





Abstract
Pulmonary arterial hypertension (PAH) is estimated to affect between 10 and 50 people per million worldwide. The lack of cure and devastating nature of the disease means that treatment is crucial to arrest rapid clinical worsening. Current therapies are limited by their focus on inhibiting residual vasoconstriction rather than targeting key regulators of the cellular pathology. Potential disease-modifying therapies may come from research directed towards causal pathways involved in the cellular and molecular mechanisms of disease. It is widely acknowledged that targeting reduced expression of the critical bone morphogenetic protein type-2 receptor and its associated signalling pathways is a compelling therapeutic avenue to explore. In this review, we highlight the advances that have been made in understanding this pathway and the therapeutics that are being tested in clinical trials and the clinic to treat PAH.
1. Introduction
Pulmonary arterial hypertension (PAH) is a rare and devastating disease associated with progressive elevation of mean pulmonary arterial pressure (mPAP) (>20–25 mmHg) and pulmonary vascular resistance (PVR) (≥3 Wood Units), in the presence of pulmonary capillary wedge pressure ≤15 mmHg, often culminating in right heart failure and premature death.1,2 Patients present with shortness of breath and pre-syncope/syncope due to a progressive decline in right heart function. In the absence of any clinical intervention, life expectancy of individuals is ∼3 years after diagnosis.3,4 Classified as Group 1 pulmonary hypertension (PH) (of five groups), PAH may occur in a heritable and idiopathic context or associated with further co-morbidities [connective tissue disease (CTD); HIV; and portopulmonary hypertension (POPH)].3,5 The most established clinical risk factor is sex, with females twice more likely to develop PAH.6,7
Our understanding of the cellular changes and key regulators responsible for PAH pathobiology has improved greatly over the past four decades. Pathologically, PAH is characterized by remodelling of small peripheral arteries within the lung vasculature (Figure 1). During disease progression, obstruction of the small arterioles results in increased PAP, PVR, and progressive decline in function. Remodelling of the vascular bed occurs through aberrant smooth cell hyperproliferation and hypertrophy leads to neomuscularization of previously non-muscular arterioles; loss of normal endothelial function and expansion of apoptosis-resistant endothelial cells disrupting homeostatic control of the vessel wall and leading to impaired barrier function.8 Latter stages of the disease are associated with recruitment of inflammatory cells and abberant immune responses, plus progressive cellular disorganization may result in the formation of complex plexiform lesions.8–10
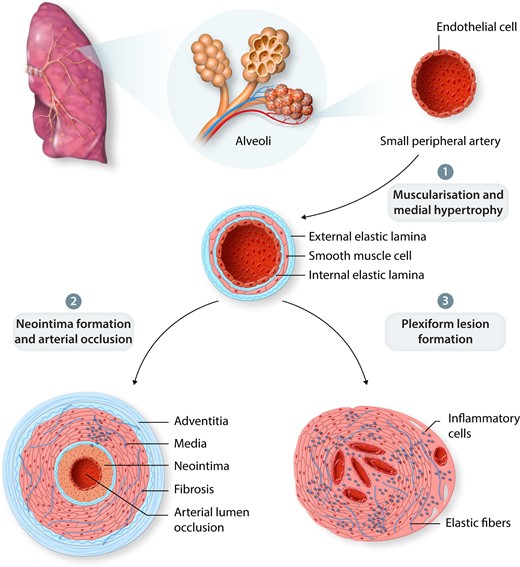
PAH Pathobiology. (1) Remodelling of small peripheral arteries. (2) Arterial lumen occlusion via neomuscularization of the arterial wall and neointimal formation. (3) Loss of normal endothelial function including vessel permeability may result in plexiform lesion formation and inflammatory cell recruitment.
Despite this knowledge, there is still debate over the initiation of PAH pathology. Multiple signalling pathways, vasoconstriction, oxidative stress, metabolic changes, inflammation, and dysregulated immunity are all implicated in PAH vascular remodelling. Promising therapeutic strategies targeting these pathways have been addressed recently in two comprehensive reviews.11,12 Current PAH pharmacotherapy can be divided into four classes aimed at reducing vasoconstriction: (i) phosphodiesterase inhibitors, including Sildenafil and Tadalafil, target the nitric oxide and cyclic guanosine monophosphate signalling pathways; (ii) the guanylate cyclase activator, Riociguat; (iii) endothelin receptor antagonists, including Bosentan, Ambrisentan and Macitentan; and (iv) prostacyclin and its analogues, including Iloprost, Epoprostenol, Treprostinil or the prostacyclin receptor agonist, Selexipag, which have potent vasodilatory activity by increasing cyclic adenosine monophosphate. However, it is worth noting that an acute vasodilator response is present in a minority of PAH patients.13 Previous meta-analyses demonstrated that targeting these vasodilatory pathways resulted in only moderate haemodynamic improvements and changes in mortality rates.14,15 Until recently only Epoprostenol has been reported to provide a clear survival benefit.13 Although, small pilot studies have suggested clinical benefit where triple combination of existing therapies has been prescribed.16,17 Nevertheless, there remains an unmet requirement for alternative therapies with disease-modifying potential.
In this review, we discuss the role of the transforming growth factor-β (TGFβ) superfamily member, bone morphogenetic protein receptor type-2 (BMPR2) in the development of PAH. Autosomal-dominant mutations in the gene encoding BMPR2 are causal of heritable PAH with mutations observed in ∼75% of familial cases, and 11–40% of idiopathic PAH.4,9,18–21 Furthermore, BMPR2 expression is also reduced in patients lacking mutations.22 BMPR2 is a serine/threonine kinase transmembrane receptor critical in the bone morphogenetic protein (BMP) signalling axis and essential for a number of key cellular processes including embryogenesis, development, and tissue homoeostasis. Multi-functional BMP signalling regulates differentiation, proliferation, and apoptosis in multiple tissues; disruption of which can lead to development of vascular diseases including PAH, hereditary haemorrhagic telangiectasia (HHT), and atherosclerosis.23 Therefore, BMPR2 deficiency or dysfunction suggests a central role in PAH pathogenesis. Thus, BMPR2 must be seen as an integral part of a complex disease and we discuss herein that treatment stratification based upon BMPR2 mutation status or downstream signalling may provide a personalized medicine approach.
2. The importance of BMPR2
2.1 BMPR2 and the BMP signalling axis
A total of 33 unique but homologous TGFβ, BMP, activin, growth and differentiation factors (GDF) and anti-mullerian hormone (AMH) ligands have been characterized and signal via heterodimeric complexes of two ‘type-1’ [activin-like kinase-1–7 (ALK1–7)] and two ‘type-2’ (activin receptor type-2a/b, ActR2a/b; BMPR2; TGFβ receptor type-2; AMH receptor type-2) receptors.24 Upon binding of a dimeric BMP complex, the constitutively active type-2 receptor forms an active heterotrimeric complex with a type-1 receptor.25 The type-2 receptor phosphorylates the type-1 receptor propagating the signal via phosphorylation of signalling intermediates initiating either canonical (small mothers against decapentaplegic; Smad-dependent) and non-canonical (Smad-independent) signalling.26 In the canonical pathway, activated BMP receptors mediate signalling proximal to the cell membrane, via C-terminal phosphorylation of the receptor-regulated Smads (R-Smads), Smad1, 5, and 8/9.27 In contrast, TGFβ receptor activation typically phosphorylates and activates Smad2/3. Upon activation, R-Smads form a complex in a 2:1 molecular ratio with the common partner Smad, Smad4.28 This active complex then translocates to the nucleus and associates with specific transcription factors on target promoters modulating the expression of downstream target genes including inhibitor of DNA binding proteins (IDs), SMAD6, SMAD7, apelin (APLN) and hairy/enhancer-of-split related with YRPW motif protein 1/2 (HEY1/2) (Figure 2A).29–32 BMP and TGFβ also transduce signals through non-canonical signalling pathways including the extracellular signal-regulated kinase (ERK), Janus kinase (JNK) and p38 mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K), and LIM kinase-1 (LIMK1) (Figure 2B).33–36 It is hypothesized that non-canonical pathways are important in diversifying/regulating BMP-induced canonical signalling pathways.37,38 Also, BMP signalling is fine-tuned by co-receptors (endoglin and betaglycan), endogenous secreted antagonists (gremlin, noggin, and chordin) modulating interactions between BMPs and their receptors, and is tightly regulated through inhibitory Smad proteins (Smad6/7) and signalling repressors, such as FK-binding protein-12 (FKBP12) (Figure 2C).38–40
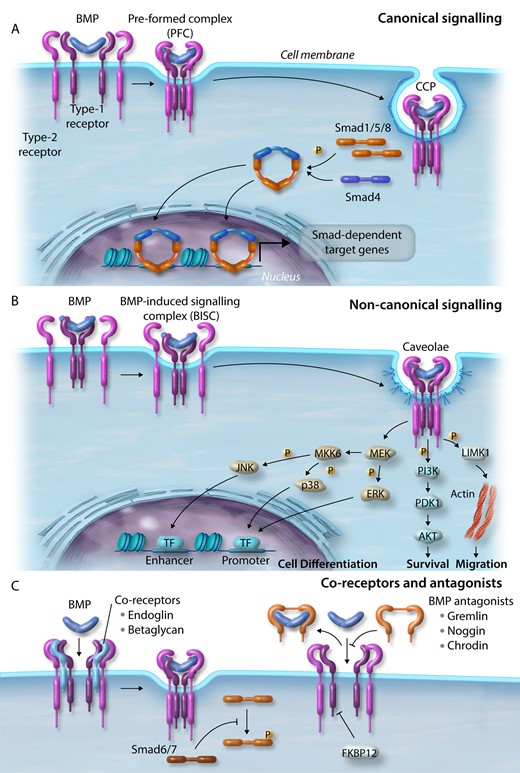
BMP signalling. (A) Canonical signalling. BMPs bind the complex of BMP type-1 and type-2 receptors and internalize via clathrin-coated pits. Activated receptors mediate signalling via phosphorylation of R-Smads (Smad1/5/8) close to the cell membrane and form a complex with Smad4. This active complex translocates to the nucleus and associates with transcription factors on target promoters. (B) Non-canonical signalling. BMPs bind to a high affinity type-1 receptor leading to subsequent recruitment of a type-2 receptor, which internalizes via caveolae to activate pathways regulating cell differentiation (ERK, JNK, and MAPK), survival (PI3K), and migration (LIMK1). (C) Co-receptors and BMP antagonists. Signalling is further regulated by co-receptors and endogenous secreted BMP signalling antagonists. Smad signalling is tightly regulated by inhibitory Smads (Smad6/7) and signalling repressors, such as FKBP12.
