





Abstract
Trastuzumab, the first humanized monoclonal antibody that targets human epidermal growth factor receptor 2 (ERBB2/HER2), is currently used as a first-line treatment for HER2 (+) tumours. However, trastuzumab increases the risk of cardiac complications without affecting myocardial structure, suggesting a distinct mechanism of cardiotoxicity.
We used medium from trastuzumab-treated human umbilical vein endothelial cells (HUVECs) to treat CCC-HEH-2 cells, the human embryonic cardiac tissue-derived cell lines, and human induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) to assess the crosstalk between vascular endothelial cells (VECs) and cardiomyocytes. Protein mass spectrometry analysis was used to identify the key factors from VECs that regulate the function of cardiomyocytes. We applied RNA-sequencing to clarify the mechanism, by which PTX3 causes cardiac dysfunction. We used an anti-human/rat HER2 (neu) monoclonal antibody to generate a rat model that was used to evaluate the effects of trastuzumab on cardiac structure and function and the rescue effects of lapatinib on trastuzumab-induced cardiac side effects. Medium from trastuzumab-treated HUVECs apparently impaired the contractility of CCC-HEH-2 cells and iPSC-CMs. PTX3 from VECs caused defective cardiomyocyte contractility and cardiac dysfunction in mice, phenocopying trastuzumab treatment. PTX3 affected calcium homoeostasis in cardiomyocytes, which led to defective contractile properties. EGFR/STAT3 signalling in VECs contributed to the increased expression and release of PTX3. Notably, lapatinib, a dual inhibitor of EGFR/HER2, could rescue the cardiac complications caused by trastuzumab by blocking the release of PTX3.
We identified a distinct mode of cardiotoxicity, wherein the activation of EGFR/STAT3 signalling by trastuzumab in VECs promotes PTX3 excretion, which contributes to the impaired contractility of cardiomyocytes by inhibiting cellular calcium signalling. We confirmed that lapatinib could be a feasible preventive agent against trastuzumab-induced cardiac complications and provided the rationale for the combined application of lapatinib and trastuzumab in cancer therapy.
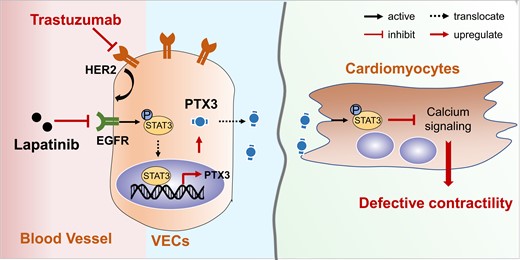
1. Introduction
Trastuzumab, a widely used humanized monoclonal antibody that targets human epidermal growth factor receptor 2 (HER2), shows dramatic therapeutic effects against HER2-overexpressing cancers in the clinic. However, trastuzumab is closely associated with the risk of cardiovascular complications. A total of 2–16% of patients treated with trastuzumab experience a decrease in left ventricular (LV) ejection fraction, and 1–4% of patients eventually develop symptomatic heart failure,1–3 which greatly limits the safe application of trastuzumab in clinical practice. Furthermore, no significant myocardial injury was observed in endomyocardial biopsy samples from patients with cardiotoxicity due to trastuzumab,4 suggesting a complicated cardiotoxic mechanism.
HER2 plays a key role in cardiac development and function,5 and inhibition of HER2 signalling causes damage to cardiomyocytes during ageing and stress overload, leading to a decrease in cardiomyocyte survival, suggesting that HER2 is crucial for cardiomyocyte survival and cardiac function. However, no significant change in mitochondrial or cardiomyocyte death was observed in ventricular-restricted HER2-deficient adult mice,6 suggesting that the role and mechanism of HER2 in the adult heart are complicated. Indeed, total HER2 expression in mouse hearts progressively decreases with age,7 and HER2 expression and function in different types of cells within the adult heart has not been detected.
Clinical studies have shown that patients who have pre-existing vascular diseases are more sensitive to trastuzumab-induced cardiovascular complications,1,8 suggesting that the vasculature might contribute to trastuzumab-related cardiotoxicity. Vascular endothelial cells (VECs) are an important factor to maintain the contractile function of cardiomyocytes, and VECs release signalling molecules to regulate the systolic response of cardiomyocytes.9,10 Therefore, HER2 inhibition in VECs might be one of the causes of trastuzumab-induced cardiac dysfunction. Nevertheless, HER2 expression and function in VECs from adult hearts remain unknown.
Here, we found that the HER2 expression level was higher in adult VECs than in cardiomyocytes and identified VECs as the main target cells of trastuzumab. Mechanistically, we found that pentraxin 3 (PTX3) from VECs causes a reduction in cardiomyocyte contractility by inhibiting calcium signalling. In addition, we provide evidence that STAT3 functions as a transcription factor of PTX3 and that EGFR-STAT3 signalling axis in VEC increases the transcription and release of PTX3. Notably, we found that lapatinib, a dual inhibitor of EGFR and HER2, rescues the trastuzumab-induced decrease in LV function, suggesting a protective strategy against trastuzumab-induced cardiomyopathy, and providing a rationale for the combined application of lapatinib and trastuzumab in cancer therapy.
2. Methods
Extended detailed methods are presented in the Supplementary material online.
2.1 Animals and drug treatment
All experimental procedures and methods were in accordance with guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes, approved by the Center for Drug Safety Evaluation and Research of Zhejiang University and performed according to the guidelines of the Institutional Animal Care and Use Committee (IACUC).
Six- to 8-week-old female Sprague Dawley rats (SLAC, Shanghai, China) were used to investigate the cardiotoxicity of anti-HER2 (neu) antibody and the rescue effects of lapatinib. Ten-week-old female Institute of Cancer Research (ICR) mice (SLAC, Shanghai, China) were used to investigate the cardiotoxic effects of the PTX3 protein. Animals were anaesthetized with 2% isoflurane and then sacrificed by cervical dislocation prior to tissue collection. Detailed information is provided in the Supplementary material online.
2.2 Blood samples from patients
The study of human blood was conducted according to the Declaration of Helsinki 2013 and approved by the institutional review board. Blood samples were obtained from 5 patients who had not been treated with trastuzumab and 14 patients after trastuzumab therapy (with and without cardiomyopathy). Written informed consent was obtained from all blood donors before the study. Detailed information of each patient is presented in the Supplementary material online, Table S1.
2.3 Measurements of human induced pluripotent stem cell-derived cardiomyocyte survival and contractility
Commercial human induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) purchased from Help Stem Cell Innovations (HELP4111) were seeded in Nanion CardioExcyte 96 Sensor Plates and treated with drugs or conditioned medium as indicated. Measurements of the survival rate (PI staining), contraction amplitude, and beat rate were taken every 30 min in CardioExcyte 96 systems from Nanion Technologies. All the parameters were normalized to the control group at the corresponding time.
2.4 Identification of secreted proteins by liquid chromatography with tandem mass spectrometry (LC-MS/MS) analysis
The concentrated conditioned medium samples were separated by SDS-PAGE. The gel was dyed with Coomassie Brilliant Blue and the most enriched band was cut out and analysed by PTM Biolabs. Briefly, the gel was digested by trypsin to obtain peptides. The tryptic peptides were dissolved in 0.1% formic acid, directly loaded onto a reversed-phase analytical column, and run on an EASY-nLC 1000 UPLC system. The peptides were subjected to nanospray ionization (NSI) source followed by tandem mass spectrometry (MS/MS) in Q ExactiveTM Plus (Thermo) coupled online to the UPLC.
