on its ‘host’ cells. Instead, in vitro adhesion assays showed that chemerin binding to CCRL2 on ECs triggers robust adhesion of CMKLR1-positive lymphoid cells through an α4β1/VCAM-1-dependent mechanism.26 Intracellular signals of chemerin action involve CMKLR1 or G protein-coupled receptor 1 (GPR1)-mediated activation of MAPK/ERK and PI3K/AKT,54 which regulates leucocyte adhesion through integrin activation.54,55 To our surprise, we showed here that THP-1 cells that have undetectable levels of the known chemerin receptors CMKLR1 or GPR1 were responsive to chemerin-induced cell adhesion, suggesting that CCRL2-concentrated chemerin may regulate monocyte adhesion via a distinct pathway from activating either CMKLR1 or GPR1.
Importantly, we found that chemerin recruited by endothelial CCRL2 induces β2 integrin activation via PDI-like activity. Integrin activation plays a critical role in monocyte adhesion over the activated endothelium. Modification of disulfide bonds in the I domain of αL and αM subunits by introducing pairs of Cys residues alters the affinity of ligand binding, suggesting that thiol exchange in integrins regulates the interaction of β2 integrin with their ligands.56,57 A recent study indicated that PDI regulates neutrophil surface αMβ2 clustering, which reveals that redox activity enables integrin activation in leucocytes. Activated αLβ2 and αMβ2 integrins bind to their ligands, such as intercellular adhesion molecule-1 (ICAM-1), triggering stable adhesion and crawling on the endothelium.58 Chemerin was reported to promote macrophage adhesion to VCAM-1 and fibronectin through clustering of VLA-4 (integrin α4β1) and VLA-5 (integrin α5β1).59 All these reports support our notion that d-flow induces endothelial expression of both cell adhesion molecules60 and CCRL2, which recruits chemerin, promoting the adhesion of circulating monocytes by activating β2 integrin via the PDI-like activity of chemerin.
Chemerin primarily circulates as inactive Chem163S in blood, which is cleaved by several proteases, causing its activation (Chem157S) and subsequent inactivation (Chem155A).41 Whether the two residues of chemerin are responsible for redox activity and whether endothelial CCRL2 exclusively recruits bioactive chemerin isoforms are open questions. When we attempted to confirm the role of chemerin C98LAC101 in its PDI-like activity by changing the cysteine to serine, the host cells could not express the recombinant protein CmutS, either in a prokaryotic or eukaryotic expression system (see Supplementary material online, Figure S7), implying that amino acid residues C98 and C101 are probably necessary for proper protein folding. This is consistent with a previous report that MIF forms inclusion bodies when its two cysteine residues of the catalytic centre ‘CLAC’ motif were mutated.61
Our preclinical data that chemerin and its receptors play a pathogenic role in atherogenesis are supported by the elevated serum chemerin levels in AAS patients. While the mechanism underlying increased serum chemerin concentrations in AAS patients is unclear, it is possible that rupture of vulnerable plaques will abruptly release EC-bound and leucocyte-derived chemerin, contributing to the elevated serum chemerin levels. Chemerin concentrations in those patients with stable atherosclerotic disease are likely to be lower than in AAS patients but may still contribute to atherosclerosis via CCRL2, as CCRL2 is able to accumulate chemerin to a higher focal concentration that may permit leucocyte recruitment. Furthermore, the identification of chemerin as an independent risk factor for AAS suggests its potential clinical application as a novel biomarker. Supportively, chemerin was reported to be expressed in human periadventitial fat depots, vascular smooth muscle cells, and foam cells in atherosclerotic lesions and positively correlated with atherosclerosis disease severity.50
The role of chemerin in atherogenesis, however, is not without controversy. A recent article reported a proinflammatory role of chemerin in ApoE−/− mice during atherogenesis,62 although there was little evidence for the mechanism and signalling pathway by which chemerin participates in atherosclerosis. We further demonstrated that chemerin activates β2 integrin through its PDI-like activity, enhancing monocyte adhesion and plaque formation in mice. On the other hand, overexpression of human chemerin via recombinant adeno-associated virus in high-fat diet-fed LDL receptor knockout mice does not significantly alter lipid levels or the severity of atherosclerotic lesions.63 This discrepancy could be due to unforeseen improper post-translational regulation of recombinant human chemerin in mice affecting its secretion or critical proteolytic processing and activation.29
In summary, we identified the atypical chemoattractant receptor CCRL2 as a novel mechanoresponsive molecule, and deletion of CCRL2 decreased monocyte recruitment to plaques and protected against the development of atherosclerosis. Mechanistically, disturbed flow-induced endothelial CCRL2 expression anchors chemerin, which activates monocytic β2 integrin via chemerin PDI-like activity, promoting downstream ERK1/2 signalling and cell adhesion. Our findings thus provide novel correlative, mechanistic, and translational insight into the role of the CCRL2-chemrin-β2-integrin axis in the initiation and progression of atherosclerosis.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Authors’ contributions
C.T., G.C., F.W., F.Y., B.A.Z., and L.Z. designed the study. C.T., G.C., F.W., Y.C., F.Y., C.L., M.L., S.H., L.R., and Q.L. performed the experiments and analysed the data. C.T., G.C., F.W., Y.C., F.Y., T.Y., Y.X., Y.Z., G.W., H.J., Y.W., B.A.Z., and L.Z. reviewed the data and wrote the manuscript. All authors critically read and commented on the manuscript.
Funding
This work was supported by the Natural Science Foundation of China (grants 81870325 and 82170466 to L.Z., 82070450 to C.T., 81900140 to T.Y.), Translational Research Grants of NCRCH (2020ZKPA01 to L.Z.), the Natural Science Foundation of Jiangsu Province (BK20190819 to L.R. and BK20200197 to S.H.), and the Priority Academic Program Development of Jiangsu Higher Education Institutions of China (to L.Z.). B.A.Z. was supported by NIH grant AI-079320.
Data availability
The data underlying this article are available in the article and in its online supplementary material. The scRNA-seq datasets for mouse carotid artery are available upon request.
References
Author notes
These authors contributed equally.
Conflict of interest: None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}