BMPs/GDFs typically bind type-1 receptors with a higher affinity than type-2 receptors, thus, expression of particular type-1/type-2 receptors dictate the selectivity of cellular responses to BMPs.41 Of the type-1 receptors, ALK1 is highly expressed by pulmonary artery endothelial cells (PAECs), ALK2 is expressed by both PAECs and pulmonary artery smooth muscle cells (PASMCs), whereas ALK3 is more restricted to PASMCs.42 Higher BMPR2 expression is observed on PAECs with lower levels expressed by PASMCs and interstitial cells.22,43 In the context of PAH pathobiology, we and others have reported that phospho-Smad1/5/8 and ALK3 expression are decreased in severe disease.43–45 With this in mind, the key ligand: receptor interactions likely to be important in maintaining pulmonary vascular homoeostasis in PAECs are BMP9/10 interacting with ALK1 (high affinity) or ALK2 (low affinity) and BMPR2/ActR2, with BMP6 interacting with ALK2: BMPR2. In PASMCs, BMP2/4 interact with ALK3: BMPR2 and BMP5/6/7 with ALK2: BMPR2.46 On a background of BMPR2 deficiency or dysfunction, normal signalling is attenuated with changes in receptor combinations potentially leading to aberrant cellular responses. For example, dysfunctional BMPR2 results in reduced BMP9/10 signalling causing increased apoptosis and reduced barrier integrity in the pulmonary endothelium.47–49 Furthermore, BMPR2 loss in PASMCs reduces BMP2/4 signalling and consequentially, gain-of-function signalling via BMP6 and ALK2: ActR2a.42,50,51
2.2 BMPR2 mutations and PAH
The importance of BMPR2 in the pulmonary vasculature has been exemplified in multiple human and animal studies and it is generally accepted that mutations and loss of signalling have a causal role in PAH. A recent meta-analysis of 1550 heritable, idiopathic, and anorexigen-associated PAH patients, 29% were identified as harbouring a BMPR2 mutation.4 Moreover, these patients were typically diagnosed at a younger age, demonstrated increased disease severity, higher mPAP, and increased PVR.4 Notably, these individuals were less responsive to acute vasodilator testing and were at an increased risk of death.4
Heterozygous germ-line mutations in BMPR2 represent the primary genetic risk factor in developing PAH.52 In 2009, 298 distinct BMPR2 mutations were identified with a subsequent study reporting a further 370 independent variants, and a recent case–control analysis of PAH index cases noted a further 80 novel mutations.10,18,52 Collectively, it is estimated that there are over 800 mutations, involving 486 distinct non-recurrent variants.53 Mutations in BMPR2 encompass all the major mutation classes, including missense mutations leading to amino acid substitutions (25%); nonsense mutations leading to nonsense-mediated decay (NMD) (27%); frame-shift mutations from nucleotide deletions or insertions (23%); gene rearrangements (14%); and splice-site variations (10%).53 Pathogenic mutations are observed in all major domains of BMPR2, spanning the extracellular ligand-binding, kinase, and cytoplasmic-tail domains.54 Clustering of mutations in certain regions appears to suggest key hotspots, with the majority of missense mutations localized to the ligand-binding domain or key catalytic regions of the kinase domain (exons 2–3, 6–9, and 11). In contrast, exons 1, 4, 10, and 13, encoding regions of unknown significance, have a relatively low mutation frequency.10 For example, most of the missense mutations within exons 2–4 of the extracellular domain (ECD) affect 10 cysteine residues, which form five disulphide bonds that are essential for the efficient tertiary folding of BMPR2.55 Of these, three disulphide bridges buried within the ECD structure are thought to control the folding rate.56 Therefore, mutation in any of these cysteines is likely to reduce folding efficiency leaving a free sulphydryl group to potentially interact with chaperone proteins of the endoplasmic reticulum (ER), thus reducing trafficking.55
In vitro studies of patient-derived PAECs, blood outgrowth endothelial cells (BOECs), and PASMCs show dysregulated canonical and non-canonical signalling pathways.27,29,30,44,47–50 Immunohistochemistry of BMPR2 mutation patient lung tissue shows decreased phospho-Smad1/5 and Id3 immunostaining, indicating an attenuation of downstream signalling.30,44 Intriguingly, BMPR2 expression is also reduced in the peripheral lung of IPAH patients lacking mutation and individuals with secondary PH, albeit to a lesser degree compared to those with mutations.22 Thus, highlighting the importance of BMPR2 in vascular homoeostasis as dysfunction is not only confined to the presence of a mutation. This is further emphasised in numerous animal studies and experimental models of PH. Although, homozygous Bmpr2−/− animals are embryonic lethal, heterozygous mice or animals harbouring a PAH causing mutation appear to develop a mild degree of PH.48,57,58 Moreover, we observed significantly reduced Bmpr2 mRNA, protein expression, and BMP signalling in experimental rat PH models.51,59,60 Furthermore, reduced Bmpr2 mRNA levels have also been reported in both chronic-hypoxia and Sugen-hypoxia (SuHx) treated mice.61
It is noteworthy that PAH-related mutations in genes other than BMPR2 have been identified, albeit at a lower frequency, in BMP signalling-related endothelial-specific proteins—ALK1, endoglin, BMP9, BMP10, and SMAD8/9.18,62–64 Consistent with attenuated BMP signalling in PAH resulting in a loss of homeostatic regulation, excessive levels of TGFβ and downstream signalling in human PAH and experimental models of the disease suggest a resulting imbalance within the superfamily.65–67 Collectively, the role of BMPR2 and BMP signalling is critical in the maintenance of normal pulmonary vascular homoeostasis.
Although BMPR2 mutations are the major genetic cause of PAH, mutations have a reduced penetrance (20–30%), which suggests that ‘second-hits,’ such as genetic modifiers, somatic events, environmental injury, or a combination of these contributes to disease.68 This review will discuss the factors that might affect penetrance by critically reducing BMPR2 expression and the potential role of this in PAH. We also discuss current findings in translational research targeted to therapeutically augment the BMPR2 pathway.
