





Abstract
Cardiomyocytes are one of the most mitochondria-rich cell types in the body, with ∼30–40% of the cell volume being composed of mitochondria. Mitochondria are well established as the primary site of adenosine triphosphate (ATP) generation in a beating cardiomyocyte, generating up to 90% of its ATP. Mitochondria have many functions in the cell, which could contribute to susceptibility to and development of cardiovascular disease (CVD). Mitochondria are key players in cell metabolism, ATP production, reactive oxygen species (ROS) production, and cell death. Mitochondrial calcium (Ca2+) plays a critical role in many of these pathways, and thus the dynamics of mitochondrial Ca2+ are important in regulating mitochondrial processes. Alterations in these varied and in many cases interrelated functions play an important role in CVD. This review will focus on the interrelationship of mitochondrial energetics, Ca2+, and ROS and their roles in CVD. Recent insights into the regulation and dysregulation of these pathways have led to some novel therapeutic approaches.
1. Introduction
Cardiovascular disease (CVD) is the leading cause of death in much of the world. Myocardial ischaemia results in damaged myocardium, and due to the poor proliferative capacity of cardiomyocytes this loss of myocardium results in cardiac hypertrophy, which leads to heart failure. Reperfusion is the main therapy for myocardial ischaemia, but reperfusion per se can initiate reperfusion injury. Mitochondria play an important role in cell death and energetics, which are key regulators of outcomes in ischaemia/reperfusion (I/R) injury and heart failure. In turn, mitochondrial calcium (Ca2+) and reactive oxygen species (ROS) are important regulators of both death and energetics. This review will focus on the roles of mitochondrial Ca2+ and ROS in regulating cell death and mitochondrial energetics and their impact on CVD.
2. Mitochondria and energetics
The role of mitochondria in regulating adenosine triphosphate (ATP) production is well established, and as it has been reviewed in several recent publications,1,2 we will limit our discussion to how mitochondrial energetics are implicated in CVD, namely I/R and heart failure.
2.1 Energetics and I/R injury
During ischaemia the lack of oxygen inhibits cytochrome c oxidase, thereby blocking the flow of electrons in the mitochondrial electron transport chain. As a result of the inhibition of electron flow, Complex I of the electron transport chain is reduced, preventing donation of electrons to Complex I from its substrate NADH, and thus NADH becomes elevated. In a perfused heart model of global ischaemia, the heart continues to beat and consume ATP for a few minutes. Creatine phosphate (CrP) breaks down to regenerate ATP, thereby buffering the fall in ATP. With the cessation of beating, ATP falls gradually until the heart goes into contracture, at which point ATP falls more rapidly3 and the mitochondrial membrane potential also falls at this time.4 Studies by Jennings et al.5 demonstrated that during anoxic conditions, the F1F0-ATP synthase (F1-ATPase) may paradoxically reverse into an energy-consuming enzyme, further favouring H+ extrusion at the expense of ATP hydrolysis. However, for the F1-ATPase to consume cytosolic ATP, the adenine nucleotide transporter (ANT) also is required to function in reverse. Theoretical calculations based on the reversal potential of the F1-ATPase and the ANT raise questions about conditions under which this process occurs.6
Cardiac metabolism during ischaemia depends on the substrates supplied to the heart and the intrinsic metabolic regulation of the heart. To examine the alterations in the intrinsic metabolism of the heart, studies have been done in ex vivo hearts in which the perfusate can be varied. The heart is highly dependent on oxidative metabolism and oxidative phosphorylation by the mitochondria, and yet the heart is omnivorous and can utilize multiple substrates.7 With global ischaemia oxidative metabolism is inhibited and the cell relies on glycolysis to generate ATP (Figure 1). Glucose is metabolized to pyruvate, generating NADH. Conversion of pyruvate to lactate regenerates NAD+, relieving the inhibition of glycolysis by NADH. Acidosis (commonly referred to as lactic acidosis) occurs during ischaemia due to the retention of protons from breakdown of the glycolytically generated ATP.8 This increase in cytosolic protons during ischaemia stimulates H+ exchange with extracellular Na+ on the plasma membrane Na+–H+ exchanger (NHE), thereby raising intracellular Na+ which leads to an increase in cytosolic Ca2+ via the plasma membrane Na+–Ca2+ exchanger (NCX).9 As discussed in Section 4.3, this increase in cell Ca2+ is thought to be a primary trigger of cell death in ischaemia–reperfusion. The increase in NADH and FADH also leads to inhibition of fatty acid oxidation leading to accumulation of acyl carnitines and acyl CoA.10
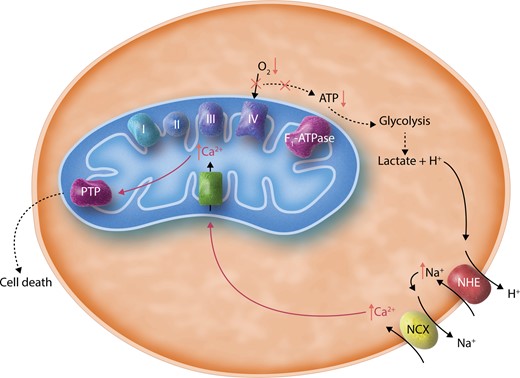
A simplified illustration of cellular changes during ischaemia. This schematic shows that the absence of oxygen (O2) inactivates Complex IV and the electron transport chain, thus preventing ATP generation through the F1-ATPase. This leads to increased glycolysis and the production of lactate, which acidifies the cytosol. NHE is thus stimulated to exchange H+ ions for Na+, and increased cytosolic Na+ in turn activates NCX, leading to increased cytosolic Ca2+. During ischaemia, mitochondrial Ca2+ is thought to increase. The rise of mitochondrial Ca2+ is canonically attributed to transport of cytosolic Ca2+ into the mitochondrial matrix via MCU, though this has not been directly proven. Overaccumulation of mitochondrial Ca2+ is thought to trigger opening of PTP, which may be inhibited by low pH during ischaemia but could become activated upon reperfusion. PTP opening is thought to lead to cell death.