2.5 RNA-sequencing
The detailed methods used for RNA-sequencing are provided in the Supplementary material online. Total RNA was isolated from PTX3-treated ventricles and purified using TRIzol reagent. After purification, the fragmented poly (A) RNA was reverse transcribed to generate the cDNA with SuperScript™ II Reverse Transcriptase (Invitrogen, 1896649). Paired-end sequencing (2 × 150 bp) was performed on an Illumina Novaseq™ 6000 (LC-BIO TECHNOLOGIES, Hangzhou, China).
2.6 Cytoplasmic and nuclear fractionation and western blot
Human umbilical vein endothelial cell (HUVEC) lysates were separated into nuclear and cytoplasmic fractions with a Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime Biotechnology, P0027). Western blot was performed as previously reported.11 Information related to the primary antibodies and HRP-labelled secondary antibodies used is provided in the Supplementary material online.
2.7 PTX3 knockout in HUVECs via CRISPR-Cas9
LV-CAS9-puro virus (Shanghai Genechem Co., Ltd) was added to HUVECs. After 72 h of infection, 2 g/mL puromycin (T19978, TargetMol) was added. The HUVECs were then cultured with a half-dose of puromycin and infected with LV-PTX3-sgRNA lentivirus (Shanghai Genechem Co., Ltd). The PTX3 protein level was detected by western blot and the PTX3 knockout cells were selected for further experiments.
2.8 HER2 or STAT3 knockdown with siRNA oligonucleotides
Cells were seeded into 6-well plates at 1 × 105 cells per well and grown to 50–60% confluence. Transfection was performed using Opti-MEM (Invitrogen, 31985-070), siRNA, and Oligofectamine (Invitrogen, 12252001) according to the manufacturer’s recommendations. siRNA oligonucleotides were transfected at a final concentration of 12 nM using Oligofectamine. The cell culture solution was changed to RPMI-1640 medium plus 10% fetal bovine serum (FBS) at 6 h after transfection. The siRNA sequences are provided in the Supplementary material online, Table S5.
2.9 Knockdown of EGFR by shRNA lentivirus
pLKO.1 vectors (Addgene, 8453; deposited by the Bob Weinberg Laboratory, Massachusetts Institute of Technology) containing EGFR shRNA were constructed by digestion with EcoRI and AgeI and subsequent T4 ligation. After transformation, monoclonal colonies were selected for sequencing identification. The correct plasmid was amplified and used for lentivirus production. HUVECs were infected with lentiviral-conditioned medium overnight and then collected for further research. The shRNA sequences are provided in the Supplementary material online, Table S5.
2.10 Statistical analysis
Statistical analysis was performed using GraphPad Prism 9. All data were expressed as the mean value ± standard deviation. When comparing the difference between two groups, Student’s t-test (unpaired, two-tailed) was applied. One-way analysis of variance (ANOVA) was performed to detect the significance of differences between multiple groups. Dunnett’s T3 and Sidak tests were used to assess the significance of differences among every mean to the control value or among selected pairs of groups, respectively. Two-way ANOVA with post hoc tests was used to assess the significance of differences among pairs of groups at different time-points.
3. Results
3.1 VECs mediate trastuzumab-induced cardiotoxicity.
To identify the cardiotoxic mechanism of trastuzumab, we first investigated the direct effect of trastuzumab on cardiomyocytes. We discovered that trastuzumab did not decrease the survival rate of cardiomyocytes (see Supplementary material online, Figure S1A). We then established a model of cell contractile force detection based on the literature12 using digoxin, which can promote cell contraction and reduce the planar surface area. We found that the planar surface area of the cells was reduced after digoxin administration in both the control and trastuzumab groups (see Supplementary material online, Figure S1B), suggesting that trastuzumab had no direct deleterious effects on cardiomyocytes contraction. In addition, we used the human iPSC-CM model and treated the iPSC-CMs with trastuzumab or adriamycin (ADR), an anticancer drug that can reduce the survival and contractility of cardiomyocytes, to generate a positive control group. After 6 h of ADR treatment, the cell survival rate significantly decreased. In contrast to ADR, trastuzumab did not significantly reduce cell survival (see Supplementary material online, Figure S1C). Furthermore, we observed that the spontaneous contraction amplitude and beat rate were not affected by trastuzumab treatment but were reduced by ADR treatment (see Supplementary material online, Figure S1D and E). These results suggested that trastuzumab had no direct effect on the survival or contraction of iPSC-CMs.
To confirm the effect of trastuzumab on the heart, a rat model was established with anti-HER2/neu (homologue of HER2 in rats) antibody. Anti-HER2/neu antibody and trastuzumab exhibit structural similarity in the third complementarity determining region and share the same antigen epitope on their targets.13 Thus, anti-HER2/neu antibody was used to mimic the cardiotoxicity of trastuzumab in vivo (see Supplementary material online, Figure S1F). Haematoxylin and eosin (H&E) staining showed no apparent cardiac injury in the hearts of anti-HER2/neu antibody-treated rats (see Supplementary material online, Figure S1G). We also noticed increased collagen deposition in heart sections from anti-HER2/neu antibody-treated rats (see Supplementary material online, Figure S1H and I), indicating that trastuzumab caused cardiac fibrosis. Wheat germ agglutinin (WGA) staining showed slightly increased cardiomyocyte size in the hearts of anti-HER2/neu antibody-treated hearts rats (see Supplementary material online, Figure S1J), suggesting the existence of slight cardiac hypertrophy. These results were consistent with the clinical findings that trastuzumab causes cardiac dysfunction in patients without features of significant cardiac structural changes or myocardial cell damage based on biopsy results.4
Considering the potential relationship between HER2 inhibition and the cardiac complications of trastuzumab and the decreased overall protein level of HER2 in adult mouse hearts, we performed an immunofluorescence assay with the hearts of adult mice and mouse embryos. We found that HER2 protein levels in cardiomyocytes were dramatically reduced in adult hearts (Figure 1A). Interestingly, the VECs in the capillary from the adult mouse hearts still maintained substantial expression of HER2 (Figure 1A), which prompted us to propose that VECs might participate in the cardiotoxicity of trastuzumab. We then detected the role of VECs in trastuzumab-related cardiotoxicity. As shown in Figure 1B, we treated HUVECs with either sterilized water (solvent control) or trastuzumab for 24 h, and the supernatants were collected as conditioned medium (control medium, Ctrl-Medium or trastuzumab medium, Tra-Medium). We found that Tra-Medium had little effect on the survival rate of CCC-HEH-2 cells (Figure 1C), but inhibited the contraction of CCC-HEH-2 cells (Figure 1D). Furthermore, we used iPSC-CMs to confirm the effects of trastuzumab-treated VECs on cardiomyocytes. As expected, Tra-Medium did not decrease the survival rate of iPSC-CMs (Figure 1E), but significantly inhibited the contraction amplitude (Figure 1F) and beat rate (Figure 1G) of iPSC-CMs, suggesting that trastuzumab-treated VECs led to defective cardiomyocyte contractility. We noticed no significant change in the mitochondrial membrane potential or protein levels of mitochondrial respiratory chain complexes in cardiomyocytes after 24 h of treatment with Tra-Medium (see Supplementary material online, Figure S2A and B). We also found that Tra-Medium increased mRNA level of NPPA, NPPB (natriuretic peptide TYPE A/B), and MYH7 (myosin, heavy peptide 7, myocardial β), and decreased mRNA level of MYH6 (myosin, heavy peptide 6, myocardial α) in CCC-HEH-2 cells (see Supplementary material online, Figure S2C), suggesting the pathological cardiac remodelling. Taken together, these data demonstrated that trastuzumab acts on VECs first and then indirectly causes defective cardiomyocyte contractility and cardiac dysfunction.