3. Targeting BMPR2
The multiple mechanisms by which pathological BMPR2 mutations cause impaired function or receptor deficiency underlying PAH have led to the exploration of potential therapeutics that target the pathway via multiple approaches. Indeed, currently available PAH treatments have been reported to impact upon BMP signalling. Previous studies showed that Sildenafil or prostacyclin analogues (Iloprost and Treprostinil) enhanced BMP signalling in PASMCs harbouring BMPR2 mutations and experimental rodent models of PAH in the absence of functional BMPR2.69,70 Although, they failed to rescue BMPR2 expression levels and it is unknown to what extent they impact the totality of downstream signalling.
Here, we discuss both novel and repurposed therapeutics aimed at restoring levels of BMPR2 and/or BMP signalling in a ‘nucleus to cell-surface’ approach dependent upon the site of action. Indeed, clinical phase assessment of many of these therapies will depend on careful stratification based on patient aetiology to ensure clinical benefit can be demonstrated, but may pave the way to a more personalized medicine approach. Figures 3 and 4 represent the mechanisms and treatments augmenting BMPR2 and BMP signalling discussed in this review.
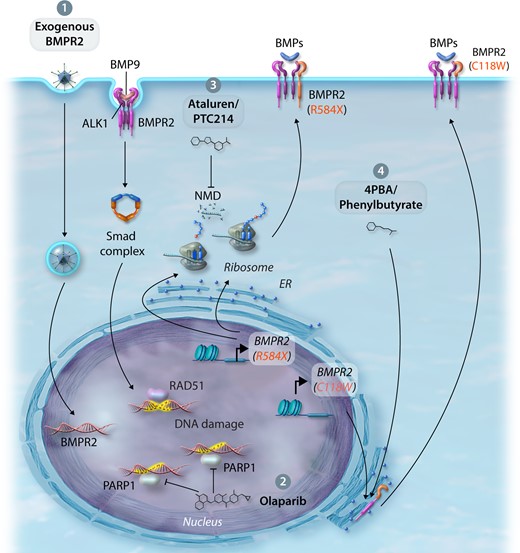
Therapeutic approaches to improve BMPR2 expression and/or BMP signalling. (1) Exogenous delivery—endothelial-targeted replacement of dysfunctional BMPR2 through viruses, exosomes, or nanoparticles. (2) Regulation of DNA damage—activation of RAD51 DNA damage repair or inhibition of PARP1 DNA repair by the small molecule inhibitor, Olaparib. (3) Promotion of translational read-through—suppression of nonsense-mediated mutations using Ataluren/PTC124 improving BMPR2 expression. (4) Rescue of BMPR2 cysteine ECD mutations from the ER—rescue BMPR2 cell-surface expression and signalling by treatment with 4PBA.
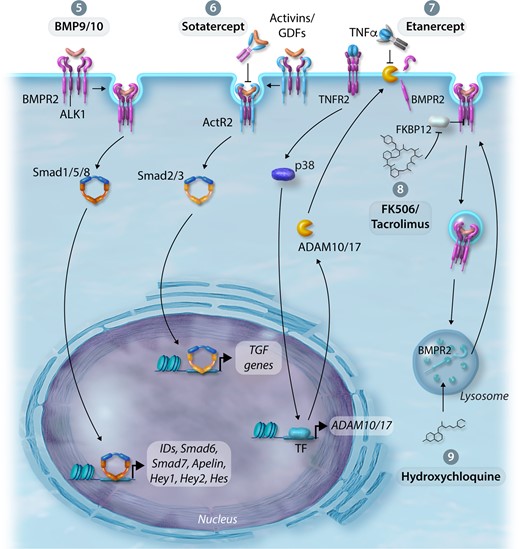
Therapeutic approaches to improve BMPR2 expression and/or BMP signalling. (5) Enhancement of BMPR2 by BMP9—increases BMPR2 expression levels, enhances downstream signalling, reduces endothelial apoptosis and permeability in the pulmonary circulation. (6) Activin/GDF Inhibition—attenuates TGFβ signalling by the activin ligand trap, Sotatercept. (7) Inhibition of TNFα-induced inflammation—ligand trap capture of TNFα by Etanercept prevents the reduction of BMPR2 expression by the sheddases ADAM10/17. (8) Inhibition of BMP signalling repression—treatment with FK506/Tacrolimus enhances BMP signalling in the context of BMPR2 deficiency. (9) Modulate BMPR2 expression by regulating lysosomal function and autophagy—treatment with hydroxychloroquine (lysosome and autophagy inhibitor) inhibits lysosomal acidification and prevents protein degradation, thus rescuing BMPR2 expression and signalling.
3.1 Nucleus
3.1.1 BMPR2 gene delivery
Replacing dysfunctional BMPR2 by delivery of exogenous wild-type receptor to the vascular cells affected in PAH was investigated using nebulized-intratracheal delivery of adenoviral BMPR2, and although targeted to the pulmonary arteries, it did not ameliorate PH in the monocrotaline-treated (MCT) rat model.71 Reynolds et al.72 adapted delivery by targeting an adenoviral-vector containing BMPR2 to the pulmonary endothelium using a bispecific-conjugated antibody to the highly expressed angiotensin-converting enzyme (ACE). In chronic-hypoxia and MCT-treated rat PH models, restored BMPR2 expression was accompanied by reduced right ventricular systolic pressure (RVSP), RV hypertrophy (RVH), and vascular remodelling, as well as increased cardiac output.72,73 Following MCT treatment, increased levels of TGFβ were attenuated by gene delivery of BMPR2.73 A subsequent study characterized the effects of endothelial-targeted adenoviral BMPR2 delivery on downstream signalling in both human cells and the MCT model. As expected, phospho-Smad1/5 was increased and phospho-Smad3 decreased in BMPR2-infected human pulmonary microvascular endothelial cells (PMECs).74 Similar to earlier studies, RVSP, RVH, and mPAP were significantly reduced by BMPR2 gene delivery in MCT-treated rats. This was associated with restored phospho-Smad1/5 and PI3K signalling along with reduced phospho-Smad3 and MAPK expression compared to the control vector.74
The limitations of targeting BMPR2 to the endothelium using viral vectors are two-fold. Firstly, adenoviruses induce only transient gene expression as the wild-type receptor is not incorporated into the host genome. Secondly, circumvention of this with stable, long-term expression using lentivirus potentially introduces unwanted, deleterious mutations upon integration. With this in mind, the same group proposed a pre-clinical cell-based study using bone-marrow-derived endothelial-like progenitor cells (BM-ELPC) isolated from the femurs of rats and transduced with adenoviral BMPR2.75 Intravenously administered BM-ELPCs localized to the lung within 1–6 h and associated with significantly elevated phospho-Smad1/5/8.75 However, retention time was short and target engagement appeared to dissipate within 24 h. As shown previously, exogenous delivery of BMPR2 reduced haemodynamics, hypertrophy, and vascular remodelling in MCT-treated rats. The authors suggest that BMPR2-expressing BM-ELPCs release exosomes containing BMPR2, which could have a protective effect on the pulmonary vasculature.76 The use of BM-ELPCs avoids potential off-target effects when using viral delivery, and transplantation could be conducted on a regular basis. Moreover, isolated and purified exosomes or engineered BMPR2-expressing nanoparticles could be developed as potential therapies.