On reperfusion, oxygen is restored allowing electron transport and oxidative metabolism to resume. If the ischaemic time was short and did not lead to substantial cell death, electron transport resumes generating the mitochondrial membrane potential which in turn generates ATP via Complex V. The relative percentage of ATP generated by oxidation of fatty acids increases due to a decrease in malonyl CoA, an inhibitor of mitochondrial fatty acid transport.11 The increase in fatty acid oxidation leads to inhibition of glucose oxidation, thereby uncoupling glycolysis from glucose oxidation. As discussed above glycolysis generates pyruvate, and if pyruvate is not oxidized by the mitochondria it is converted to lactate. This can lead to an increase in generation of protons and hence via NHE and NCX increased entry of Ca2+ into the cell. Dichloroacetic acid (DCA), which activates pyruvate dehydrogenase (PDH) and thereby activates glucose oxidation has been shown to be protective in I/R injury (Figure 2). The details of altered substrate selection and its role in I/R injury have been discussed in detail elsewhere.12,13

Mitochondrial pathways and potential mitochondrial therapeutic targets in ischaemia/reperfusion (I/R). This schematic illustrates in a simplified manner the interconnections of mitochondrial metabolism, Ca2+, and ROS, and potential therapeutic targets in I/R. The activation of the permeability transition pore (PTP) by Ca2+ and ROS is shown by arrows. Two potential PTP candidates, ANT and the F1-ATPase, are shown. Interventions are shown with gray arrows. Stimulation of PDH with DCA is protective presumably due to enhancing glucose oxidation. Inhibitors of PTP, such as Cyclosporin A (CsA), are also protective. Ca2+ and ROS are activators of PTP and it is proposed that reducing mitochondrial Ca2+, either by blocking mitochondrial Ca2+ uptake through MCU (by Ru360 or recently characterized small molecules) or by enhancing efflux through NCLX (for which there is currently no drug), might be beneficial. Reducing ROS, either by inhibiting SDH (Complex II) and reverse electron transfer (RET), by inhibiting electron leak at Sites I and III, or by mitochondrial targeted antioxidants have all shown benefit. SS-31 which binds to cardiolipin (CL) has also been shown to be beneficial, possibly by modulating the binding of cardiolipin to ANT and potentially inhibiting PTP. Other effects of SS-31, for instance against ROS via interactions of cardiolipin with cytochrome c and the electron transport chain, are not shown for clarity, but likely contribute to SS-31’s efficacy.
2.2 Energetics and heart failure
The failing heart has been described as an energy starved heart, and studies have shown that failing human hearts have a decrease in CrP/ATP.14 Depending on the type and severity of heart failure, an increase, decrease, or no change in fatty acid oxidation has been reported.2 In severe heart failure associated with ischaemia or hypertension there is a transition from beta-oxidation to increased glycolysis, accompanied by a return to a more fetal metabolic profile. The increase in glycolysis during heart failure occurs often without an increase in glucose oxidation. Inhibitors of fatty acid oxidation, such as trimetazidine (a ketothiolase inhibitor) as well as etomoxir and perhexiline (CPT1 inhibitors) have been generally shown to be protective in heart failure models, which are proposed to be due, at least in part, to an increase in glucose oxidation (Figure 3). Similarly, inhibitors of malonyl CoA decarboxylase, an enzyme which catabolizes malonyl CoA, leads to an increase in malonyl CoA and thereby decreases fatty acid oxidation because malonyl CoA inhibits fatty acid uptake. Activation of PDH and hence glucose oxidation by DCA has also been shown to be protective in heart failure models.
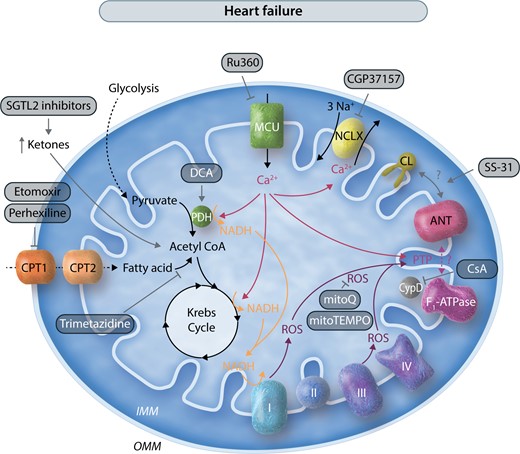
Mitochondrial pathways and potential mitochondrial therapeutic targets in heart failure. This schematic illustrates in a simplified manner the interconnections of mitochondrial metabolism, Ca2+, and ROS, and potential therapeutic targets in heart failure. As in Figure 2, interventions are shown with gray arrows. Many of the targets are the same as in Figure 2, including PDH, PTP, MCU, and ROS. Activation of PTP is proposed to play a role in heart failure, thus inhibitors of PTP and ROS have also been shown to be beneficial. SS-31 has also been shown to improve heart failure outcomes. In some heart failure models in which mitochondrial Ca2+ is insufficient, inhibiting NCLX with CGP37157 has had efficacy. In heart failure, inhibitors of fatty acid oxidation including CPT inhibitors are protective, and increased ketone metabolism to presumably stimulate glucose oxidation is also beneficial.
An increase in ketone metabolism has also been shown in heart failure,15,16 and studies were performed to evaluate whether the increase in ketone utilization is adaptive or maladaptive. Recent studies suggest that enhanced ketone oxidation increases energy production but does not improve cardiac efficiency.17 An increase in ketone metabolism can be accomplished by increasing circulating ketones. Interestingly, sodium-glucose transport protein 2 (SGLT2) inhibitors, which were originally given as glucose-lowering drugs have been shown to have additional cardioprotective benefit in heart failure patients in addition to their ability to lower serum glucose.18 Although the exact mechanism by which SGLT2 inhibitors confer this protection is debated, they have been shown to increase circulating ketone levels, which could be involved in the protection. Insulin19 and GLP-120,21 have also been shown to be beneficial. Finally as discussed elsewhere22–24 supplementation with nicotinamide or other NAD precursors has been shown to be protective in heart failure.