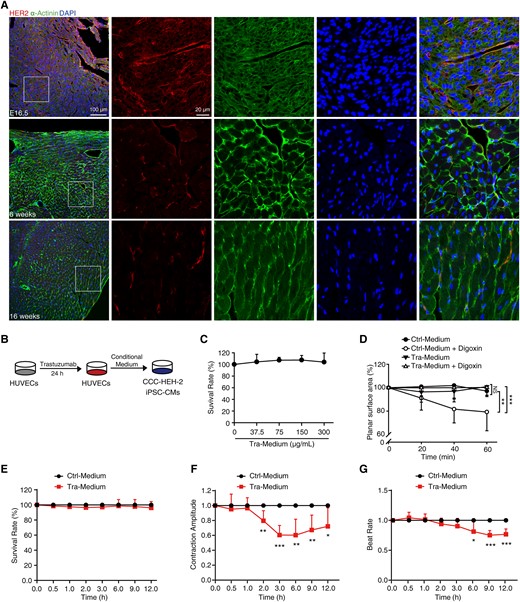
Vascular endothelial cells participate in trastuzumab-induced cardiotoxicity. (A) Representative confocal images of the heart of mouse embryos at Day 16.5 (n = 3 embryos) and of 6-week-old and 16-week-old mice (n = 3 mice) stained with HER2 (red), α-Actinin (green) antibody, and DAPI (blue). Scale bar, 100 μm and 20 μm. (B–G) Effects of conditioned medium on CCC-HEH-2 cells and iPSC-CMs. (B) The schematic diagram of the preparation and application of conditioned medium. (C) SRB colorimetric assay showed the effect of conditioned medium on the survival of CCC-HEH-2 cells (n = 3). (D) Digoxin was used to stimulate the contraction of CCC-HEH-2 cells, and the contractility was calculated by planar surface area (n = 8). (E–G) The normalized survival rate, contraction amplitude, and beat rate of iPSC-CMs (n = 6 individuals). The P-value was calculated by one-way ANOVA (Dunnett’s or Sidak multiple comparisons tests). NS, no significance; *P < 0.05, **P < 0.01, ***P < 0.001.DAPI, 4’,6-diamidino-2-phenylindole; iPSC-CMs, induced pluripotent stem cell-derived cardiomyocytes; Ctrl-Medium, control medium; Tra-Medium, trastuzumab medium.
3.2 PTX3 released from VECs promotes trastuzumab-induced cardiac dysfunction
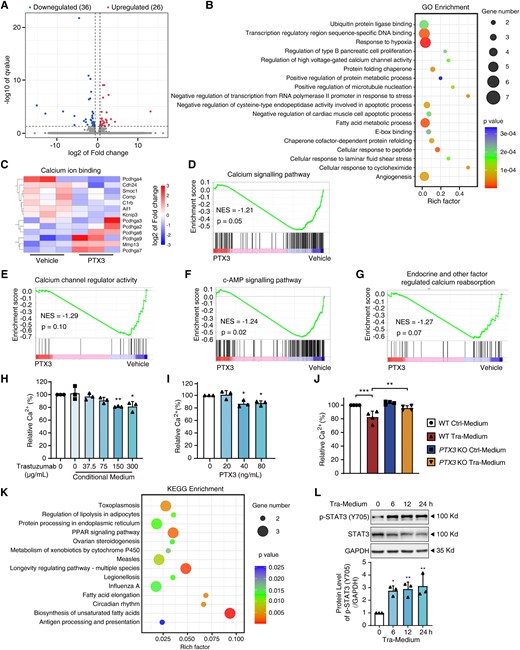
To identify the key factors from VECs that inhibit cardiomyocyte contractility, the substances in the medium of trastuzumab-treated HUVECs were analysed by LC-MS/MS (see Supplementary material online, Figure S3A). One hundred and forty proteins were identified, suggesting that many factors were secreted by HUVECs upon trastuzumab treatment. To catch the key secreted protein(s), we analysed the top 10 enriched proteins (see Supplementary material online, Figure S3B), wherein PTX3 is only one protein that can be released by VECs and plays key functions in endothelial dysfunction and cardiomyocyte injury. Thus, we focused on PTX3 in our later study and confirmed the function of PTX3 in trastuzumab-induced cardiac dysfunction. The levels of PTX3 in the cell lysates and medium were detected by western blot and ELISA. We found that trastuzumab treatment led to an increase in PTX3 levels in the medium in a time- and dose-dependent manner (Figure2A and B, see Supplementary material online, Figure S3C), suggesting the release of PTX3 from HUVECs. We also found that CCC-HEH-2, human aorta vascular smooth cells, and primary macrophages induced from human peripheral blood monocytes cells could not release PTX3 upon trastuzumab treatment (see Supplementary material online, Figure S3D), further confirming that the increase in secreted-PTX3 was from VECs other than other cell types. To further investigate the release of PTX3, we measured the plasma from patients diagnosed with/without cardiac complications after trastuzumab therapy. The ELISA results showed that patients with cardiac complications, like reduced LV systolic function, reduced LV diastolic function, tricuspid regurgitation, mitral regurgitation, or tachycardia, had higher plasma PTX3 levels than patients without cardiac complications (Figure 2C and see Supplementary material online, Table S1). These data indicated that PTX3 might be the key factor that mediates the crosstalk between VECs and cardiomyocytes in trastuzumab-induced cardiac complications.

PTX3 governs trastuzumab-induced cardiotoxicity. (A and B) The level of PTX3 in HUVECs medium. (A) Western blot and quantification of PTX3 (n = 3). (B) ELISA analysis of PTX3 (n = 3). (C) ELISA analysis of PTX3 on the plasma from patients as indicated. (D–G) The conditioned medium and PTX3-neutralizing antibody were used to treat CCC-HEH-2 cells and iPSC-CMs. (D) The contractility of CCC-HEH-2 cells (n = 8). (E–G) The normalized survival rate, contraction amplitude, and beat rate of iPSC-CMs (n = 6 individuals). (H) The transcriptional level of PTX3 was measured by qPCR in HUVECs (WT and PTX3 KO, n = 5). (I) The secretion level of PTX3 was measured by western blot (n = 3). (J–M) The conditioned medium from HUVECs (WT and PTX3 KO) were treated with CCC-HEH-2 cells and iPSC-CMs. (J) The contractility of CCC-HEH-2 cells (n = 8). (K–M) The normalized viability, contraction amplitude, and beat rate of iPSC-CMs (n = 6 individuals). The P-value was calculated by one-way ANOVA (Dunnett’s or Sidak multiple comparisons tests). NS, no significance; *P < 0.05, **P < 0.01, ***P < 0.001. Ctrl-Medium, control medium; Tra-Medium, trastuzumab medium; PTX3 Neu-Antibody, PTX3-neutralizing antibody.