3.1.2 DNA Damage
The link between BMPR2 expression and DNA damage is an intriguing phenomenon. Endothelial cells play a key role in maintaining normal vascular homoeostasis, though their proximal location often leaves them exposed to pathological factors that induce vascular injury including shear stress, reactive oxygen species, and inflammatory cytokines present in the bloodstream.77 Hence, the integrity of blood vessels is reliant upon endothelial repair mechanisms after exposure to acute insults. However, these may become impaired during chronic exposure as failure in DNA damage repair can lead to mutations and chromosomal rearrangements.77 PAECs of vascular lesions observed in PAH patients have exaggerated mutations and microsatellite instability in genes controlling proliferation and apoptosis, as well as somatic chromosomal abnormalities.78
More directly, DNA damage sensitivity in PMECs from BMPR2-deficient IPAH patients was associated with decreased expression of breast cancer type-1 susceptibility protein (BRCA1) and DNA topoisomerase II-binding protein; well-characterized DNA damage sensors.79,80 Moreover, repair of highly damaging double-stranded DNA breaks via homologous recombination involves RAD51, which interacts with BRCA1.81 Vattulainen-Collanus et al.82 showed that BMPR2 depletion was associated with decreased RAD51 expression and increased DNA damage; and BMPR2 knockdown or chemical-induced DNA damage in PMECs was accompanied by reduced BRCA1 and RAD51 expression. Furthermore, BMPR2 was essential for maintaining RAD51 expression and genomic integrity as PMECs were more sensitive to chemical-induced DNA damage after BMPR2 knockdown.82 Crucially, BMP9 restored BMPR2 and RAD51 protein expression and decreased the number of double-stranded DNA breaks.82 The potential for BMP9 as a therapeutic in PAH is multi-factorial and discussed further in this review. Finally, inhibitory targeting of the small non-coding microRNA-96 (miR-96), which is normally suppressed by BMPR2, was proposed as a potential therapeutic intervention. PMECs transfected with a miR-96-mimic exhibited reduced RAD51 expression and increased DNA damage.82 In PMECs deficient in BMPR2 expression, treatment with an anti-miR96 partially restored RAD51, but had no effect on BMPR2 protein levels.82
Restoration of DNA damage repair pathways in PAH might not be appropriate on a global scale. Distal PAs and PASMCs from PAH patients exhibit increased DNA damage and poly(ADP-ribose)polymerase-1 (PARP1) expression.83 Ordinarily, PARP1 actively contributes to DNA repair but in some contexts is considered tumourigenic.84–86 In this context, PARP1 may propagate NFκB-mediated inflammatory responses and secretion of potentially pro-tumourigenic cytokines.87 In fact, cells with activated PARP1 produce high levels of interleukin-6 (IL6), an inflammatory cytokine implicated in PAH.88,89 Meloche et al.83 proposed that the PARP inhibitor, ABT-888, reversed PH in two experimental PH models. This research has translated into a phase-1b trial using the selective PARP1 inhibitor, Olaparib with results expected shortly (ClinicalTrials.gov identifier NCT03782818). Therefore, understanding the balance of DNA damage and repair will be important in the PAH context.
3.2 Ribosome
3.2.1 Ataluren/PTC124—promoting translational read-through
Around 75% of BMPR2 mutations causal in PAH are heterozygous nonsense mutations resulting from frame-shift deletions or insertions. Nonsense mutations introduce premature translation codons (PTC) into the DNA sequence and the unstable mutant mRNA transcript is rapidly removed by NMD; leading to complete absence of mutant protein.90 The lack of two functional alleles results in haploinsufficiency where translation of the remaining functional allele is inadequate for proper function. Interestingly, ∼12% of all known disease-causing mutations in the Human Gene Mutation Database are nonsense mutations.91 However, the functional impact of the PTC can be difficult to predict with variants close to the 3' end of a transcript predicted to avoid NMD.92 A large-scale RNA-seq study suggested that ∼68% of variants predicted to cause NMD actually escape RNA surveillance.93
Proof-of-concept studies aimed at correcting nonsense mutations by inducing read-through of PTCs have focussed on the treatment of cystic fibrosis (CF) and Duchenne muscular dystrophy (DMD) using repurposed aminoglycosides.94–96 Aminoglycosides, a family of antibiotics including gentamicin, overcome PTCs by binding to ribosomal RNA, incorporating an extra amino acid and generating a full-length protein.97 Significantly, normal translation remains unaffected due to the presence of regulatory sequences around the normal termination codon.98 In a nonsense BMPR2 mutation model, gentamicin successfully initiated translation, but the risk of renal and otic toxicities due to the requirement for high doses precludes its use.99 A high-throughput screen of compounds reported to suppress nonsense mutations identified Ataluren (PTC124), a non-aminoglycoside oxadiazole that promotes read-through without these side effects.100 Clinical trials in CF and DMD, initially showed promising efficacy and a good safety profile.101,102 Moreover, Drake et al.103 investigated the ability of PTC124 to improve BMPR2 expression, BMP-induced signalling, and reverse proliferation in vascular cells isolated from PTC-mutant BMPR2 patients. In support of these findings, we undertook a pre-clinical study of PTC124 in PAH-patient BOECs and a knock-in mouse-model both harbouring a nonsense BMPR2 mutation (R584X).104 PTC124 treatment increased both BMPR2 protein expression and ID1 signalling in the mutant BOECs.104 The increased endothelial vascular permeability, apoptosis, and proliferation associated with BMPR2 mutations was also significantly reduced by PTC124.104 Furthermore, oral delivery of PTC124 increased lung BMPR2 protein expression and reduced hyperproliferation in PASMCs isolated from mice harbouring the R584X mutation.104 However, it is noteworthy that PTC124 could not rescue BMPR2 expression or BMP signalling in all PTC mutants. Further supporting the need for future pre-clinical studies, examining the feasibility of treating specific nonsense BMPR2 mutation patients with PTC124, before the drug is considered therapeutically beneficial.
3.3 Endoplasmic reticulum
3.3.1 Sodium 4-phenylbutyrate/4PBA—chemical chaperones
Missense mutations in BMPR2 constitute 25% of cases, which in the context of disease are deleterious when occurring in conserved domains or regions of the protein leading to loss of function.10,52–54 We reported that missense mutations causing substitutions of ECD cysteine residues resulted in the retention of BMPR2 in the ER, thus reducing the trafficking of the receptor to the cell-surface and consequently, reducing Smad signalling.105,106 Furthermore, the ECD C118W missense mutation in BMPR2 causes co-retention of type-1 receptors in the ER as part of a functional receptor complex, thus exerting a dominant-negative effect on BMP signalling.106 Small molecule chemical chaperones, such as sodium 4-phenylbutyrate (4PBA), thapsigargin, glycerol, and tauroursodeoxycholic acid have been proposed to restore BMPR2 trafficking to the cell-surface and enhance signalling in human and mouse studies.106,107
In particular, 4PBA has emerged as a promising candidate. Principally used for the treatment of urea cycle disorders as an ammonia scavenger it was proposed as a treatment for CF, as the most common mutation in the cystic fibrosis transmembrane regulator results in retention in the ER.108–110 We previously demonstrated that cell lines transiently or stably expressing the C118W BMPR2 mutation were defective in trafficking and signalling, but could be corrected by 4PBA treatment.106 Moreover, as cysteine residues of the ECD are located in exons 2–4, Frump et al.107 investigated the effects of 4PBA on PMECs isolated from a mouse-model possessing an in-frame deletion of exon 2 in Bmpr2. As observed in our studies, 4PBA rescued defective BMPR2 trafficking and downstream signalling. We subsequently showed that 4PBA rescues dysfunctional BMP signalling in human dermal fibroblasts isolated from individuals harbouring the C118W mutation, and the hyperproliferative phenotype of PASMCs isolated from heterozygous C118W mice.111 Mutant animals treated with 4PBA exhibited increased BMPR2 expression, corrected downstream signalling, and improved vascular remodelling.111 Further supporting evidence is observed in two animal models of PH where 4PBA treatment improved haemodynamics and vascular remodelling.112,113 The above evidence provides proof-of-concept for restoration of BMPR2 expression, trafficking, and signalling. Therefore, 4PBA represents an example of drug-repurposing and precision medicine for patients with mutations in cysteine residues of the BMPR2 ECD.