3. Mitochondrial Ca2+
3.1 Mitochondrial Ca2+ and energetics
Mitochondrial Ca2+, which is altered in many CVDs, has been shown to regulate ATP production.25 Denton and McCormack26 demonstrated that an increase in cytosolic Ca2+, as occurs with an increase in energy demand, leads to an increase in mitochondrial Ca2+ levels. Ca2+ enters mitochondria through the mitochondrial Ca2+ uniporter (MCU) complex27 and in the matrix activates three mitochondrial NAD(P)H-generating dehydrogenases26: PDH, isocitrate dehydrogenase (ICDH), and α-ketoglutarate dehydrogenase. Ca2+ activation of PDH occurs primarily via Ca2+ activation of PDH phosphatase, which results in dephosphorylation and activation of PDH. Ca2+ activation of these dehydrogenases stimulates increased NADH production, which provides electrons to the electron transport chain at Complex I to enhance ATP production. Brandes and Bers28 showed in cardiac trabeculae that with an increase in pacing, there was a rise in mitochondrial Ca2+ and transient elevation of NADH. Liu and O’Rourke29 studied adult guinea pig cardiomyocytes and found that the relationship between an increase in mitochondrial Ca2+ and a change in NADH with increased pacing was dependent on cellular Na+ concentration. If the experiment was done in the presence of 15 mM Na+ (compared with 5 mM Na+), upon pacing there was less of an increase in mitochondrial Ca2+ and an oxidation in NADH, suggesting that Na+-dependent mitochondrial Ca2+ efflux plays a role in regulating mitochondria Ca2+ during increased work. In the presence of Ru360, an inhibitor of MCU, the pacing-induced increase in mitochondrial Ca2+ was reduced and net NADH oxidation was observed.
In addition to Ca2+ activation of the mitochondrial dehydrogenases, Ca2+ has also been suggested to activate mitochondrial electron transport Complexes III and V.25 However, Wescott et al.30 recently reported that an increase in matrix Ca2+ only activates ATP production from NADH-linked substrates via Ca2+-activated dehydrogenases, thereby questioning Ca2+ activation of Complex III or V. Additional studies will be needed to resolve this discrepancy.
3.2 Mitochondrial Ca2+ and cell death pathways
Cardiac cell death can be initiated by multiple pathways and the relative importance of these pathways depends upon a number of context-dependent factors. Autophagy, mitophagy, and ferroptosis can initiate cell death and are reviewed elsewhere.31–34 This review will focus on Ca2+ activation of necroptosis and opening of the mitochondrial permeability transition pore (PTP). Under pathological conditions of Ca2+ overload, Ca2+ can enter mitochondria presumably via the MCU leading to an increase in mitochondrial Ca2+ and opening of the PTP.35 The PTP is a large conductance channel in the mitochondrial inner membrane. Studies in isolated mitochondria showed that cyclosporin A (CsA) can inhibit Ca2+-activated PTP opening and the resultant mitochondrial swelling. Cyclophilin D (CypD), a mitochondrial matrix protein, was shown to be the target of CsA. An increase in matrix Ca2+ and ROS, which both occur with ischaemia and reperfusion, triggers PTP opening. Sustained opening of PTP leads to loss of mitochondrial cofactors and proteins up to ∼1.5 kDa. Thus, PTP opening leads to loss of mitochondrial membrane potential and loss of mitochondrial cofactors such as NAD/NADH. Low pH inhibits PTP opening.36,37 However, this effect is complex, as low pH inhibits PTP opening only in de-energized mitochondria.37
Although CypD has been shown to be a regulator of the PTP, it is not the pore-forming unit. Unfortunately, the identity of the PTP is still debated, limiting progress on devising treatments to block PTP opening during I/R. There are a number of recent reviews on the PTP38–41 so that topic will not be discussed in detail here. Briefly, recent data have suggested that the F1F0-ATP synthase (F1-ATPase) can, under certain circumstances, form the PTP. Bernardi’s group has proposed that dimers of the F1-ATPase can form the pore-forming unit of the PTP.42 Jonas’ group has suggested that delipidation of the c-ring of the F1-ATPase can form the PTP.43 Although there are strong data in support for both hypotheses, there are also data in opposition. Data from Walker’s group have shown a CsA-inhibitable PTP-like swelling is still present in cells lacking the c-ring and other components of the F1-ATPase that are proposed to be involved in PTP formation.44,45 One explanation for the discrepancy is that there are two or more CsA-inhibitable pores. Data from over 30 years ago proposed a role for ANT as a key component of PTP.46,47 CypD was shown to bind to ANT and adenine nucleotides were known inhibitors of PTP. Bongkrekic acid (BKA) and carboxyatractyloside (CA), two inhibitors of ANT that lock ANT in different conformations, were shown to regulate PTP.47 BKA locks ANT in the matrix facing conformation and inhibits PTP, whereas CA binds to the cytosolic facing conformation and activates PTP. The hypothesis that ANT was the PTP fell out of favour in 2005 when Wallace’s group reported that a CsA-inhibitable PTP, albeit requiring higher Ca2+ for activation, was present when ANT1 and 2 were deleted.48 Recent studies have revisited a role for ANT as a pore component of PTP.49 Karch et al.49 showed that mitochondria with deletion of ANT1, 2, and 4 were desensitized to Ca2+-induced PTP opening, and PTP opening was completely inhibited by the further addition of CsA. They proposed that there are two distinct PTPs: the ANT and a second pore. Both components may be activated by CypD, although the non-ANT component has a strict requirement for CypD.
Although a role for Ca2+ in PTP opening has been clearly demonstrated, the mechanism by which Ca2+ activates PTP is unclear. Much of the difficulty with deciphering how Ca2+ activates PTP is related to the lack of clarity regarding the identity of PTP. Ca2+ binding to T163 of the beta-subunit of the F1-ATPase alters its conformation to promote pore opening at the dimer/tetramer interface of the Fo domain.50 Mutation of T163 alters Ca2+- and Mg2+-dependent ATP hydrolysis. It was further demonstrated in HeLa cells that when T163 is mutated, the Ca2+ sensitivity of the PTP is attenuated and almost twice as much Ca2+ is required to activate PTP.50
Moreover, Ca2+ can bind to cardiolipin, which is known to be in close proximity with several major transport proteins including ANT. It has been proposed that Ca2+ binding to cardiolipin might regulate the conformation of ANT and thus promote PTP.51 Alternatively, glutamate 152/3 and aspartate 167/8 of ANT have been proposed to play a role in regulating Ca2+ binding to ANT to promote PTP47; however, this has not been confirmed experimentally.
CypD, a key activator of PTP, has been shown to possess an acylation site at C202. Acylation is the attachment of a fatty acid to a cysteine via a thioester bond. Interestingly, high Ca2+ has been shown to lead to de-acylation of CypD at C202.52 It is proposed that de-acylation of CypD might be important in its targeting to PTP; perhaps acylation targets it away from PTP or alternatively a free C202 might be important to activate PTP. Clearly, conclusive identification and better mechanistic understanding of PTP will be important in elucidating the mechanism by which Ca2+ and other regulators of the PTP mediate their effects.