To test this hypothesis, a PTX3-neutralizing antibody was applied to block PTX3 function. We found that PTX3 neutralization reversed Tra-Medium-induced defective cardiomyocyte contractility in the digoxin model (Figure 2D). Moreover, the PTX3-neutralizing antibody was sufficient to inhibit the decreased contraction amplitude (Figure 2E) and beat rate (Figure 2F) of iPSC-CMs, without affecting the survival rate (Figure 2G). In addition, we investigated the role of PTX3 in VEC-mediated cardiac dysfunction by knocking out the PTX3 expression in HUVECs via CRISPR-Cas9 and the knockout efficiency was confirmed by qPCR (Figure 2H). The release of PTX3 caused by trastuzumab was remarkably ablated in HUVECs (PTX3 KO) (Figure 2I). Notably, trastuzumab-containing medium from HUVECs (PTX3 KO) failed to inhibit the contractility of cardiomyocytes in the digoxin model (Figure 2J) or decrease the contraction amplitude or beat rate of iPSC-CMs (Figure 2K–M).
Next, we applied recombinant PTX3 protein to mimic the release of PTX3. We found that the survival rate of CCC-HEH-2 cells remained unaltered with recombinant PTX3 protein treatment (Figure 3A). Notably, in PTX3-treated CCC-HEH-2 cells, we observed a reduction in digoxin-stimulated contractility (Figure 3B). Moreover, 40 ng/mL PTX3 led to a significant decrease in contraction amplitude (Figure 3C) but could not reduce the survival rate (Figure 3D) or beat rate (Figure 3E) of iPSC-CMs. Then we examined the role of PTX3 in vivo and treated the ICR mice with 20 or 100 ng of recombinant mouse PTX3 protein daily for 14 days via intravenous injection. We found a decreased ejection fraction and fractional shortening in the PTX3-treated mice (Figure 3F and Supplementary material online, Table S2), suggesting cardiac dysfunction in the mice treated with PTX3. H&E staining results showed no apparent cardiac injury in the PTX3-treated hearts (Figure 3G). However, we observed some collagen deposition in heart sections from PTX3-injected mice, indicating slight cardiac fibrosis (Figure3H and I). WGA staining showed a slightly increased cardiomyocyte size in hearts from PTX3-injected mice (Figure 3J), indicating that PTX3 caused cardiac hypertrophy. Meanwhile, the injection of PTX3 had little effect on heart weight/body weight ratio or heart rate (see Supplementary material online, Figure S4A and B). In addition, we found that PTX3 failed to increase the levels of serum creatine kinase myocardial band (CK-MB) (see Supplementary material online, Figure S4C), ALT or AST (Supplementary material online, Figure S4D), which are biochemical markers of cardiac and hepatic injury, respectively. These results indicated that PTX3 is sufficient to cause cardiac dysfunction directly without damaging the cardiac structure. It has been reported that PTX3 could induce endothelial dysfunction14 and we found that trastuzumab showed nearly no effects on the proliferation and migration but slight effects on the survival of HUVECs (see Supplementary material online, Figure S5), suggesting that endothelial dysfunction might be involved in the cardiomyocyte contractility defects. Collectively, our data suggest that PTX3 is the key factor released from VECs that mediates trastuzumab-induced cardiac dysfunction.

PTX3 reduces the contractility of cardiomyocytes and causes left ventricular dysfunction in mice. (A–E) Effects of recombinant PTX3 protein on CCC-HEH-2 cells and iPSC-CMs. (A) Survival rate of CCC-HEH-2 cells (n = 3). (B) Contractility of CCC-HEH-2 cells (n = 8). (C–E) The normalized contraction amplitude, survival rate, and beat rate of iPSC-CMs (n = 5 in control and n = 4 in PTX3). (F–J) Mice were treated with saline or PTX3 (20 ng or 100 ng/mouse) once a day by i.v. for 14 days (n = 8 mice). (F) Cardiac function was examined by echocardiography. (G–I) Heart tissues were collected for histopathological analysis. (G) Haematoxylin and Eosin staining. Scale bar, 100 μm and 50 μm. (H) Sirius Red staining and quantification. Scale bar, 100 μm. (I) Masson’s Trichrome staining. Scale bar, 25 μm. (J) WGA staining and quantification of the cross-sectional area of cardiomyocytes. The P-value was calculated by one-way ANOVA (Dunnett’s or Sidak multiple comparisons tests) or Student’s t-test (unpaired, two-tailed, two groups). *P < 0.05, **P < 0.01, ***P < 0.001.
3.3 PTX3 causes aberrant calcium signalling in cardiomyocytes
Next, we investigated the mechanisms by which PTX3 or Tra-Medium caused defective contractility by performing RNA-sequencing of hearts from PTX3-treated mice. We found that the expression of 26 genes was significantly up-regulated in the hearts of PTX3-treated mice compared with control mice (≥2- or ≤0.5-fold, false discovery rate ≤0.05), whereas the expression of 36 genes was down-regulated (Figure 4A and Supplementary material online, Tables S3 and S4). Gene ontology pathway analysis of biological processes showed enrichment of genes in processes related to the response to hypoxia, regulation of high voltage-gated calcium channel activity, and angiogenesis (Figure 4B). Notably, gene set enrichment analysis further confirmed that signalling pathways closely related to cardiac function, including the calcium signalling pathway, calcium channel regulator activity, c-AMP, and endocrine- and other factor-regulated calcium reabsorption, were more enriched in vehicle-treated hearts (Figure 4C–G), suggesting that dysregulated calcium signalling might contribute to the cardiac dysfunction caused by trastuzumab or PTX3. To further assess the function of PTX3 in calcium signalling, we detected the intracellular calcium content in CCC-HEH-2 cells. Flow cytometry with the calcium dye Fluo-4 AM revealed that Tra-Medium and recombinant PTX3 protein led to a decrease in intracellular calcium content in CCC-HEH-2 cells (Figure4H and I). Moreover, deletion of the PTX3 gene in HUVECs rescued the decreased intracellular calcium content in CCC-HEH-2 cells caused by trastuzumab-containing medium (Figure 4J).
PTX3 reduces the Ca2+ content and impairs the contraction of cardiomyocytes. (A–G) RNA-sequencing of the three mice hearts treated with vehicle or PTX3. (A) The volcano plot of log2-fold change of gene expression. (B) Gene ontology (GO) analysis. (C) Heatmap of different gene expression patterns related to calcium ion binding. (D–G) Gene set enrichment analysis (GSEA). (H and I) Fluo-4 AM staining and flow cytometry showed the effects of conditioned medium and recombinant PTX3 protein on the Ca2+ content of CCC-HEH-2 cells (n = 3). (J) CCC-HEH-2 cells were treated with conditioned medium from HUVECs (WT and PTX3 KO) and measured for the Ca2+ content (n = 4). (K) KEGG pathway enrichment analysis. (L) CCC-HEH-2 cells were treated with conditioned medium at various times as indicated. The protein levels of P-STAT3 (Y705) of CCC-HEH-2 cells were measured by western blot (n = 3). The P-value was calculated by one-way ANOVA (Dunnett’s or Sidak test). *P < 0.05, **P < 0.01; ***P < 0.001. GO, Gene Ontology; NES, normalized enrichment score; Tra-Medium, trastuzumab-medium; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Moreover, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis and the resulting heatmap showed the enrichment of pathways related to the pathogen infection and immune response (Figure 4K and Supplementary material online, Figure S6A), suggesting activation of inflammation response in the PTX3-treated hearts, which was consistent with the occurrence of inflammatory infiltration in PTX3-treated hearts (Figure 3G) and anti-HER2/neu antibody-treated hearts (see Supplementary material online, Figure S1G) and the canonical role of PTX3 in regulating immune activity. Considering that PTX3 promotes inflammatory response partially by STAT3 signalling15 and that STAT3 plays key roles in the regulation of cardiac calcium signalling,16,17 we detected the activation of STAT3 signalling by PTX3. We found that Tra-Medium apparently increased the phosphorylation of STAT3 (Y705) in CCC-HEH-2 cells in a time- and dose-dependent manner (Figure 4L and Supplementary material online, Figure S6B). In addition, PTX3 treatment increased the expression of p-STAT3 (Y705) in the hearts (see Supplementary material online, Figure S6C). Notably, trastuzumab had a little direct effect on the phosphorylation of STAT3 (Y705) in CCC-HEH-2 cells (see Supplementary material online, Figure S6D). These data supported the hypothesis that PTX3 from VECs regulates cardiac homoeostasis and function by STAT3 signalling in cardiomyocytes.