3.4 Cell membrane
3.4.1 BMP9/10—receptor and signalling rescue
In vitro studies using PASMCs showed that increasing concentrations of BMP ligand could overcome BMP signalling deficiency and induced growth suppression in the presence of a BMPR2 mutation.44 However, given the number of diverse BMP ligands and receptors, it was unclear which BMP ligand signalling would be most disrupted by BMPR2 mutation. This is unsurprising given the multiple receptor: ligand combinations, the range of target cell types and the pleiotropic effects of many BMPs. It was subsequently discovered that BMP9 and BMP10 specifically induce a BMPR2/ActR2: ALK1 signalling complex in endothelial cells.114,115 Also, unlike the local tissue expression of many BMP ligands, BMP9 (primarily secreted by the liver) and BMP10 (secreted by the right atrium) circulate at active concentrations in the bloodstream regulating endothelial cell function.48,116–119
This section predominantly focuses upon BMP9, but the findings might also apply to BMP10 given their similarities in activity.118 In PAECs, siRNA ablation of either BMPR2 or ACVR2A (activin receptor type-2a) was insufficient to impact BMP9-induced Smad1/5 phosphorylation or downstream transcriptional responses.27 However, knockdown of both receptors completely abolished phospho-Smad1/5 and ID1/2 expression in the presence of BMP9.27 Interestingly, only BMPR2 perturbation affected BMP9-induced E-selectin and interleukin-8 transcription implying that BMPR2 may serve a specific transcriptional programme relevant to PAH.27 Also, BMP9 does not alter endothelial ACVR2A or ACVRL1 (ALK-1) expression, but induces BMPR2 expression, suggesting plasma BMP9/10 shifts the balance of signalling in the endothelium towards BMPR2.27,48,114
As a vascular quiescence factor noted for inhibiting angiogenesis, the therapeutic effects of BMP9 in PAH were examined.115,117 In BMPR2-deficient ECs, treatment with BMP9 prevented apoptosis, enhanced monolayer integrity, and in three experimental PH models, BMP9 rescued BMPR2 expression, downstream signalling, and reversed the disease phenotype.48 Further highlighting their importance, reduced plasma BMP9/10 levels have been reported in some PAH patients, both in the presence and absence of BMP9 mutations.18,116 Furthermore, BMP9/10 levels were decreased in human studies and rodent models of POPH and liver cirrhosis.120–122 Moreover, treatment with a BMP9/10 ligand trap, ALK1 ECD-Fc fusion protein (ALK1-Fc), induced a PH phenotype and vascular remodelling in hypoxic conditions.120
However, Tu et al.123 proposed that inhibition of BMP9 protected against the development of PAH. In the chronic-hypoxia PH mouse-model, BMP9 knockout animals and neutralizing BMP9 antibodies prevented PH progression, similarly in MCT or SuHx-treated rats ALK1-Fc attenuated the development of PH.123 The authors proposed that BMP9 played an important role in balancing vasoconstriction and vasodilation, as BMP9 knockout mice in chronic-hypoxia exhibited lower gene expression of the vasoconstrictor peptide, endothelin-1, and higher expression of vasodilators apelin and adrenomedullin.123 However, this study remains unexplained in the face of the genetic and clinical data available. One potential explanation is that BMP9 inhibition promotes an HHT-like phenotype in rodents that leads to a microvasculature that is hyporesponsive to vasoconstrictors and lacking mural cell coverage, as described in patients with HHT.62,124,125
3.4.2 ActR2a-Fc/Sotatercept—targeting aberrant BMP/activin/GDF signalling
Another potential target would be to address the imbalance between activin/GDF and BMP signalling. As members of the TGFβ superfamily, activin and GDF share many of the same receptors as BMPs and TGFβ, but preferentially activate Smad2/3 signalling.59 In human PAH, circulating levels of activin-A are increased with elevated levels reported in the lungs of mice with hypoxia-induced PH.126 Consistent with this study, activin-A neutralizing antibodies reduced the pro-proliferative phenotype of PAH PASMCs, suggesting an imbalance in favour of activin-A.127 In MCT-treated rat PASMCs, expression of ActR2a is enhanced while Bmpr2 expression is significantly reduced suggesting a relationship between these type-2 receptors.128 Elevated ActR2a expression may result from dysregulated responses as a consequence of BMPR2 reduction. Indeed, where BMPR2 insufficiency exists, ActR2a expression is substantially and abnormally induced by TNFα.51 Furthermore, ActR2a is the main type-2 receptor in activin and GDF8/11 signalling, but can also transduce BMP9 signalling in conjunction with ALK1.27
Recent studies highlight that targeting activin/GDF signalling using a ligand trap might be a viable treatment strategy in PAH. Yung et al.129 used the ActR2a ECD-Fc fusion protein (ActR2a-Fc/Sotatercept) in both in vitro and in vivo models of PH. Purportedly rebalancing activin: BMP, Sotatercept successfully inhibited activin and GDF8/11 activation of Smad2/3 and attenuated PASMCs and PMECs proliferation.129 Administration in established experimental PH models improved vascular remodelling and hemodynamics whilst reducing Smad2 signalling. However, this study provided no evidence for restoration of BMP signalling, because ActR2a-Fc did not restore the BMPR2: Smad1/5 axis in lung tissues or vascular cells.129
The clinical use of Sotatercept has been assessed in two open-label trials for the treatment of anaemia and multiple myeloma, and is apparently well-tolerated, at least for the duration of these studies.130–132 A phase-2 trial in PAH (PULSAR; ClinicalTrials.gov identifier NCT03496207) met its clinical end-points after 106 patients received either placebo, 0.3 or 0.7 mg/kg of Sotatercept every 3 weeks for a 24-week period. Treatment with Sotatercept reduced PVR (>18% reduction) and improved the 6MWD (>24 m) with 97 of the 106 patients now enrolled on a long-term phase-2a trial, SPECTRA (ClinicalTrials.gov identifier NCT03738150).
3.4.3 Etanercept—inflammation inhibits BMPR2
As discussed above, the ‘second-hit’ hypothesis is an important aspect of PAH pathogenesis given the low penetrance of mutations. Inflammation has long been considered a trigger for disease with inflammatory cells observed in plexiform lesions of PAH lungs.133 Several inflammatory mediators have been studied with respect to PAH, including macrophage inflammatory protein-1α, interleukin-1β, IL6, and the 5-lipoxygenase pathway.134–138 In fact, Bmpr2 heterozygous mice were more susceptible to the development of PAH when 5-lipoxygenase was overexpressed.139 In accordance, heightened circulating levels of inflammatory cytokines are observed in PAH patients correlating with poor survival.88 Of these cytokines, elevated IL6 correlates with adverse prognosis, and is implicated in pulmonary vascular remodelling in mice.88,140,141 Furthermore, overexpression of the IL6-receptor (IL6R) is associated with PASMC hyperproliferation and apoptosis inhibition, reversal of which was successfully achieved following treatment with a non-peptide IL6R antagonist, ERBF (20S, 21-epoxy-resibufogenin-3-formate), in animal models.142 Treatment with Tocilizumab, a potent IL6R-neutralizing antibody, was assessed in a phase-2 open-label trial for the treatment of PAH (TRANSFORM-UK; ClinicalTrials.gov Identifier: NCT02676947).143 Whilst adequately powered, treatment failed to meet primary and secondary clinical outcomes despite demonstrating target engagement.144 The authors noted a moderate signal in CTD-associated PAH, recently identified as having the highest serum IL6 levels of the PAH aetiologies, highlighting the need for patient stratification within the umbrella diagnosis of PAH.144,145
TNFα is also elevated in PAH.88 Transgenic mice overexpressing TNFα in the lung spontaneously develop PH, but unlike IL6, TNFα treatment has been shown to directly down-regulate BMPR2 mRNA and protein expression in ECs, and PASMCs where sheddases, ADAM10 and ADAM17 are implicated.51,146,147 Suppression of BMPR2 by TNFα modified BMP signalling, leading to a pro-proliferative state in the presence of BMP6 upon activation of the ALK2: ActR2a and NOTCH2 axis.51 TNFα inhibitors have been utilized in a variety of inflammatory diseases including rheumatoid arthritis (RA), psoriasis, psoriatic arthritis, inflammatory bowel disease, and heart failure.148–151 Etanercept, an FDA-approved TNFα receptor type-2 ECD-Fc fusion protein used in the treatment of RA and psoriasis, ameliorated vascular remodelling and reduced haemodynamics in MCT-treated rats, as well as lowering PVR in pigs with acute endotoxin-induced PH.152–154 Findings in our laboratory revealed that Etanercept reduced RVSP and vascular remodelling in the SuHx-treated rat PH model.51 This was associated with normalized BMPR2 protein expression, restored Smad signalling, and reduced NOTCH2 expression in the lungs of treated animals.51 As yet there are no clinical trials examining the effects of TNFα inhibitors in PAH, but a strong safety profile suggests that these could be investigated in the future.