Mitochondrial Ca2+ may influence cell death and survival by mechanisms that go beyond triggering of PTP. One of these mechanisms could be the activation of mitochondrial calpains, which have been shown to play a role in cardiomyocyte injury and cell death.53,54 In addition, mitochondrial Ca2+ uptake during I/R could buffer cytosolic Ca2+ overload, a trigger of reperfusion-induced hypercontraction and death. Indeed, in certain conditions such as ageing in which mitochondrial Ca2+ uptake is reduced or impaired cytosolic Ca2+ overload is aggravated, and this effect is associated with an increased cardiomyocyte hypercontracture and sarcolemmal rupture.55
3.3 Mitochondrial Ca2+ at baseline and during ischaemia and reperfusion
3.3.1 Baseline pre-ischaemic Ca2+
There is some variability in measurements of the baseline level of mitochondrial Ca2+, but in general it is reported to be 100–200 nM or lower.56–58 Andrienko et al.56 reported that for cytosolic free Ca2+ levels at or below 475 nM, mitochondrial Ca2+ is lower than cytosolic Ca2+. As discussed elsewhere, there has been some disagreement about whether mitochondrial Ca2+ can respond on a beat-to-beat basis or whether mitochondrial Ca2+ acts to integrate increases in the cytosolic Ca2+ transient.59,60
3.3.2 Ca2+ during ischaemia
Studies done over two decades ago showed that an increase in cellular Ca2+ was an important trigger of necrotic cell death.9 As mentioned above, it was shown that during myocardial ischaemia there is an increase in acidosis that promotes plasma membrane NHE activity, leading to an increase in cytosolic Na+, which in turn leads to an increase in cytosolic Ca2+ via NCX9 (Figure 1). The importance of these ionic changes in triggering cell death was demonstrated by showing that inhibitors of NHE attenuated the rise in Na+ and Ca2+ during ischaemia and were cardioprotective.9 However, these NHE inhibitors were not protective in a large clinical trial.61 Reasons for the lack of protection are discussed elsewhere62 and include an increase in stroke. However, the lack of protection is consistent with the preclinical data, which showed that the rise in Na+ and Ca2+ occur during ischaemia, before patients can be treated.9 Although a number of drugs have been reported in animal studies to be beneficial when added on reperfusion,63,64 few if any have shown benefit in clinical trials. Possible reasons for this have been discussed elsewhere.65,66 The field has shifted focus to try to understand the downstream pathways by which cytosolic Ca2+ initiates cell death. One current hypothesis is that during ischaemia and reperfusion Ca2+ enters the mitochondria via the MCU to activate the PTP. The PTP is reported to be inhibited by acidic conditions that are present during ischaemia and is therefore not activated until reperfusion when the pH is restored.67 The inhibition of PTP during ischaemia provides a window of opportunity to get PTP inhibitors to cells at the start of reperfusion to inhibit PTP opening. Addition of CsA or deletion of CypD has been shown to reduce cell death following I/R in isolated heart68 and animal studies.69,70 An initial small clinical trial demonstrated CsA protection in acute myocardial infarction (MI)71; however, a recent clinical trial did not find CsA to protect in patients with MI.72 A possible reason could be that CsA does not inhibit the PTP itself; rather, it inhibits CypD, an activator of the PTP. It has been shown that with higher Ca2+ or ROS levels, PTP opening can occur in the presence of CsA.73–76 Thus, future development of direct inhibitors of the PTP itself could potentially be beneficial.
An alternative to inhibiting PTP is to block the upstream increase in mitochondrial Ca2+. As MCU is the only documented mitochondrial Ca2+ import channel,77,78 it has been suggested that inhibiting MCU in I/R might be a strategy to reduce PTP opening.79 It should be noted that if mitochondrial Ca2+ increases during ischaemia this would limit the protective effect of MCU inhibitors added at the start of reperfusion. To test the role of MCU in regulating mitochondrial Ca2+ in CVD, several lines of mice with MCU deletion were developed. In the first model, MCU was deleted globally using a gene trap method in the germline80 (Mcu–/– mice). In another model MCU was conditionally deleted in adult cardiac cells using Mcu floxed mice crossed with the tamoxifen-inducible α-MHC-driven Mer-Cre-Mer81,82 (Mcufl/fl–MCM mice). Cardiac mitochondria from both lines were studied and shown not to take up Ca2+ or undergo PTP. An α-MHC-driven dominant negative MCU (DN-Mcu) was also developed,83,84 and similar to the Mcu–/– and Mcufl/fl–MCM mitochondria, mitochondria from this mouse did not take up Ca2+ and did not undergo PTP.
As mitochondria from the Mcu–/–, Mcufl/fl–MCM, and DN-Mcu mice do not take up Ca2+ and do not undergo PTP, it was expected that they would be protected from I/R. Surprisingly, there was no protection following I/R in the germline Mcu–/– mice,80 and CsA provided no protection in the Mcu–/– mice. Furthermore, there was no protection in the mice with cardiac specific expression of DN-Mcu.83 In contrast, when MCU was conditionally deleted in adult cardiac cells, the hearts were protected from I/R injury.81,82 The reason for this discrepancy is not clear, but it has been proposed that adaptive changes are likely to occur when MCU activity is lost (by deletion or a DN) at or before birth.85 The precise nature of these adaptations is under study, but it appears that in the germline Mcu–/– mitochondria less Ca2+ is required for PTP activation.85 It is also unclear whether the adaptations are related to development or whether they would occur in adult mice with additional time after acute MCU deletion. As mentioned above, CsA provides no protection in the germline Mcu–/– mice80; perhaps the adaptations, which occur in the Mcu–/– mice might provide some insight into why CsA did not provide protection in a recent clinical trial.72
It should also be noted that the studies showing protection in the adult Mcufl/fl–MCM hearts did not directly confirm that the protection was due to blocking an increase in mitochondrial Ca2+ during I/R.81,82 A recent abstract reported that simulated ‘ischaemia’ in neonatal mouse cardiomyocytes isolated from the conditional Mcufl/fl mouse and treated with Cre-expressing adenovirus to delete MCU led to similar mitochondrial Ca2+ increases as in control cardiomyocytes.86 Clearly, additional studies are needed to fully address the role of mitochondrial Ca2+ in I/R. The development of new methodology to measure mitochondrial Ca2+ in beating perfused hearts should be helpful in addressing this question.87
Inhibition of MCU to block the rise in mitochondrial Ca2+ and hence activation of PTP is proposed to be beneficial in I/R. Therefore, drugs targeting MCU might be beneficial in CVD, though existing inhibitors of MCU such as Ru360 are limited in membrane permeability. Thus, recent screening has identified new small molecule inhibitors of MCU activity.79 However, one such molecule, MCU-i11, was shown to be ineffective against cell death in a cardiomyocyte-based in vitro hypoxia/reoxygenation model79 and was suggested to potentially be of use in chronic mild Ca2+ overload conditions, though this remains to be tested.
