





This editorial refers to ‘NOS1AP polymorphisms reduce NOS1 activity and interact with prolonged repolarization in arrhythmogenesis’ by C. Ronchi et al., pp. 472–483.
The long QT syndrome (LQTS) is a primary arrhythmic disorder that is characterized by a prolonged QT-interval on the surface electrocardiogram and an increased risk of sudden cardiac death that is typically precipitated by a polymorphic ventricular tachycardia known as torsade de pointes.1 LQTS has genetic and acquired forms that are exacerbated by a multitude of environmental stressors. At the tissue level, arrhythmias arising as a consequence of acquired or drug-induced LQTS share common electrophysiological mechanisms with their inherited channelopathy counterparts because they originate from alterations in the same ionic currents.2
Since the pioneering works of Keating et al.3 in the early 1990s, the molecular genetics of LQTS has come into focus. This has enabled the development of genetic screening strategies to identify patients at risk of LQTS-related sudden cardiac death. Despite this major advance, clinical decision-making processes associated with managing LQTS patients have remained highly challenging. Indeed, a major hurdle in the quest towards effective personalized medicine has been the poor ability to identify exactly whom amongst the carriers of a given LQTS mutation will succumb to sudden cardiac death and whom will go on to live a symptom-free life.4 As such, achieving a comprehensive risk assessment profile among genotype positive individuals to guide the application of ‘draconian’ measures in young individuals, such as implantable cardioverter-defibrillator implantation, is of utmost clinical importance.
The highly variable expressivity of heritable LQTS even among carriers within the same family emphasizes the multi-factorial nature of arrhythmias, even in their simplest form, when they are caused by a monogenic disorder such as LQTS. For one, extrinsic elements, such as environmental factors, stress, exercise, drugs, lifestyle, and acquired disease play a critical role in unmasking the arrhythmic phenotype of the primary channelopathy often in a gender-specific manner. Adding to this complexity, so-called modifier genes that act either synergistically with or in opposition to the primary LQTS causing mutations have gained considerable attention for their role as critical modulators of the arrhythmic substrate.4
Among those modifier genes, an unlikely candidate has emerged as a major predictor of QT-interval prolongation and associated arrhythmia. Specifically, NOS1AP, an adaptor protein of the neuronal isoform of the nitric oxide (NO) synthase (nNOS) was shown in genome-wide association studies to exhibit multiple common nucleotide variants that are highly associated with QT-interval duration even in the general population.4–6 A focused search in LQTS families ultimately yielded multiple NOS1AP variants that are highly predictive of the severity of the disease phenotype.7 Remarkably, not only did these variants predict pro-arrhythmic risk in familial LQTS, they were also found to correlate with drug-induced forms of the disorder.8 This highlights their potential relevance as universal modifiers of arrhythmia susceptibility in settings of QT-interval prolongation whether stemming from inherited channelopathies or acquired disease. Despite the strong genetic evidence linking NOS1AP variants to the severity of the LQTS phenotype, the underlying cellular and molecular mechanisms by which this neuronal adaptor protein promotes QT-interval prolongation and associated arrhythmogenesis have remained largely unknown.
In addition to its presence in the brain, NOS1AP is also expressed in the heart where it interacts with nNOS within specific cellular microdomains, including the sarcoplasmic reticulum (SR) and mitochondria to modulate myocyte function,6,9–11 but how? This important question has thus far been very difficult to answer particularly because studies investigating the effects of its main cellular target, nNOS, have yielded discrepant findings. While Sears et al.9 demonstrated that genetic nNOS knockdown or pharmacological inhibition increased myocyte contractility, L-type Ca current (ICaL) density, and SR Ca load, Wang et al.12 found contrary evidence of decreased contractility and impaired rate of Ca transient decay in ventricular myocytes from nNOS knockout mice, consistent with negative ionotropy and lusitropy. Similarly, studies using overexpression and knockdown strategies to modulate the myocardial levels of NOS1AP itself have also yielded discrepant findings. Chang et al.6 reported that adenoviral-mediated overexpression of NOS1AP in the guinea pig heart shortened action potential duration (APD) by inhibiting ICaL. In contrast, studies in zebrafish models by Milan et al.13 documented qualitatively similar APD shortening but this time in response to morpholino-mediated NOS1AP knockdown rather than overexpression.13 These species differences in the response of myocytes to altered nNOS and NOS1AP expression prompted thought leaders in this field to affirm the need for a human cardiomyocyte model system to uncover the mechanisms by which LQTS-linked NOS1AP variants promote arrhythmias.4
The case against social distancing between NOS1AP and its primary cellular target
Ronchi et al.14 used a multi-faceted approach to fill this critical knowledge gap by addressing these discrepancies. They combined studies in cardiomyocytes that were freshly isolated from guinea pig hearts with those that were differentiated from human inducible pluripotent stem cells (hiPSC-CM). In the former, they modelled drug-induced LQTS using pharmacological agents to block the slowly activating delayed rectifier K channels, while activating β-adrenergic receptors with isoproterenol. For the latter, they studied hiPSC-CM lines that were either differentiated from symptomatic LQTS patients harbouring a disease-causing minor NOS1AP allele or their asymptomatic major allele counterparts. Ronchi et al.14 hypothesized that loss of nNOS function in the cardiomyocyte subpopulation exacerbates the LQTS phenotype. Indeed, in their guinea pig myocyte studies, they found that pharmacological nNOS inhibition resulted in marked APD prolongation consistent with a cellular phenotype of LQTS. They also reported increased densities of ICaL and the late Na current along with enhanced predisposition to afterdepolarization-mediated triggers driven by SR Ca instability. Remarkably, these cellular arrhythmogenic triggers were reversed when a shorter action potential was forcefully imposed on the myocyte in action potential clamp recordings. This highlighted the synergistic interactions between Ca-mediated triggers and the electrophysiological substrate formed by APD prolongation in response to a unifying mechanism, namely nNOS inhibition.