3.5 Cytoplasm
3.5.1 Tacrolimus/FK506—indirect targeting via FKBP12 inhibition
In the search for novel therapeutic approaches that could activate BMPR2 signalling, Spiekerkoetter et al.155 performed a large-scale drug screen (3756 FDA-approved drugs and bioactive compounds) and identified the immunosuppressive drug tacrolimus (FK506) as an activator of BMP/SMAD signalling. FK506 had a dual mode of action independent of ligand activation, both promoting BMP signalling and abolishing the interaction between the BMPR type-1 receptor kinase domain and the endogenous intracellular BMP signalling repressor, FKBP12.155 In experimental PH rat models, low-dose FK506 therapy appeared to prevent disease and ameliorated PH in a mouse-model of Bmpr2 depletion in the endothelium.155 Complimentary findings in IPAH patient-derived PMECs demonstrated rescue of endothelial dysfunction, even in the presence of BMPR2 mutation or ablation.155 Interestingly, FK506 treatment increased ALK1 and endoglin gene and protein expression in ECs.156
On the basis of these findings, two clinical trials attempted to determine the efficacy of FK506 in PAH. First, a small case study suggested that low-dose FK506 showed improvements in three patients with end-stage PAH via stabilization of cardiac function and significantly improved BMPR2 gene expression.157 A phase-2a trial with a larger patient cohort (23 individuals) showed no dose response in any clinical end-points, and overall BMPR2 and ID1 levels were not statistically increased by FK506.158 However, sub-cohort analyses suggested that increased BMPR2 expression correlated with RV function in a subset of patients. In conclusion, the authors reported that low-dose FK506 (<2 ng/mL) treatment had a good safety profile but a large multi-centre trial is required in BMPR2-deficient PAH patients before conclusions on efficacy can be drawn.158
3.6 Lysosome
3.6.1 Hydroxychloroquine—regulating degradation and autophagy
Viral infections are implicated as potential aetiological agents representing a ‘second-hit’ in the development of PAH. Infection with the human herpesvirus-8 [Kaposi sarcoma-associated herpes virus (KSHV)], were thought to be associated with PAH, although these data were not replicated in other patient cohorts.159–161 KSHV expresses a membrane-associated RING E3 viral ubiquitin ligase, K5 and is known to preferentially infect ECs, reportedly ubiquitinating and degrading endothelium-specific receptors.162,163 Our own research revealed that KSHV infection of ECs down-regulated cell-surface BMPR2 through K5 ubiquitination of the membrane-proximal lysine in the cytoplasmic domain, resulting in lysosomal degradation that was sensitive to the lysosomal inhibitor, concanamycin-A.164
Chloroquine and hydroxychloroquine are therapeutic lysosomal inhibitors that have widely been used as malarial prophylaxis, and repurposed as anti-inflammatory therapies for RA and systemic lupus erythematosus (SLE).165 Treatment with chloroquine/hydroxychloroquine represents a dual action approach in the effort to increase BMPR2 signalling, as it not only prevents protein degradation but also inhibits autophagy due to increased lysosomal pH.166 Autophagy is increased in MCT-treated rats and is accompanied by decreased lung BMPR2 expression, increased PASMC proliferation, and inhibition of apoptosis, corresponding to a reduction in vascular remodelling.60 Treatment with chloroquine/hydroxychloroquine inhibited the development or progression of PH, rescued BMPR2 expression and reduced aberrant vascular cell responses.60 Chloroquine also prevented the degradation of p62, a key protein that is degraded in autophagy.60,In vitro, chloroquine significantly increased cell-surface BMPR2 expression in PAECs abrogating the rapid turnover of the receptor and rescuing expression following siRNA knockdown. Moreover, chloroquine restores BMPR2 cell-surface expression in BOECs isolated from patients with BMPR2 mutations and enhances BMP9-BMPR2 signalling targets in BMPR2-mutant ECs.167
Gomez-Puerto et al.168 confirmed many of the above findings using a BMPR2 HaloTag® system, demonstrating that inhibition of the lysosome in PAECs and PASMCs increased total and plasma-membrane BMPR2 expression. Intriguingly, the authors also delineated that BMPR2 heterozygosity and inflammation triggered autophagy, which in turn was associated with decreased BMPR2 expression levels. As inflammation represents a potential ‘second-hit’, treatment with hydroxychloroquine could provide further beneficial effects in inhibiting inflammatory pathways.46 As a note of caution, as with other cell-based manipulations, the consequence of off-target effects should be strongly considered and weighted against potential benefits. Therefore, understanding the molecular mechanisms underpinning BMPR2 turnover and degradation would allow the development of more specific pharmacological therapies.
Emerging research highlights the role of human E3 ligases in the regulation of BMPR2 ubiquitination and degradation. In a siRNA screen of NEDD4-like E3 ligases, knockdown of ITCH significantly increased BMPR2 protein expression in PAECs, however, whether this is a direct interaction remains to be elucidated.164 Similarly, the NEDD4-like E3 ligase, SMURF1 has been implicated in regulating TGFβ and BMP signalling.169 Murakami et al.170 proposed that SMURF1 not only targets Smad1/5 but also reduces BMPR2 protein levels. SMURF1 was significantly elevated in MCT and hypoxia-induced PH models and overexpression of BMPR2 appeared to be reduced by heterologous delivery of SMURF1.170 The same study reported that both proteasomal and lysosomal inhibitors elevated BMPR2 levels in an overexpression system.170 However, we would urge caution with interpretation of overexpression studies as we have observed that neither knockdown of SMURF1 nor inhibition of the proteasome affects endogenous levels of BMPR2.164,168
4. Conclusions
Although clear advances have been made in the treatment of PAH in the past 40 years, life expectancy is still markedly reduced in patients. Discovery of prostacyclin analogues and other vasodilator therapies, coupled with the establishment of expert centres, has improved the survival rate, but the need for therapies that directly target the underlying mechanisms and molecules are clearly needed. We maintain that targeting BMPR2, either through restoration or rebalancing of related signalling pathways, provides a highly validated drug target to develop new approaches (Figure 5). However, many of the therapeutic advances, we describe are still at the pre-clinical stage and require careful stratification of patient cohorts that will be benefited most in future clinical trials (Table 1).
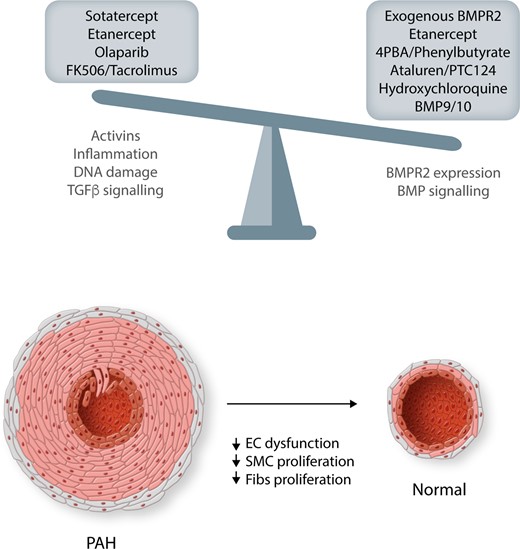
Maintaining the balance of BMPR2/BMP and TGFβ pathways is critical for normal vascular homoeostasis. Shifting the balance in disease in these pathways will be critical in reducing/reversing the vascular remodelling associated with PAH pathobiology. The central pathways targeted by the therapeutics in this review are detailed, but these may not be exclusive as many will have multiple benefits. EC, endothelial cells; SMC, smooth muscle cells; Fibs, fibroblasts.