In addition to the MCU-mediated Ca2+ uptake pathway, there are Ca2+ efflux pathways.88,89 In steady state, the Ca2+ that enters mitochondria through the MCU must exit via a Ca2+ efflux pathway. Data from the 1970s suggested that there are Na+-dependent and Na+-independent Ca2+ efflux mechanisms. The protein responsible for mitochondrial Na+-dependent Ca2+ efflux (Na+–Ca2+–Li+ exchanger; NCLX) was identified in 2010.89 NCLX is thought to be electrogenic; therefore, in the presence of a typical large negative mitochondrial Δψ, it will only function to extrude Ca2+. A mouse with conditional deletion in NCLX in adult heart was developed and used to show that acute loss of NCLX was associated with high mortality as most mice died within days after NCLX deletion.90 These data suggest that NCLX is a major pathway for Ca2+ efflux in the heart. Mice with cardiac specific overexpression of NCLX were generated and it was shown that hearts from these mice are protected from I/R injury.90 These data are consistent with the concept that increased NCLX-dependent Ca2+ efflux during I/R reduces mitochondrial Ca2+ and can mediate cardioprotection.
3.3.3 Mitochondrial Ca2+ during reperfusion
What happens to mitochondrial Ca2+ on reperfusion depends on a number of factors with the length of ischaemia being an important factor. In perfused rat heart after a short (20–25 min) period of ischaemia, cytosolic Ca2+ returns close to baseline Ca2+ by ∼30 min of reperfusion.9 It should also be noted that the length of ischaemia that leads to more irreversible changes varies depending on the species. Studies in isolated cardiomyocytes report oscillations in cytosolic Ca2+ on reperfusion, which appear to be due to oscillations in sarcoplasmic reticulum (SR) Ca2+ release and re-uptake. As reviewed elsewhere,91,92 a number of mechanisms were identified by which cytosolic Ca2+ can modulate injury on reperfusion. There are few if any reports of how mitochondrial Ca2+ responds during reperfusion. It might be expected to follow the changes in cytosolic Ca2+ with perhaps some lag. Future studies will be needed to address the changes that occur in mitochondrial Ca2+ on reperfusion. As mitochondrial Ca2+ can regulate metabolism and ATP production, prolonged alterations in mitochondrial Ca2+ could have important metabolic and energetic effects.
3.4 Mitochondrial Ca2+ and heart failure
A great deal of controversy exists surrounding to what degree mitochondrial Ca2+ uptake increases or decreases in the progression of heart failure. Some of the discrepancies in the results can be attributed to different animal species (for instance, mouse vs. guinea pig) and different types of induced heart failure. Studies have used various models, including transverse aortic constriction (TAC), post-MI heart failure, or isoproterenol-induced heart failure.
Among the studies pointing to excessive mitochondrial Ca2+ in heart failure, one study showed that elevated mitochondrial Ca2+ levels caused by Ca2+ leak from the SR contributes to heart failure.93 The mouse model of post-MI heart failure examined in this study was induced by permanent occlusion of the proximal left anterior descending (LAD) coronary artery. Direct measurement of mitochondrial Ca2+ levels revealed a more than two-fold increase in post-MI heart failure samples relative to sham. Concurrently, SR Ca2+ leak and production of ROS increased in post-MI isolated cardiomyocytes. Mice harbouring a constitutively leaky mutation of the Type 2 ryanodine receptor (RyR2) channels (termed RyR2-S2808D mice) were used to demonstrate that the SR Ca2+ leak causally increased mitochondrial Ca2+, resulting in dysmorphic and malfunctioning mitochondria as well as loss of membrane potential. RyR2-S2808D mice exhibited further elevation of mitochondrial Ca2+ levels in sham and post-MI conditions, and these mice also showed exacerbated progressive decline in ejection fraction measured in the weeks post-MI. Although this study examined only post-MI heart failure, it overall supports the view that increased mitochondrial Ca2+, in this case due to SR Ca2+ leak, contributes to the progression of heart failure. A similar mechanism for increased SR Ca2+ leak leading to mitochondrial Ca2+ overload has been shown to occur with ageing in both mice and humans.94
In a study mentioned above, elevated mitochondrial Ca2+ induced by blocking export via NCLX acutely in adulthood led to severe heart defects and a high rate (87%) of spontaneous death.90 Tamoxifen-induced, heart-specific deletion of Slc8b1, the gene encoding NCLX, in adult mice led to hallmarks of heart failure including left ventricular (LV) dilation and decreased fractional shortening, hypertrophy, and increased fibrosis.90 These effects were attributed to opening of the PTP, a known consequence of mitochondrial Ca2+ overload, and could be rescued by deletion of CypD, an activator of PTP opening. As discussed above, transgenic overexpression of NCLX in the heart increased Ca2+ efflux from the mitochondria and protected against I/R injury.90 In addition, overexpression of NCLX also reduced pathological heart remodelling after permanent occlusion of the LAD coronary artery, the same post-MI heart failure model used in the above-mentioned study.93 Interestingly, increased mRNA expression of SLC8B1 was observed in LV tissue of patients with ischaemic heart failure, which the authors suggest represents a compensatory mechanism during heart failure to limit pathological mitochondrial Ca2+ overload. Taken together, these studies suggest that increased mitochondrial Ca2+ is a driver of heart pathology and that decreasing mitochondrial Ca2+ would be a potential therapeutic avenue.