3.4 Activation of EGFR signalling led to the release of PTX3 from VECs
Given that the release of PTX3 from VECs was the key cause of trastuzumab-induced cardiac dysfunction, we next investigated the mechanisms by which trastuzumab promotes PTX3 release. We first examined the protein secretion pathways that are responsible for trastuzumab-induced PTX3 release using brefeldin A (BFA), which blocks transport from endoplasmic reticulum to Golgi and is used as an inhibitor of the conventional protein secretion pathway. Western blot results showed that BFA inhibited the increase in the level of extracellular PTX3 caused by trastuzumab and led to the intracellular accumulation of the PTX3 protein (see Supplementary material online, Figure S7A). Notably, 3-methyladenine (3-MA), an autophagy inhibitor, did not affect the level of PTX3. These results indicated that trastuzumab promotes PTX3 secretion from VECs through the conventional pathway. In the conventional secretory pathway, the signal peptides located in the N-termini of secreted proteins are essential for secretion.18 Therefore, we predicted the signal peptides of PTX3 using the SignalP 5.0 server. As shown in Figure 5A, the peptide from amino acids 1–16, which was highly conserved in diverse mammalian species (Figure 5B), may be a signal peptide. To confirm this signal peptide, we constructed a full-length PTX3 plasmid and a truncated PTX3 (Δ 2–16) plasmid, and examined the release of PTX3 from HUVECs (PTX3-KO). We found that the truncation of amino acids 2–16 apparently impeded the trastuzumab-induced release of PTX3 (Figure 5C), indicating that this peptide is the signal peptide in PTX3 that is required for its release from VECs.
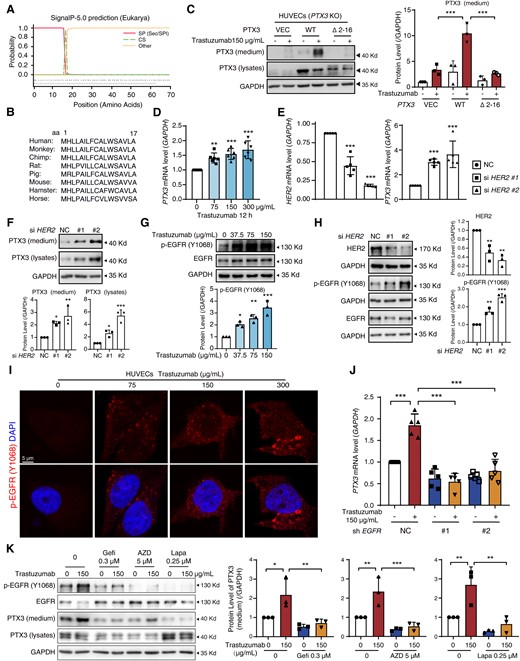
Activation of EGFR in vascular endothelial cells promotes the transcription and secretion of PTX3. (A) The signal peptide prediction for PTX3 via SignalP 5.0. (B) Phylogenetic conservation of the N-terminal amino acids 2–16 in PTX3 protein. (C) HUVECs (PTX3 KO) were transfected with PCDH, PTX3 (WT), or PTX3 harbouring a deletion of amino acids 2–16 (Δ2–16) for 12 h and treated with trastuzumab. The protein levels of secreted-PTX3 were measured by western blot (n = 3). (D) The transcription levels of PTX3 measured by qPCR (n = 6). (E and F) HUVECs were transfected with NC or HER2 siRNA. (E) The transcription levels of HER2 and PTX3 were measured by qPCR (n = 5). (F) The protein levels of PTX3 of medium and lysates were measured by western blot (n = 3). (G and H) Western blot of p-EGFR (Y1068) levels in HUVECs treated with trastuzumab or transfected with NC or HER2 siRNA (n = 3). (I) Immunofluorescence assays of p-EGFR (Y1068) levels in HUVECs (n = 3). Scale bar, 5 μm. (J) The transcription levels of PTX3 in HUVECs that were transfected with EGFR shRNA (#1, #2) or NC lentivirus (n = 5). (K) HUVECs were exposed to trastuzumab with or without gefitinib, AZD3759, or lapatinib for 24 h. The protein levels of secreted-PTX3 were measured by western blot (n = 3). The P-value was calculated by one-way ANOVA (Dunnett’s or Sidak test). *P < 0.05, **P < 0.01, ***P < 0.001. VEC, Vector; DAPI, 4’,6-diamidino-2-phenylindole; Gefi, gefitinib; AZD, AZD3759; Lapa, lapatinib.
The increased transcription of secretory proteins promoted their release to the extracellular space. As expected, 75, 150, and 300 μg/mL trastuzumab increased the mRNA level of PTX3 (Figure 5D). Therefore, we then investigated how trastuzumab regulates the transcription of PTX3. We first explored the involvement of the inhibition of HER2, the target of trastuzumab, in PTX3 transcription and release. Compared with non-targeting siRNA (NC), HER2 siRNA increased the mRNA level of PTX3 (Figure 5E). Notably, HER2 siRNA triggered the release of PTX3 into the extracellular medium (Figure 5F). These results suggested that inhibition of HER2 in VECs leads to the increased transcription and release of PTX3.
HER2 inhibition induces compensatory activation of the EGFR signalling pathway19 and the overexpression/activation of EGFR promotes cardiovascular diseases and injuries, including cardiac hypertrophy, fibrosis, endothelial dysfunction, and atherogenesis,20,21 which prompted us to investigate whether EGFR signalling is involved in trastuzumab-induced PTX3 release and cardiac complications. We found that trastuzumab treatment or silencing of the HER2 gene promoted the phosphorylation of tyrosine (Y) 1068 of EGFR in HUVECs (Figure5G and H). Immunofluorescence and immunohistochemistry assays further confirmed the increased phosphorylation of EGFR in HUVECs and in VECs of the hearts, respectively (Figure 5I and Supplementary material online, Figure S7B). These data suggested the activation of EGFR by trastuzumab-mediated HER2 inhibition in VECs. We then examined the effect of EGFR activation on the trastuzumab-induced increase in the transcription and release of PTX3. We found that EGF, the ligand of EGFR, increased the transcription level of PTX3 and promoted PTX3 secretion, accompanied by the increased phosphorylation of EGFR (see Supplementary material online, Figure S7C and D). We also constructed an active EGFR plasmid by mutating tyrosine 1068 to aspartic acid (D) or glutamic acid (E), termed Y1068D or Y1068E, to mimic tyrosine phosphorylation. Overexpression of the Y1068D or Y1068E plasmid markedly increased the mRNA level, and intracellular and secreted protein levels of PTX3 (see Supplementary material online, Figure S7E and F). These data indicated that activated EGFR contributes to the increased transcription and secretion of PTX3.