Summary of proposed therapeutics targeting BMPR2 and BMP signalling in PAH
| Drug | Mechanism of action | Pre-clinical human studies | Pre-clinical animal studies | Route of admin | Clinical studies | Safety profile (adverse effects) |
|---|---|---|---|---|---|---|
| Ataluren/PTC124 (Translarna®) | Promotes translational read-through of premature stop codons in mutant BMPR2 | PAECs and BOECs103,104; Bmpr2 R584X PASMCs106 | Bmpr2 R584X mice104 | Oral | Cystic fibrosis101; Duchenne muscular dystrophy102 | |
| BMP9/10 | Promotes BMP signalling and increased BMPR2 expression | PAECs and BOECs27,48 | MCT rats, SU-5416 hypoxia rats and Bmpr2 R899X mice48 | |||
| Etanercept (Embrel®) | Inhibits TNF-alpha-induced inflammation and BMPR2 down-regulation by acting as a TNF-alpha ligand trap | PAECs and PASMCs51 | MCT rat153; endotoxaemic pigs154; SU-416 hypoxia rats51 | SC injection | RA149; heart failure151 | |
| Exogenous BMPR2 | Delivery of wild-type BMPR2 via virus, exosomes or nanoparticles | NMuMGsa and HUVECsb; 72 PMECs74; BM-ELPCs75 | Chronic-hypoxia rats72; Chronic-hypoxia and MCT rats73; MCT rats74,75 | |||
| Hydroxychloroquine sulfate (Plaquenil®) | Prevents lysosomal degradation of BMPR2 | PASMCs and PAECs60; PAECs and BOECs167; PAECs, PASMCs and HMEC-1sc, 168 | MCT rats60 | Oral | Malaria, RA, SLE165 (narrow safety margin) | |
| Olaparib (Lynparza®) | Inhibits PARP1-induced DNA repair in the absence of BRCA1 expression | PASMCs83 | MCT rats and SU-5416 hypoxia rats83 | Oral | Olaparib for PAH— NCT03782818 (Phase-1b) | Ovarian cancer171 (severe anaemia) |
| Sodium 4-phenylbutyrate/4PBA or glycerolf phenylbutyrate (Ravicti®) | Releases endoplasmic reticulum trapped BMPR2 | HeLas and MRC-5sd; 106 Bmpr2 ΔEx2 PMECs107; BMPR2 C118W HDFse and Bmpr2 C118W PASMCs111 | Bmpr2 ΔEx2 mice107; Bmpr2 C118W mice111; chronic-hypoxia mice112; chronic-hypoxia mice and MCT rats113 | Oral | Urea cycle disorders173 | |
| Sotatercept (ACE-011) | Blocks TGF-beta signalling by acting as an activin ligand trap | PMECs and PAMSCs129 | MCT rats and SU-5416 hypoxia rats129 | SC injection | PULSAR— NCT03496207 (Phase-2) SPECTRA— NCT03738150 (Phase-2a) | Anaemia and multiple myeloma130–132 |
| Tacrolimus/FK506 | Restores BMP signalling by prevention of FKBP12 signalling suppression | PAECs155 | MCT rats, SU-5416 hypoxia rats and EC-Bmpr2−/− mice | Oral | Low-dose FK506 in end-stage PAH157; TransformPAH— NCT01647945 (Phase-2a)158 | Safety and tolerability trial for PAH158 |
| Drug | Mechanism of action | Pre-clinical human studies | Pre-clinical animal studies | Route of admin | Clinical studies | Safety profile (adverse effects) |
|---|---|---|---|---|---|---|
| Ataluren/PTC124 (Translarna®) | Promotes translational read-through of premature stop codons in mutant BMPR2 | PAECs and BOECs103,104; Bmpr2 R584X PASMCs106 | Bmpr2 R584X mice104 | Oral | Cystic fibrosis101; Duchenne muscular dystrophy102 | |
| BMP9/10 | Promotes BMP signalling and increased BMPR2 expression | PAECs and BOECs27,48 | MCT rats, SU-5416 hypoxia rats and Bmpr2 R899X mice48 | |||
| Etanercept (Embrel®) | Inhibits TNF-alpha-induced inflammation and BMPR2 down-regulation by acting as a TNF-alpha ligand trap | PAECs and PASMCs51 | MCT rat153; endotoxaemic pigs154; SU-416 hypoxia rats51 | SC injection | RA149; heart failure151 | |
| Exogenous BMPR2 | Delivery of wild-type BMPR2 via virus, exosomes or nanoparticles | NMuMGsa and HUVECsb; 72 PMECs74; BM-ELPCs75 | Chronic-hypoxia rats72; Chronic-hypoxia and MCT rats73; MCT rats74,75 | |||
| Hydroxychloroquine sulfate (Plaquenil®) | Prevents lysosomal degradation of BMPR2 | PASMCs and PAECs60; PAECs and BOECs167; PAECs, PASMCs and HMEC-1sc, 168 | MCT rats60 | Oral | Malaria, RA, SLE165 (narrow safety margin) | |
| Olaparib (Lynparza®) | Inhibits PARP1-induced DNA repair in the absence of BRCA1 expression | PASMCs83 | MCT rats and SU-5416 hypoxia rats83 | Oral | Olaparib for PAH— NCT03782818 (Phase-1b) | Ovarian cancer171 (severe anaemia) |
| Sodium 4-phenylbutyrate/4PBA or glycerolf phenylbutyrate (Ravicti®) | Releases endoplasmic reticulum trapped BMPR2 | HeLas and MRC-5sd; 106 Bmpr2 ΔEx2 PMECs107; BMPR2 C118W HDFse and Bmpr2 C118W PASMCs111 | Bmpr2 ΔEx2 mice107; Bmpr2 C118W mice111; chronic-hypoxia mice112; chronic-hypoxia mice and MCT rats113 | Oral | Urea cycle disorders173 | |
| Sotatercept (ACE-011) | Blocks TGF-beta signalling by acting as an activin ligand trap | PMECs and PAMSCs129 | MCT rats and SU-5416 hypoxia rats129 | SC injection | PULSAR— NCT03496207 (Phase-2) SPECTRA— NCT03738150 (Phase-2a) | Anaemia and multiple myeloma130–132 |
| Tacrolimus/FK506 | Restores BMP signalling by prevention of FKBP12 signalling suppression | PAECs155 | MCT rats, SU-5416 hypoxia rats and EC-Bmpr2−/− mice | Oral | Low-dose FK506 in end-stage PAH157; TransformPAH— NCT01647945 (Phase-2a)158 | Safety and tolerability trial for PAH158 |
SC, subcutaneous; RA, rheumatioid arthritis; SLE, systemic lupus erythematosus.
NMuMGs—non-transformed mouse mammary gland epithelial cells.
HUVECs—human umbilical vein endothelial cells.
HMEC-1s—human dermal microvascular endothelial cells.
MRC-5s—human lung fibroblasts.
HDFs—human dermal fibroblasts.
Glycerol phenylbutyrate has replaced sodium phenylbutyrate in the treatment of urea cycle disorders, as it is supplied as a tasteless/colourless liquid rather than powder, which has to be mixed into patient food.
Summary of proposed therapeutics targeting BMPR2 and BMP signalling in PAH
| Drug | Mechanism of action | Pre-clinical human studies | Pre-clinical animal studies | Route of admin | Clinical studies | Safety profile (adverse effects) |
|---|---|---|---|---|---|---|
| Ataluren/PTC124 (Translarna®) | Promotes translational read-through of premature stop codons in mutant BMPR2 | PAECs and BOECs103,104; Bmpr2 R584X PASMCs106 | Bmpr2 R584X mice104 | Oral | Cystic fibrosis101; Duchenne muscular dystrophy102 | |
| BMP9/10 | Promotes BMP signalling and increased BMPR2 expression | PAECs and BOECs27,48 | MCT rats, SU-5416 hypoxia rats and Bmpr2 R899X mice48 | |||
| Etanercept (Embrel®) | Inhibits TNF-alpha-induced inflammation and BMPR2 down-regulation by acting as a TNF-alpha ligand trap | PAECs and PASMCs51 | MCT rat153; endotoxaemic pigs154; SU-416 hypoxia rats51 | SC injection | RA149; heart failure151 | |
| Exogenous BMPR2 | Delivery of wild-type BMPR2 via virus, exosomes or nanoparticles | NMuMGsa and HUVECsb; 72 PMECs74; BM-ELPCs75 | Chronic-hypoxia rats72; Chronic-hypoxia and MCT rats73; MCT rats74,75 | |||
| Hydroxychloroquine sulfate (Plaquenil®) | Prevents lysosomal degradation of BMPR2 | PASMCs and PAECs60; PAECs and BOECs167; PAECs, PASMCs and HMEC-1sc, 168 | MCT rats60 | Oral | Malaria, RA, SLE165 (narrow safety margin) | |
| Olaparib (Lynparza®) | Inhibits PARP1-induced DNA repair in the absence of BRCA1 expression | PASMCs83 | MCT rats and SU-5416 hypoxia rats83 | Oral | Olaparib for PAH— NCT03782818 (Phase-1b) | Ovarian cancer171 (severe anaemia) |
| Sodium 4-phenylbutyrate/4PBA or glycerolf phenylbutyrate (Ravicti®) | Releases endoplasmic reticulum trapped BMPR2 | HeLas and MRC-5sd; 106 Bmpr2 ΔEx2 PMECs107; BMPR2 C118W HDFse and Bmpr2 C118W PASMCs111 | Bmpr2 ΔEx2 mice107; Bmpr2 C118W mice111; chronic-hypoxia mice112; chronic-hypoxia mice and MCT rats113 | Oral | Urea cycle disorders173 | |
| Sotatercept (ACE-011) | Blocks TGF-beta signalling by acting as an activin ligand trap | PMECs and PAMSCs129 | MCT rats and SU-5416 hypoxia rats129 | SC injection | PULSAR— NCT03496207 (Phase-2) SPECTRA— NCT03738150 (Phase-2a) | Anaemia and multiple myeloma130–132 |
| Tacrolimus/FK506 | Restores BMP signalling by prevention of FKBP12 signalling suppression | PAECs155 | MCT rats, SU-5416 hypoxia rats and EC-Bmpr2−/− mice | Oral | Low-dose FK506 in end-stage PAH157; TransformPAH— NCT01647945 (Phase-2a)158 | Safety and tolerability trial for PAH158 |
| Drug | Mechanism of action | Pre-clinical human studies | Pre-clinical animal studies | Route of admin | Clinical studies | Safety profile (adverse effects) |
|---|---|---|---|---|---|---|
| Ataluren/PTC124 (Translarna®) | Promotes translational read-through of premature stop codons in mutant BMPR2 | PAECs and BOECs103,104; Bmpr2 R584X PASMCs106 | Bmpr2 R584X mice104 | Oral | Cystic fibrosis101; Duchenne muscular dystrophy102 | |