Results using TAC rather than post-MI heart failure in mice have been unclear on the utility of deleting MCU. In both a global knockout mouse model95 and cardiac-specific tamoxifen-inducible mouse model,81 MCU deletion did not alter the course of pathology after TAC. However, it is not completely clear whether MCU deletion can prevent the chronic accumulation of mitochondrial Ca2+; without MCU, the acute Ca2+ response to adrenergic stimulation seems to be blunted,87 but in pathological or chronic conditions, more investigation is needed. In fact with 60 min of adrenergic stimulation an increase in mitochondrial Ca2+ was noted in the cardiac-specific tamoxifen-inducible Mcufl/fl–MCM mouse model.81 There is evidence, however, that germline deletion of MICU3, a regulator of the uniporter that enhances mitochondrial Ca2+ uptake, attenuates isoproterenol-induced hypertrophy.96 Post-isoproterenol, the Micu3–/– mice concurrently displayed increased phospho-PDH to total PDH ratio relative to wild-type controls, indicative of lower mitochondrial Ca2+. Hence, limiting mitochondrial Ca2+ uptake remains a potential avenue to ameliorate heart failure.
Another substantial body of evidence supports a role for decreased mitochondrial Ca2+ in worsening heart failure. A number of studies have proposed that heart failure-associated changes in intracellular Ca2+ and Na+ handling lead to decreased mitochondrial Ca2+, impairing mitochondrial energy production and depleting mitochondrial antioxidative capacity. Mimicking heart failure conditions including reduced systolic SR Ca2+ release by patch clamp in guinea pig myocytes, one study showed that MCU activity decreased and failed to stimulate Ca2+-sensitive Krebs cycle enzymes.97 Consistent with these results, blocking NCLX with the inhibitor CGP-37157, thus presumably increasing mitochondrial Ca2+, prevented imbalance in energetics (NADH oxidation).29 Indeed, chronic CGP-37157 in a guinea pig model of heart failure and sudden cardiac death (SCD) using aortic constriction and adrenergic receptor stimulation was beneficial and attenuated hypertrophic remodelling and dysfunction.98 Further in support of the view that heart failure is linked to depletion of mitochondrial Ca2+, increased intracellular Na+ concentration in heart failure was shown to drive increased NCLX activity, accelerating Ca2+ export from the mitochondria.99 The consequences of reduced mitochondrial Ca2+ are thought to be not only bioenergetic impairment but also increased ROS. Because regeneration of NADPH, an important component of mitochondrial antioxidant capacity, relies on the Krebs cycle, lower mitochondrial Ca2+ depletes NADPH and leads to increased oxidative stress. Again in guinea pig myocytes, NCLX inhibition via CGP-37157 also prevented increased ROS formation.100 More recently, a study showed that consistent with these results, overexpression of MCU increased mitochondrial Ca2+, decreased ROS, and improved contractility in a guinea pig model of hypertrophy via aortic banding and isoproterenol.101
It should also be noted that cardiac myocytes have several mitochondrial populations (subsarcolemmal, interfibrillar, perinuclear), with distinct energetic traits and Ca2+ tolerance properties. These traits may be relevant in pathological conditions, such as heart failure and I/R injury. On the other hand, subsarcolemmal mitochondria differentially contain connexin 43 (Cx43)102 that has been shown to have a pathophysiological role in I/R injury.103
Summed together, the evidence suggests there may be an optimal physiological range for mitochondrial Ca2+ and that straying either too low or too high is detrimental.104 Further studies are needed to determine under what circumstances mitochondrial Ca2+ increases or decreases excessively, and whether a change in either direction is a cause or effect of heart failure. Our current understanding does not exclude the possibilities that elevation or decline in mitochondrial Ca2+ could be associated with different types of heart failure or that during different stages of heart failure mitochondrial Ca2+ shifts from one extreme to the other.
4. Mitochondrial ROS
4.1 Mechanisms involved in regulating ROS
ROS can lead to oxidative stress and are inextricably linked to mitochondrial function, as oxidative phosphorylation inherently generates ROS as by-products. Oxidative stress is defined as an imbalance of ROS production and scavenging. Electron leaks from at least 11 distinct sites in the electron transport chain can generate ROS in the form of superoxide anions (O2–) and hydrogen peroxide (H2O2).105 The capacities of these sites to produce O2– and H2O2 vary substantially,105 and the capacity of a given site can vary among tissues106 and even between cardiac subsarcolemmal and interfibrillar mitochondria.107 Canonically, Complexes I and III are recognized as the primary sites of mitochondrial production of ROS,108,109 but under different substrate and exercise conditions other sites also contribute.110 In addition to sites within the electron transport chain, several other mitochondrial proteins associated with the inner and outer mitochondrial membranes can contribute to ROS production,111 including p66Shc, monoamine oxidase isoforms A and B (MAO-A and MAO-B), and NADPH Oxidase 4 (NOX4).
The impact of mitochondrial ROS on cellular and heart function is complex. Though evidence exists that ROS can also serve as molecular signals112 as will be discussed later, ROS cause damage to macromolecules including proteins and DNA. Thus, it is generally accepted that oxidative stress plays an important pathological role in CVDs.113 For instance, ROS overload is thought to contribute to opening of the PTP, which as mentioned above is damaging to mitochondria and cells. In a phenomenon known as ROS-induced ROS release, opening of the PTP upon ‘triggering’ ROS leads to a further burst of ROS, thus increasing oxidative stress and leading to more damage.114,115
The chemical reactivities of O2– and H2O2 are different, and thus they target distinct types of molecules.116 Superoxide is highly reactive and oxidizes positively charged Fe–S centres in proteins including the TCA cycle enzyme aconitase.117 Hydrogen peroxide is reactive with thiolates, such as cysteine residues on PDH Kinase 2, and being membrane-permeant can also diffuse to the cytosol and impact kinase and transcription factor signalling.112 To mitigate ROS-mediated oxidative stress, cells employ antioxidant defence systems that consume O2– and H2O2. O2– can be converted into H2O2 by superoxide dismutases (SOD family), of which one member is mitochondrial (SOD2, also known as Mn-SOD), or by spontaneous dismutation. Conversion of H2O2 to water in the mitochondrial matrix can be mediated by matrix thiol systems, including members of the peroxiredoxin (Prx) family, such as mitochondrial Prx3, and the glutathione peroxidase (Gpx) family.118 Prx and Gpx rely on the cofactors thioredoxin and glutathione, respectively, which require NADPH for regeneration. The peroxisomal protein catalase can also consume H2O2, and a limited number of studies show evidence that it can localize to mitochondria in cardiomyocytes.119,120 Despite some controversy in this regard, mitochondria-targeted catalase is nonetheless a useful experimental tool to link matrix H2O2 to a phenotype.116 These ROS scavenging systems each play important roles in maintaining cell function and experimental loss or gain of function can be instructive in differentiating between the contributions of O2– and H2O2 to various pathologies.