In contrast, we applied lentivirus containing shRNA targeting EGFR to knock down EGFR expression in HUVECs and found that EGFR knockdown reversed the increase in the transcription (Figure 5J), and secreted protein levels of PTX3 caused by trastuzumab (see Supplementary material online, Figure S7G). In addition, we detected the effects of EGFR inhibitors on the transcription and release of PTX3. The EGFR inhibitors, gefitinib and AZD3759, and the EGFR/HER2 dual inhibitors, lapatinib, neratinib, and pyrotinib, also directly decreased the mRNA level of PTX3 in HUVECs (see Supplementary material online, Figure S7H–L). Moreover, gefitinib, AZD3759, and lapatinib blocked the effects of trastuzumab on PTX3 transcription (see Supplementary material online, Figure S7M–O) and release (Figure 5K). Taken together, our data showed that the activation of EGFR by trastuzumab led to the increased transcription and release of PTX3 from VECs.
3.5 EGFR signalling mediates transcription and release of PTX3 by STAT3
The EGFR signalling pathway, in which several classic downstream effectors, including STAT3, AKT, IkBα, ERK, and JNK1/2, regulate signal transduction from EGFR activation, is one of the most thoroughly studied signalling pathways. Therefore, we next proceeded to identify the downstream effector of EGFR that is required for the transcription and release of PTX3. We found that the level of only phosphorylated STAT3 (Y705) among the tested proteins was increased by trastuzumab (Figure 6A and see Supplementary material online, Figure S8A). Phosphorylation of STAT3 at Y705 promotes the translocation of STAT3 from cytoplasm to nucleus, where STAT3 functions as a transcription factor to regulate the transcription of target genes.22 Immunofluorescence assay and subcellular fractionation analysis revealed that trastuzumab promoted the nuclear localization of STAT3 (Figure 6B and see Supplementary material online, Figure S8B), suggesting that trastuzumab promotes the nuclear localization of STAT3 in HUVECs, which was further confirmed by immunohistochemistry assay with trastuzumab-treated heart sections (Figure 6C). We then investigated the role of STAT3 in the transcription and release of PTX3. Overexpression of the STAT3 plasmid increased the mRNA and secreted protein level of PTX3 in HUVECs (Figure6D and E). In addition, an increase in the mRNA and secreted protein levels of PTX3 caused by trastuzumab was abolished by STAT3 knockdown (Figure6F and G). Luciferase assay showed that PTX3 luciferase activity was up-regulated upon trastuzumab treatment (Figure 6H). Overexpression of the STAT3 plasmid increased PTX3 luciferase activity 1.73-fold (Figure 6I). Conversely, PTX3 deletion had little effect on the phosphorylation of STAT3 (see Supplementary material online, Figure S8C). Taken together, these data suggest that STAT3 is required for EGFR-mediated PTX3 transcription and release and that PTX3 is a target gene of STAT3.
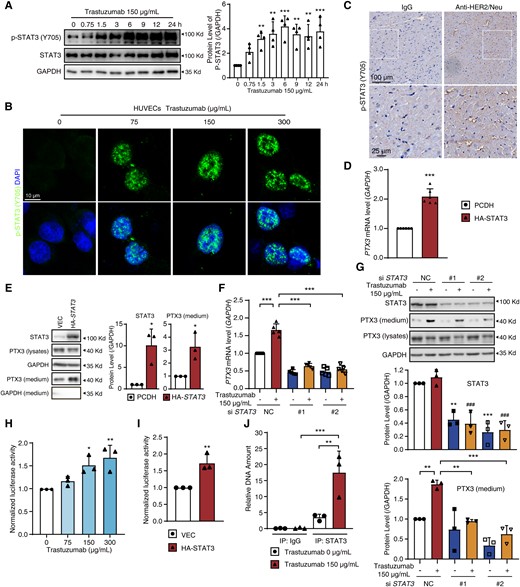
STAT3 functions as a transcription factor of PTX3 in HUVECs. (A and B) HUVECs were treated with trastuzumab at various times or various concentrations of trastuzumab as indicated. (A) The level of p-STAT3 (Y705) measured by western blot (n = 4). (B) Representative images of HUVECs with p-STAT3 (Y705, green) and DAPI (blue) (n = 3). Scale bar, 10 μm. (C) Sprague Dawley rats were treated with IgG or anti-HER2/neu antibody by i.v. for 4 weeks. Heart sections were stained with p-STAT3 (Y705) antibody by immunohistochemistry (n = 3). Scale bar, 100 μm and 25 μm. (D and E) HUVECs were transfected with PCDH or HA-STAT3 plasmid. (D) The PTX3 mRNA level was measured by qPCR (n = 6). (E) The protein levels of STAT3 and PTX3 were measured by western blot (n = 3). (F and G) HUVECs were transfected with NC or STAT3 siRNA for 12 h and then treated with or without trastuzumab. (F) The PTX3 mRNA level (n = 5). (G) The protein levels of STAT3 and secreted-PTX3 (n = 3). (H) Relative luciferase activity in HUVECs treated with various concentrations of trastuzumab for 12 h (n = 3). (I) Relative luciferase activity in HUVECs transfected with PCDH or HA-STAT3 plasmid (n = 3). (J) ChIP-qPCR analysis of STAT3 binding to the promoter of the PTX3 gene in HUVECs (n = 3). The P-value was calculated by one-way ANOVA (Dunnett’s or Sidak test) or Student’s t-test (unpaired, two-tailed, two groups). *P < 0.05, **P < 0.01, ***P < 0.001 (comparing with the indicated group or lane one) and ###P < 0.001 (comparing with lane two in Figure 6G). DAPI, 4′,6-diamidino-2-phenylindole; VEC, Vector.
We then examined the direct regulation of PTX3 transcription by STAT3 by detecting the binding of STAT3 to the PTX3 promoter region via chromatin immunoprecipitation (ChIP) assay. A previous study identified the 1 kb region upstream of the transcription start site (TSS) as a potential promoter region of the PTX3 gene.23 We analysed the PTX3 promoter region with the NCBI database and found that it contains several conserved STAT3-binding motifs [TTC(N)2-4GAA and TT(N)4-6AA].24,25 We designed a pair of PCR primers to amplify the region upstream of TSS that also contains the STAT3-binding motifs to detect the enrichment of DNA fragments by STAT3 pull-down. As shown in Figure 6J, the ability of trastuzumab to promote the enrichment of STAT3 on the promoter region of PTX3 was confirmed by STAT3 ChIP-qPCR. Overall, we demonstrated that STAT3 is recruited to the PTX3 promoter region to directly promote PTX3 transcription upon trastuzumab-induced activation of EGFR signalling.
3.6 Lapatinib could be an intervention agent for trastuzumab-induced cardiotoxicity by inhibiting EGFR/STAT3/PTX3 signalling
Given that EGFR activation in VECs led to cardiac dysfunction caused by trastuzumab and that lapatinib inhibited EGFR-mediated PTX3 release, we investigated the rescue effects of lapatinib on trastuzumab-induced cardiotoxicity. The addition of lapatinib reversed the effects of Tra-Medium on calcium content in CCC-HEH-2 cells (Figure 7A). Moreover, we found that lapatinib abolished the decreases in contraction amplitude and beat rate caused by Tra-Medium without affecting the survival fraction of iPSC-CMs (Figure 7B–D).