| BMP9/10 | Promotes BMP signalling and increased BMPR2 expression | PAECs and BOECs27,48 | MCT rats, SU-5416 hypoxia rats and Bmpr2 R899X mice48 | |||
| Etanercept (Embrel®) | Inhibits TNF-alpha-induced inflammation and BMPR2 down-regulation by acting as a TNF-alpha ligand trap | PAECs and PASMCs51 | MCT rat153; endotoxaemic pigs154; SU-416 hypoxia rats51 | SC injection | RA149; heart failure151 | |
| Exogenous BMPR2 | Delivery of wild-type BMPR2 via virus, exosomes or nanoparticles | NMuMGsa and HUVECsb; 72 PMECs74; BM-ELPCs75 | Chronic-hypoxia rats72; Chronic-hypoxia and MCT rats73; MCT rats74,75 | |||
| Hydroxychloroquine sulfate (Plaquenil®) | Prevents lysosomal degradation of BMPR2 | PASMCs and PAECs60; PAECs and BOECs167; PAECs, PASMCs and HMEC-1sc, 168 | MCT rats60 | Oral | Malaria, RA, SLE165 (narrow safety margin) | |
| Olaparib (Lynparza®) | Inhibits PARP1-induced DNA repair in the absence of BRCA1 expression | PASMCs83 | MCT rats and SU-5416 hypoxia rats83 | Oral | Olaparib for PAH— NCT03782818 (Phase-1b) | Ovarian cancer171 (severe anaemia) |
| Sodium 4-phenylbutyrate/4PBA or glycerolf phenylbutyrate (Ravicti®) | Releases endoplasmic reticulum trapped BMPR2 | HeLas and MRC-5sd; 106 Bmpr2 ΔEx2 PMECs107; BMPR2 C118W HDFse and Bmpr2 C118W PASMCs111 | Bmpr2 ΔEx2 mice107; Bmpr2 C118W mice111; chronic-hypoxia mice112; chronic-hypoxia mice and MCT rats113 | Oral | Urea cycle disorders173 | |
| Sotatercept (ACE-011) | Blocks TGF-beta signalling by acting as an activin ligand trap | PMECs and PAMSCs129 | MCT rats and SU-5416 hypoxia rats129 | SC injection | PULSAR— NCT03496207 (Phase-2) SPECTRA— NCT03738150 (Phase-2a) | Anaemia and multiple myeloma130–132 |
| Tacrolimus/FK506 | Restores BMP signalling by prevention of FKBP12 signalling suppression | PAECs155 | MCT rats, SU-5416 hypoxia rats and EC-Bmpr2−/− mice | Oral | Low-dose FK506 in end-stage PAH157; TransformPAH— NCT01647945 (Phase-2a)158 | Safety and tolerability trial for PAH158 |
SC, subcutaneous; RA, rheumatioid arthritis; SLE, systemic lupus erythematosus.
NMuMGs—non-transformed mouse mammary gland epithelial cells.
HUVECs—human umbilical vein endothelial cells.
HMEC-1s—human dermal microvascular endothelial cells.
MRC-5s—human lung fibroblasts.
HDFs—human dermal fibroblasts.
Glycerol phenylbutyrate has replaced sodium phenylbutyrate in the treatment of urea cycle disorders, as it is supplied as a tasteless/colourless liquid rather than powder, which has to be mixed into patient food.
The mechanisms for the therapeutic delivery of exogenous BMPR2 require refinement. In particular, the potential to generate mutations upon integration by the viral delivery system must be minimized, with a further drawback being the short duration of transgene expression. Promisingly, improvements in recombinant vector technology have provided an increase in transient gene expression and no discernible long-term side effects.172 Transplantation of BM-ELPCs or delivery of exosomes with augmented BMPR2 expression provides a promising approach of directly targeting the endothelium. Alternatively, nanoparticles that specifically target lung endothelium using antibodies to specific proteins, such as ACE, represent a potential delivery system.173
Detailed patient genotyping/phenotyping will help in matching patients with the appropriate pre-clinical study. This will be particularly important in the use of PTC124 or 4PBA where targeting of specific mutations in BMPR2 will likely provide the most benefit. The safety profiles and tolerability of any therapy will also dictate use and efficacy. Careful consideration applies in the use of FK506, an immunosuppressive drug used after allogeneic organ transplant, as the efficacy of a low-dose was unremarkable in a recent phase-2a trial in PAH. However, target engagement of BMPR2 was reported in a subset of patients and correlated with improved RV function, again highlighting the need for appropriate patient cohort identification. When administering hydroxychloroquine, careful profiling and long-term monitoring will be required to mitigate any non-specific off-target effects. However, as the drug is approved for the long-term treatment of RA and SLE, a wealth of knowledge is available. Furthermore, 4PBA will most likely be replaced by treatment with glycerol phenylbutyrate (Ravicti) as this has been shown to be better tolerated in urea cycle disorder patients.174 Recently, a phase-2a randomized placebo-controlled trial was approved in the UK with the aim of assessing both hydroxychloroquine and phenylbutyrate treatment in BMPR2 target engagement and disease modification. The trial will follow a Bayesian stratified adaptive design providing a personalized approach in treatment.
With the widespread adoption of genetic testing in PAH patients, increasing emphasis in clinical trials will be on whether PAH patients with mutations in specific genes respond differently to those lacking mutations. Genetic testing also increases the possibility of precision medicine approaches targeting specific mutational consequences. Another important consideration in early phase trials is the demonstration of target engagement. In other words, does the drug achieve the predicted and desired impact on the pathway in question? Measuring cellular responses to BMPR2 modifying agents can be challenging. Approaches to date have relied on changes in gene expression of BMP target genes in peripheral blood monocytes, but the approach has not been fully validated. There is a need for robust and reliable readouts of, e.g. endothelial BMP signalling to aid drug development in this field.
Perhaps the most promising approaches might be to combine BMPR2 restoration with active reduction in inflammation or enhanced BMP signalling. Several anti-inflammatory/immune therapies have been or are currently being tested in clinical trials for PAH including, Anakinra (anti-IL1), Tocilizumab (anti-IL6), and Rituximab (anti-CD20). Another approach might be the repurposing of anti-TNF therapies to provide the dual approach of inhibiting inflammation and rescuing BMPR2. Inhibition of TNFα in pre-clinical models seemingly restores BMPR2 levels, Smad and Notch signalling as well as ameliorating experimental PH. As yet no clinical trial for anti-TNF therapies has been undertaken for PAH, but a strong safety profile underlies these drugs as suitable for assessment. In addition to inflammatory processes, inhibition of TGFβ or activin/GDF signalling pathways are promising clinical approaches to target these aberrant pathways in PAH. The ligand trap, Sotatercept, has shown promise in pre-clinical models and clinical trials to date, indicating it may represent a viable therapy. However, the potential inhibitory effect of Sotatercept on BMP9/10 signalling has not been fully explored and this should be addressed in terms of the safety profile. Dual targeting of BMPR2/BMP signalling and endothelial dysfunction by exogenous delivery of BMP9/10 could restore vascular homoeostasis. Both in vitro and in vivo delivery of BMP9 increased BMPR2 expression whilst inhibiting endothelium apoptosis and permeability resulting in reduced PH in pre-clinical models. Therefore, BMP9-based therapies may represent a pathway-specific approach to rebalance BMP signalling and therefore, reduce the susceptibility to aberrant growth factor responses and inflammatory mediators in PAH.
The modulation of BMPR2/BMP and TGFβ pathways clearly remains a promising alternative or combinatorial approach to the current therapies in PAH. Nonetheless, it is also obvious that stratifying the correct treatment to the patient based upon genotype/phenotype will also be beneficial.
Conflict of interest: M.R.T. has received personal fees from Actelion Pharmaceuticals Ltd., GSK and Bayer AG, and is on the scientific advisory board of Morphogen-IX Ltd. P.D.U. is a founder of, and scientific advisor to Morphogen-IX Ltd. N.W.M. is a founder and CEO of Morphogen-IX Ltd. P.D.U. and N.W.M. have published US (US10336800) and EU (EP3166628B1) patents entitled: ‘Therapeutic Use of Bone Morphogenetic Proteins’. All other authors declare no conflicts of interests.
Funding
B.J.D., P.D.U. and N.W.M. are funded by the British Heart Foundation (RG/13/4/30107).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}