It is difficult to define a general set of conditions that necessarily lead to increased ROS generation. Rates of electron leak to O2– or H2O2 do not depend on respiration rate per se but on whether the particular site becomes more oxidized or reduced when electron flow into or out of that site changes.105 Hence, increased respiration rate can lead to either increased or decreased ROS production: if the increased respiration rate is driven by increased substrate supply, further reducing a site, ROS production at that site will increase, but if driven by increased ATP demand or proton leak, oxidizing the site, ROS production will decrease. Indeed, ATP synthase was hyperacetylated in heart failure patients, which was associated with decreased activity.121 In principle, decreased ATP synthesis would pose a roadblock to electron flow and tend to increase ROS production in failing hearts. Even so, identifying the exact sites and mechanisms contributing to increased ROS production in CVDs is complicated. Therefore, many studies rely on methods to scavenge ROS and assess if they mitigate pathological outcomes to help identify whether O2– or H2O2 are responsible.
4.2 ROS and I/R injury
Many studies demonstrate an important role for ROS in I/R injury. Hearts from SOD2+/– mice showed impaired functional recovery in a Langendorff perfusion model of I/R,122 and correspondingly, SOD2 overexpression in transgenic mice was protective both in ex vivo and in vivo I/R models.123 These data indicate that mitochondrial O2– contributes to tissue damage. Interestingly, in simulated I/R in embryonic chick cardiomyocytes, death was attenuated by overexpression of SOD2, but not by catalase, mitochondrial expressed catalase (mCAT), or primarily cytosolic SOD1.124 While more studies are needed, ideally in vivo in a mammalian model, this suggests that O2– might play a more significant role than H2O2 in I/R injury.
I/R has also been studied using other means of targeting the generation of ROS, including the use of small molecules that suppress electron leak to oxygen without inhibiting electron transport in the respiratory chain. Named S1QELs (suppressors of site IQ electron leak) and S3QELs (suppressors of site IIIQo electron leak), these molecules selectively decrease formation of O2– and possibly H2O2 at specific sites.110 Discovered via high-throughput screening, S1QEL1.1 improved cardiac functional recovery and decreased infarct size in a Langendorff-perfused mouse heart model of I/R injury.125 Additionally, the drug OP2113 was shown to act as an S1QEL, and in ex vivo Langendorff-perfused rat hearts OP2113 also improved recovery of contractile performance and reduced infarct size.126 OP2113 was also effective in a rat in vivo post-MI I/R model. When given preventively prior to coronary artery occlusion, OP2113 significantly reduced infarct size.127
The importance of O2– and Complex I in I/R injury is consistent with a recently elucidated metabolic pathway linking I/R to ROS generation. Comparative metabolomics identified accumulation of succinate during ischaemia in multiple metabolically diverse tissues.128 Simulations predicted that upon reperfusion, succinate would rapidly be oxidized by succinate dehydrogenase (SDH; Complex II), driving reverse electron transport through Complex I and generating O2–. In line with this hypothesis, infusion of dimethyl malonate to inhibit SDH prevented a burst of mitochondrial ROS at the onset of reperfusion and reduced infarct size after in vivo MI-induced I/R injury in mice128 and in pigs.129 While this model of the relationship between I/R and ROS is not without controversy,130 it fits with the above observations of a critical role in I/R for O2– generated by Complex I.
Additionally, though its mechanism of action against ROS is less direct, the mitochondria-targeted compound SS-31 (Elamipretide, Bendavia) also has been shown to have efficacy in I/R. SS-31 binds to cardiolipin, a phospholipid enriched in the mitochondrial inner membrane, by electrostatic and hydrophobic interaction. Cardiolipin interacts with ANT and the electron transport complexes and has been reported to play a role in cristae structure. Interaction of cardiolipin with cytochrome c has been reported to regulate the cytochrome c switch between an electron carrier and a peroxidase. Cardiolipin oxidation occurs in many CVDs, and Barth syndrome is characterized by loss of the tafazzin gene, which is needed to produce cardiolipin. Loss or oxidation of cardiolipin can alter bioenergetics, ROS, and redox and mitochondrial transporters. By binding cardiolipin, SS-31 has been shown to have beneficial effects, including against I/R injury. SS-31 was protective and reduced infarct size in ex vivo and in vivo I/R models in a range of animals, including mice, rats, guinea pigs, rabbits, and sheep.131–134 Notably, however, when SS-31 was administered to rabbits even 10 min after the onset of reperfusion in vivo, protection was lost.134 Furthermore, even though SS-31 was administered more than 15 min prior to reperfusion, the EMBRACE STEMI trial in 118 patients with MI revealed that SS-31 did not reduce estimated infarct area or produce improvements in clinical outcomes.135
4.3 ROS and heart failure
Multiple studies deleting or decreasing SOD2 have linked O2– to cardiac disease outcomes. SOD2 homozygous deficiency in multiple mouse models led to postnatal death at differing ages and degrees of severity of dilated cardiomyopathy depending on genetic background.136–138 Heart and muscle specific knockout of SOD2 led to hypertrophy, dilatation, and reduced contractility, which were partially rescued by the SOD mimetic MnTBAP.139 In mice heterozygous for SOD2, though myocardial ultrastructural damage was not always observed, cardiomyocytes were shown to be sensitized towards PTP opening.140,141 Strikingly, in humans a genetic defect in SOD2 was associated with severe neonatal cardiomyopathy.142 Conversely, overexpression of SOD2 in mice increased ejection fraction.143 Experiments using the SOD mimetic mitoTEMPO to rescue heart failure phenotypes also provide good evidence that O2– is involved in pathology. In guinea pigs undergoing ascending aortic constriction/beta-adrenergic stimulation to induce nonischaemic heart failure, accompanied by SCD, mitoTEMPO reduced ROS production and prevented and reversed heart failure and SCD.144 Additionally, in mice, pressure overload via TAC induced decreased contractility, which improved with infusion with mitoTEMPO.145 Taken together, all these lines of evidence point to O2– contributing significantly to heart failure.