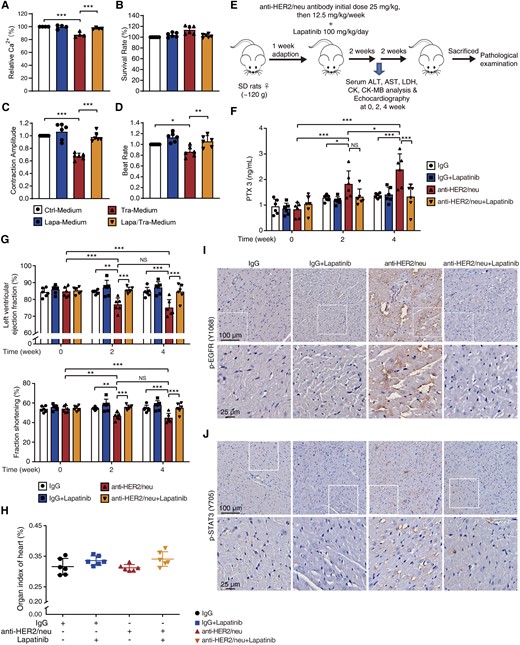
The dual EGFR/HER2 inhibitor lapatinib confers functional protection from trastuzumab-induced cardiotoxicity. (A–D) HUVECs were exposed to trastuzumab with or without lapatinib for 24 h and the conditioned medium was added to CCC-HEH-2 cells and iPSC-CMs. (A) The Ca2+ content of CCC-HEH-2 cells measured by flow cytometry (n = 4). (B–D) The normalized survival rate, contraction amplitude, and beat rate of iPSC-CMs (n = 6 individuals). (E–J) Sprague Dawley rats were treated with saline, anti-HER2/neu antibody, lapatinib, or lapatinib plus anti-HER2/neu antibody for 4 weeks (n = 6 rats). (E) The schematic diagram of animal experiment. (F) Serum was analysed for PTX3 level by ELISA. (G) Cardiac function examined by echocardiography. (H) Organ index of the heart. (I and J) Heart sections were stained with P-EGFR (Y1068) and p-STAT3 (Y705) antibody by immunohistochemistry (n = 3 rats). Scale bar, 100 μm and 25 μm. The P-value was calculated by one-way ANOVA (Sidak test) or two-way ANOVA (Tukey’s test). NS, no significance; *P < 0.05, **P < 0.01, ***P < 0.001. Ctrl-Medium, Control medium; Tra-Medium, trastuzumab medium; Lapa-Medium, lapatinib medium; Lapa/Tra-Medium, lapatinib-plus-trastuzumab medium.
Our in vitro data prompted us to examine the feasibility of lapatinib as an intervention strategy for trastuzumab-induced cardiac complications in vivo. A rat model was established with anti-HER2/neu antibody combined with lapatinib and used to investigate the effects on the heart (Figure 7E). Considering that PTX3 is the key factor underlying trastuzumab-induced cardiotoxicity, we then detected the change in the PTX3 content in rat serum. Compared with the baseline level at Week 0, the level of PTX3 in the anti-HER2/neu group was significantly increased at Weeks 2 and 4 and lapatinib reduced the increased PTX3 level caused by anti-HER2/neu to the level of the IgG group (Figure 7F). The echocardiography results showed that anti-HER2/neu antibody caused cardiac dysfunction, as evidenced by the decreased LV ejection fraction and fraction shortening (Figure 7G and Supplementary material online, Table S5). Notably, the addition of lapatinib improved cardiac function, suggesting a significant protective effect (Figure 7G). We found no apparent change in markers of the myocardial injury, including the heart index; histological features; and lactate dehydrogenase, creatine kinase, and CK-MB levels (Figure 7H, Supplementary material online, Figure S9A and B), which was consistent with clinical observations. We also confirmed that lapatinib could mitigate the activation of EFGR/STAT3 signalling (Figure7I and J) in the heart. Taken together, these results indicated that lapatinib protected against trastuzumab-induced cardiac complications by preventing EFGR/STAT3-mediated PTX3 release and provided a potential strategy to improve the cardiac safety of trastuzumab in the clinic.
4. Discussion
Cardiovascular complications, such as heart failure, caused by trastuzumab dramatically decrease the survival quality of cancer patients, but an effective therapeutic strategy is lacking. Here, we reveal that VECs express a substantial amount of the HER2 protein and are the main target cells of trastuzumab. We confirmed that the activation of EGFR/STAT3 signalling in VECs by trastuzumab promotes the release of PTX3, which leads to the decreased contractility of cardiomyocytes by the disruption of calcium homoeostasis and increased immune activity. Furthermore, we identified lapatinib as a feasible preventive and protective agent for trastuzumab-induced cardiac complications.
Decreased LV ejection fraction and/or congestive heart failure, which are common cardiotoxic effects caused by trastuzumab, are observed in 2–16% of patients treated with trastuzumab, though the incidence has varied in different trials.1,3,26 Typically, this cardiotoxicity further increases with exposure to anthracyclines,27 further complicating the cardiotoxic mechanism. To obtain clear insights into the mechanism underlying trastuzumab-induced cardiac dysfunction, we mainly investigated the effects of trastuzumab alone on the heart in this study, to mimic the clinical practice wherein trastuzumab is used as a single therapeutic agent. Indeed, it has been well established that anthracyclines cause cardiomyocyte death and myocardial injury,28,29 which might increase the sensitivity of cardiomyocytes to trastuzumab or the PTX3 protein. Future studies on the effects of combined trastuzumab and anthracyclines treatment on the heart and the effects of trastuzumab treatment on anthracycline-damaged hearts are warranted.
Kitani et al.30 reported that 7 days of treatment with trastuzumab caused mitochondrial dysfunction and defective biogenesis in iPSC-CMs, so these effects might underly the mechanism by which trastuzumab induces cardiac dysfunction. One possible explanation for these phenotypes is HER2 inhibition in cardiomyocytes, since in vitro results showed that the NRG1-HER2 signalling pathway plays a protective role against mitochondrial dysfunction and apoptosis in cardiomyocytes.31,32 However, endomyocardial biopsy of the ventricles from patients who suffered from the trastuzumab cardiotoxicity showed no features of significant cardiac structural changes or myocardial cell damage,4 in contrast to the phenotypes caused by HER2 inhibition in vitro. Cardiomyocyte-specific HER2-deficient adult mice that were induced by MLC2v-Cre showed dilated cardiomyopathy, but no significant change in mitochondrial function or structure was observed, which is contrary to the conclusion that HER2 is essential for mitochondria and cardiomyocytes, suggesting that the role and mechanism of HER2 in the heart are complicated. Notably, the Myl2 gene, which encodes MLC2v, starts to express in the mouse ventricular cardiac primordia at embryonic Day 7.5. Therefore, dilated cardiomyopathy caused by MLC2v-Cre-driven HER2 deletion might not precisely reflect the role of HER2 in the adult heart. These findings indicated that clarification of the function of HER2 in the adult heart using inducible cardiac-specific Cre mice would promote the understanding of the cardiotoxicity of HER2 inhibitors.