Because previous studies in animal model systems yielded discrepant findings, the authors sought to verify some of their key observations in hiPSC-CMs.14 In concordance with their guinea pig myocyte studies, these authors elegantly demonstrated that cell lines from symptomatic patients exhibited longer APDs and greater ICaL likely driven by reduced expression of nNOS and its adaptor protein NOS1AP.14 This seemingly contradicted elegant observations of a negative correlation between QT-duration and NOS1AP transcript expression previously measured in myocardial samples obtained from extracted device leads.15 It is important to note, however, that the aforementioned study was not carried out in LQTS patients nor did it allow the assessment of NOS1AP expression specifically in the myocyte sub-population.15
Ronchi et al.14 further demonstrated that in addition to reduced expression, the colocalization between nNOS and its adaptor protein was reduced in ‘symptomatic’ hiPSC-CMs. This is particularly notable because the PDZ domain interaction between NOS1AP and nNOS has been shown to play a critical role in guiding the local subcellular distribution of nNOS, and therefore, the compartmentalization of NO signalling. Indeed, dual immunogold electron microscopy and co-immunoprecipitation assays have shown tight co-localization and interaction between NOS1AP with nNOS in critical subcellular loci, including the SR and mitochondria.10 Therefore, the ‘social distancing’ between nNOS and NOS1AP observed by Ronchi et al.14 may have caused aberrant trafficking or mistargeting of nNOS away from these cellular microdomains. This, in turn, may have promoted the arrhythmogenic triggers that these authors observed. The elegant work by Ronchi et al. reaffirms the ‘multi-hit’ nature of arrhythmias in LQT1. In this case, the arrhythmogenic phenotype of a mutation in the pore forming KCNQ1 subunit was unmasked by a minor loss-of-function NOS1AP variant which further prolonged repolarization and destabilized intracellular calcium cycling. As such, the destructive synergistic interactions between this LQTS causing mutation and a minor NOS1AP variant set the conditions needed for the arrhythmogenic triggers and substrate required for torsade de pointes (Figure 1).
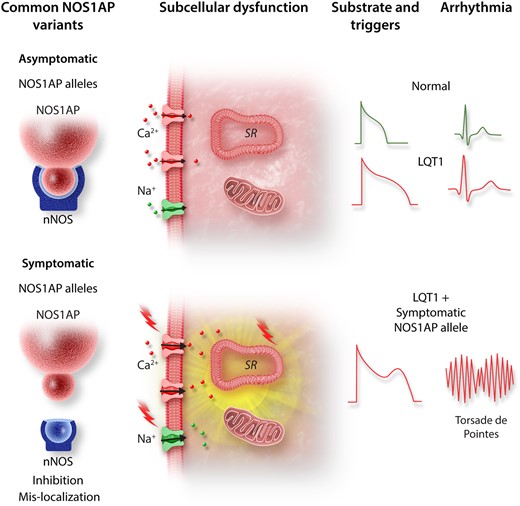
Schematic illustrating the proposed mechanism by which a minor NOS1AP allele unmasks the arrhythmogenic nature of a LQT1-causing KCNQ1 mutation. The minor NOS1AP variant exhibits reduced co-localization with its primary cellular target nNOS resulting in loss of nNOS function. This causes aberrant nNOS/NO signalling at key cellular compartments, thereby causing action potential prolongation and destabilization of intracellular calcium cycling. Combined, these loss of nNOS function effects form the triggers and substrate required for torsade de pointes.
As with any good study, the paper by Ronchi et al.14 leaves us with far more questions than answers. For example, to what extent does subcellular nNOS mistargeting alter fundamental myocyte properties including SR–mitochondria interactions, mitochondrial reactive oxygen species generation, and overall cardiac bioenergetics? Moreover, is the impact of NOS1AP variants on electrophysiological properties mediated exclusively by altered nitrosylation of ion channels and Ca cycling proteins or are other NO-dependent and independent mechanisms involved? Do LQTS-causing NOS1AP variants redistribute away from the SR to the plasma membrane to alter cellular repolarization by directly interacting with ion channel complexes? Ronchi et al.14 have left us with the motivation to ask these challenging but important questions as well as with a roadmap on how to do so.
Funding
This work was supported by National Institutes of Health grants R01 HL149344 and R01 HL137259 to FGA.
Conflict of interest: none declared.
The opinions expressed in this article are not necessarily those of the Editors of Cardiovascular Research or of the European Society of Cardiology.

{kind=link}