Mitochondrial targeted antioxidants such as MitoQ have been developed to reduce mitochondrial-generated ROS. In animal studies, MitoQ has shown great promise in reducing both I/R injury146,147 and heart failure.148–150 Although MitoQ showed no improvement over placebo in a patient study for Parkinson’s disease,151 a small human trial for hepatitis C did show a reduction in markers of liver damage in the MitoQ-treated patients.152 It should be mentioned that ROS also has signalling roles in the heart and that prolonged treatment with drugs such as MitoQ could interfere with beneficial ROS signalling. For example, it has been shown that antioxidants can block cardioprotective signalling, as will be discussed below.
Analogous to targeting SOD2 to focus on O2–, studies targeting PRDX3, or using mCAT have implicated H2O2 damage in multiple contexts. Transgenic expression of the rat Prx-3 gene in a mouse post-MI heart failure model attenuated MI-induced dilatation of the LV and contractile dysfunction.153 In a pressure overload mouse model using TAC, ROS and protein carbonylation increased and led to decline in contractility, which was partially rescued by expression of mitochondrial catalase.145,154 Taken together, PRDX3 or mCAT rescue of multiple heart failure models strongly demonstrate a causal role for H2O2 in pathologies resembling heart failure with reduced ejection fraction (HFrEF).
The beneficial effects of mitochondrial suppression of ROS are not limited to HFrEF. One study showed that mice fed a high-fat, high-sucrose (HFHS) diet developed cardiac diastolic dysfunction while maintaining systolic function, resembling conditions of heart failure with preserved ejection fraction (HFpEF). Expression of mCAT in transgenic mice ameliorated HFHS diet-induced diastolic dysfunction and prevented an increase in the rate of mitochondrial H2O2 production.155 Hence, while studies are scarcer, combating H2O2 may also prove to be therapeutically beneficial in HFpEF. In further contexts, mCAT has been shown to reduce cardiac disease in aged mice156 and a progeroid mouse model.157 Ageing seems to involve not only H2O2 but O2– as well, as mitoTEMPO also improves systolic and diastolic function in old mice158 and rats.159 Treatment with mitoTEMPO decreased oxidative stress, cell death, and hypertrophy, and led to improved function in diabetic cardiomyopathy.160 Conditions leading to oxidative stress also can lead to glycation of RyR94 that can contribute to the development of heart failure.
The results of SS-31 intervention in the context of heart failure have been more encouraging than the results in I/R. In animal experiments, SS-31 also improved outcomes in multiple heart failure models including TAC161 and angiotensin II treatment162 in mice and in a canine heart failure model.163 Recently, SS-31 appeared to improve cardiac function in a double-blind and placebo-controlled trial of human patients presenting with HFrEF.164 These results provide considerable motivation for moving SS-31 into the clinical setting, though challenges in translating promising results in animal studies to human patients remain.
Collectively these studies, by manipulating various antioxidant proteins, have demonstrated that ROS-mediated damage is a major contributor to the development of heart failure. Alongside these data, a series of studies has explored the molecular underpinnings of oxidative stress observed in heart failure.165 It was observed that in heart failure, rising cytosolic Na+ leads to decreased mitochondrial Ca2+, thus depressing Krebs cycle flux.100 As a result, because reducing NADP+ to NADPH depends on substrates derived from the Krebs cycle, the NADPH pool declines, inhibiting the regeneration of glutathione and thioredoxin, which are required for H2O2 elimination. Overall, the antioxidant capacity of mitochondria decreases, leading to oxidative stress. Furthermore, nicotinamide nucleotide transhydrogenase (NNT), which normally regenerates NADPH from NADH, was reported to work in reverse in conditions of increased afterload and insufficient mitochondrial Ca2+. The reversal of NNT to regenerate NADH for ATP production at the expense of ADP leads to oxidative stress, PTP activation, and cell and tissue damage.166 Overall, targeting the multiple pathways involved in increased ROS generation and decreased ROS scavenging in heart failure could be a promising therapeutic approach.
Despite the abundance of evidence for the negative effects of ROS in CVD, pushing to eliminate ROS entirely can also be harmful. Studies have shown that increasing glutathione pharmacologically or genetically can paradoxically increase oxidation and lead to cell death.167 This phenomenon known as reductive stress, in which excess reducing equivalents induce deleterious effects, has been linked to cardiomyopathy.168 Moreover, ROS could be essential signalling molecules in the heart as they are in other tissues,169 and heightened suppression of ROS could disrupt these signalling pathways. One study found that accumulation of damaged mitochondria, leading to heart failure, in mice lacking cardiac expression of Mitofusin 2 could be rescued by moderate levels of mCAT, but not high levels.170 High mCAT expression was not beneficial because it prevented ROS-mediated activation of autophagic mitochondrial quality control pathways.170 These data support the theory that ROS contribute to mitohormesis, a term referring to mild mitochondrial stresses than can induce a cytoprotective state that is beneficial in the long term.171 Blocking of mitohormetic responses could be a possible explanation for the lackluster results of global antioxidants in treating CVD.111 Indeed, in one study, vitamin E was associated with an increased risk of heart failure in patients with vascular disease or diabetes.172 Overall, although excess ROS are pathological, low levels of ROS can prevent reductive stress and stimulate mitohormesis, thereby providing functional value to the heart.
5. Summary and conclusion
The unique reliance of the heart on mitochondria for energy production puts an interconnected network of mitochondrial functions front and centre in cardiomyocyte health and disease. Mitochondria house multiple metabolic pathways, as well as the electron transport chain. Mitochondrial Ca2+ impinges on the Krebs cycle and the electron transport chain, and it also is a primary activator of the PTP. Electron transport generates ROS, which in addition to causing oxidative stress can also contribute to PTP activation. The regulation of all these mitochondrial pathways are critical for cardiomyocyte homeostasis, and thus it comes as no surprise that their dysregulation is associated with CVD, including I/R injury and heart failure. We have reviewed a growing number of studies that demonstrate the links between various mitochondrial functions and CVD, as well as some approaches that seek to target them for therapeutic benefit. The pace of research in this field promises innovative mitochondria-targeted interventions in CVD in the near future.
Funding
J.C. Liu was supported by K22HL137901 from the National Institutes of Health. EM was supported by the intramural program of NHLBI-NIH.
References
Author notes
Conflict of interest: None declared.

{kind=link}
{kind=link}
{kind=link}