Nevertheless, ErbB2 expression in mouse hearts progressively decreases with age.7 However, the role of HER2 inhibition in trastuzumab-induced cardiac complications cannot be excluded and has yet to be investigated. Therefore, we sought to investigate the expression of HER2 in different cell types within embryonic and adult hearts. Our data revealed that VECs in adult hearts maintained HER2 expression but most of the cardiomyocytes showed low expression of HER2. We then confirmed that VECs contributed to the decreased contractility of cardiomyocytes. These findings might explain why patients who have pre-existing vascular diseases more easily undergo trastuzumab-induced cardiovascular complications.1,8
VECs can release signalling factors to promote myocardial development and control the contractility of cardiomyocytes in the normal adult heart. Thus, the identification of the key factors that mediate the crosstalk between VECs and cardiomyocytes is of key importance for the study of the pathology of cardiac diseases. We identified PTX3 as a factor that contributes to decreased contractility of cardiomyocytes. Nevertheless, the direct effect of PTX3 on cardiomyocytes is much less understood. Increased plasma PTX3 levels were observed in heart failure patients and PTX3 levels were positively correlated with the severity of heart failure.33,34 Somatic deletion of Ptx3 in mice attenuates the cardiac remodelling and decreased cardiac function induced by transverse aortic constriction, suggesting the potential deleterious role of PTX3 in heart failure. We found that PTX3 decreased the contractility of iPSC-CMs in vitro and led to cardiac dysfunction in vivo, confirming the undiscovered deleterious effects of PTX3 on cardiomyocytes in drug-induced cardiomyopathy. Notably, PTX3 can also induce vascular endothelial dysfunction,14 which might secondarily affect cardiomyocytes. In our study, we found that recombinant PTX3 protein had a direct effect on cardiomyocytes, and we noticed that this effect was slightly less pronounced than that of trastuzumab-conditioned medium, suggesting that other factors related to endothelial dysfunction might also contribute to cardiomyocyte injury. However, all these results indicated the initial and key role of PTX3 secretion in trastuzumab-induced cardiotoxicity and that PTX3 could be an ideal therapeutic target in trastuzumab-induced cardiotoxicity.
The heart is composed of various cell types, including cardiomyocytes, fibroblasts, endothelial cells, pericytes, smooth muscle cells, immune cells, and other cells,35 and crosstalk between these various cell types occurs in the heart and is important for heart function.36 Here, we applied cardiomyocytes, endothelial cells, smooth muscle cells, and macrophages, the main cell types in the heart that are commercially available or can be easily used in the laboratory, to confirm the source of PTX3. One of the limitations of this study is that the involvement of other types of cells was not detected. However, we focused on the effect on only cardiomyocytes, given that cardiomyocytes are the most important functional cells. Understanding changes that occur in other cell types could provide a deeper understanding of this cardiotoxicity mechanism.
Heart failure is characterized by altered calcium signalling37,38 and the production of proinflammatory cytokines and inflammatory mediators.39 Here, we preliminarily found that aberrant calcium signalling and immune activity might contribute to defective cardiomyocyte contractility. The regulation of immune activity is the canonical role of PTX3, and PTX3 treatment significantly enhances STAT3 phosphorylation. In addition, STAT3 plays key roles in the regulation of cardiac calcium signalling. Indeed, we noticed the recruitment of inflammatory cells in the hearts of rats treated with the anti-Her2/neu antibody or recombinant PTX3 protein. Therefore, PTX3-mediated immune activity or immune cells might also play role in trastuzumab-induced cardiotoxicity.
Our data showed that the EGFR-STAT3 signalling pathway in VECs is required for PTX3 expression and release and that STAT3 directly binds the promoter region of the PTX3 gene, which contains the canonical STAT3-binding motifs TTC(N)2-4GAA and TT(N)4-6AA,24,25 revealing that the STAT3-PTX3 signalling axis plays a key role in the crosstalk between VECs and cardiomyocytes and that EGFR and STAT3 could be potential targets in the treatment of trastuzumab-induced cardiomyopathy. Considering the lack of clinical STAT3 inhibitors and that several EGFR inhibitors with high cardiac safety have been used in the clinic, we focused on EGFR inhibitors. We confirmed that the combined use of lapatinib and trastuzumab could rescue trastuzumab-induced cardiotoxicity. Clinical trials found that the combination of lapatinib and trastuzumab resulted in a lower risk of cardiotoxicity, which confirms our finding that lapatinib could rescue the cardiotoxicity of trastuzumab.40,41 However, sub-analysis of the Adjuvant Lapatinib And/Or Trastuzumab Treatment Optimisation (ALTTO) trial found that trastuzumab plus lapatinib increased the risk of toxicity in older patients (≥65 years old)42 and here, we used 6-week-old rats to detect the protective effect of lapatinib on the cardiotoxicity of trastuzumab. Given that physiological characteristics differ with age, there must be another mechanism underlying this clinical finding and the application of trastuzumab plus lapatinib should potentially be avoided in older patients.
Taken together, our results identified a distinct model of trastuzumab-induced cardiomyopathy, wherein trastuzumab promotes EGFR/STAT3 signalling-mediated PTX3 secretion from VECs and then indirectly decreases cardiomyocyte contractility by regulating calcium homoeostasis. Our data showed that targeting EGFR with lapatinib rescued the cardiovascular complications caused by trastuzumab. Our findings provided the rationale for the improved clinical cardiac safety of combined treatment with lapatinib and trastuzumab, which could further expand the clinical application of this combined therapeutic strategy.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Authors’ contributions
Z.X., X.C., X.Z., B.Y., Q.H., and P.L. designed the study. Z.X., Z.G., and H.F. performed in vitro studies and statistical analysis. Y.J., B.X., and YT.Z. checked the data and statistical analysis. Y.Z., Y.J., Z.P., H.Y., and X.Y. performed animal studies and data analysis. Z.X. and Y.Z. generated and analysed the LC-MS/MS and RNA-Seq data. X.C., X.Z., and X.W. provided reagents, plasma samples from patients, and clinical views. Z.X., H.F., and H.Y. wrote the raw manuscript and drew the figures. Z.X., H.Y., and P.L. received funding. Z.X., B.Y., Q.H., and P.L. revised the manuscript and figures. All authors interpreted the data and approved the final manuscript.
Acknowledgements
We would like to thank experimenter Wei Yin from the Image Center, Core Facilities, School of Medicine, Zhejiang University for the confocal laser scanning microscope images. We also would like to thank the staff members of LC-BIO TECHNOLOGIES (HANGZHOU) CO., LTD. for their technical support and expertise in RNA-Sequencing.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 81872941 to P.L., 82003862 to Z.X., and 82104315 to H.Y.), Postdoctoral Foundation of Zhejiang Province (ZJ2020038 to Z.X.), and the Fundamental Research Funds for the Central Universities.
Data availability
The data and methods for data analysis are available from the corresponding author upon reasonable request. The RNA-seq data generated in this study are publicly available in the Gene Expression Omnibus (GEO) in entry GSE200325.
References
We identified PTX3 as a potential biomarker and target for the treatment of trastuzumab-induced cardiac complications and demonstrated that lapatinib can prevent cardiac dysfunction caused by trastuzumab by blocking EFGR/STAT3-mediated PTX3 release from VECs, which provided a mechanistic rationale for the combined application of lapatinib and trastuzumab in cancer.
Author notes
Conflict of interest: None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}