





This editorial refers to ‘Inducibility, but not stability, of atrial fibrillation is increased by NOX2 overexpression in mice’, by A.S. Mighiu et al., pp. .
Despite enormous research efforts for decades, the underlying mechanisms of atrial fibrillation (AF), the most frequent arrhythmia, are poorly understood.1 AF is increasingly considered the consequence of a complex atrial cardiomyopathy due to the combined effects of genetics, risk factors, and comorbidities. Thus, a better understanding of the AF-promoting atrial cardiomyopathy and its thrombo-embolic complications is required for improved patient care, as current anti-arrhythmic therapies have limited efficacy and off-target effects. Accumulating evidence has implicated a potential role of oxidative stress in AF-pathophysiology.2,3 There are several possible reactive oxygen species (ROS) pathways that may induce AF by causing electrical, structural, and Ca2+-handling remodelling that supports the formation of AF-promoting ectopic activity and re-entry (Figure 1). Although NOX has been considered as one major enzymatic source for ROS-production in AF, detailed molecular mechanisms by which specific NOX-isoform(s) are contributing to AF and the extent to which activation of NOX plays a causal role in AF development remain to be determined.
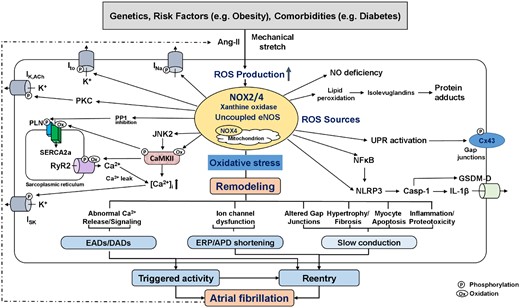
Putative role of reactive oxygen species (ROS) in atrial fibrillation (AF). ROS-production from activated NADPH-oxidase (NOX) isoforms, xanthine oxidase, and downstream ROS-generating systems including mitochondria and uncoupled eNOS. The sustained oxidative stress leads to abnormal Ca2+ signalling with potential occurrence of AF-triggering EADs and DADs, ion-channel dysfunction with ERP/APD-abbreviation, and to altered gap-junctions, fibrosis, myocyte apoptosis, and inflammation, which produce a heterogeneous slowing in impulse conduction. Shorter ERP and conduction slowing support the formation of AF-maintaining reentry. Examples of key mediators downstream of increased ROS-production include oxidized CaMKII (CaMKII), activated PKC, inhibited PP1, and activated NFkB and NLRP3-inflammasome. Activated CaMKII increases ryanodine-receptor type-2 (RyR2) hyperphospohrylation and causes sarcoplasmic reticulum Ca2+-overload, both of which increases diastolic Ca2+-leak. NFkB is a redox-sensitive transcription factor that activates NLRP3-inflammatory signalling and causes structural remodelling by increasing the priming of NLRP3, interleukin-1β (IL-1β), and matrix metalloproteinases. ROS-generation also increases lipid peroxidation and formation of highly reactive isolevuglandins that rapidly adduct and crosslink proteins causing myocyte proteotoxicity. AF further increases the activity of ROS-generating systems by increasing both angiotensin-II (Ang-II) and atrial stretch, creating a vicious cycle of atrial cardiomyopathy-driven NOX-activation and AF-mediated oxidative stress amplification, ultimately leading to AF-promoting atrial remodelling. APD, action-potential duration; CaMKII, Ca2+-calmodulin-dependent protein kinase-II; Cx43, connexin-43; DAD, delayed afterdepolarization; EAD, early afterdepolarization; eNOS, endothelial nitric oxide; ERP, effective refractory period; INa, Na+-current; ISK, small-conductance Ca2+-activated K+-current; Ito, transient-outward K+-current; JNK2, janus kinase-2; NFkB, nuclear-factor kappa-B; NLRP3, NLR family pyrin-domains containing protein-3; PKC, protein kinase-C; PP1, protein phosphatase type-1; UPR, unfolded protein response.
Mighiu et al.4 studied whether myocardial NOX2-overexpression in mice (NOX2-Tg) is sufficient to generate a proarrhythmic AF-substrate and NOX2-inhibition with atorvastatin prevents atrial ROS-production and AF-susceptibility. NOX2-Tg had a two-fold increase in ROS-production and a modest increase (∼28%) in burst-pacing induced AF, with no changes in AF-duration, surface electrocardiogram-parameters, action-potential duration (APD) or conduction velocity, and left ventricular-mass or function. There were no indices of fibrosis, inflammatory signalling or cellular triggered activity. Although treatment with atorvastatin significantly inhibited atrial ROS-production in NOX2-Tg, it has no effect on AF-induction. The authors concluded that atrial NOX2-derived ROS is likely a biomarker of AF-risk rather than a primary AF-driver, and that NOX2-inhibition is unlikely to prevent new-onset AF. The results with statins in this model are similar to previous human clinical trials showing no efficacy.5
This study raises several important questions. The present data suggest that NOX2 overexpression in the absence of atrial remodelling could slightly increase AF-induction but is insufficient to maintain AF, pointing to NOX2 as a potential trigger of AF. Conversely, recent findings from a canine model with AF-mimicking rapid-atrial pacing, which creates a substrate for AF-maintenance, show that the rapid-atrial rate increases NOX2 and ROS-generation, and enhances protein kinase-C with subsequent activation of constitutively active IK,ACh.6 This causes APD-abbreviation that supports AF-maintenance. Genetic knockdown of NOX2 in canines with atrial tachycardia remodelling prevented onset and maintenance of atrial electrical remodelling, supporting the notion that the presence of atrial remodelling or atrial cardiomyopathy due to AF-promoting comorbidities and risk factors is required for NOX2 to causally contribute to AF-maintenance.6 Of note, the sources of ROS-generation dynamically change with AF-progression and predominantly locate in the left atrium.2 Thus, it will be important to assess whether the sources of ROS and their location also change with the progression of risk factors and comorbidities that promote AF-progression and maintenance. Finally, ROS are short-lived species typically acting within their microdomain and NOX2-overexpression might cause a cellular microdomain distribution of NOX2 other than atrial remodelling induced NOX2-upregulation. It has been shown that targeting specific sources of oxidative stress can be anti-arrhythmic while a generalized antioxidant is ineffective, suggesting that the source of ROS is critical to arrhythmic risk.7 NOX2 is only one source of AF-promoting ROS-production and key mediators and other sources of oxidative stress such as other NOXs (e.g. NOX4), uncoupled nitric oxide synthase, CaMKII,8 and inflammation9,10 might trigger AF independently from a NOX2-contribution (Figure 1). Extensive work employing animal models with atrial cardiomyopathy due to isolated atrial dysfunction or due to comorbidities and risk factors that were subjected to atrial-selective NOX2 knockdown is required to define the distinct ROS sources and to validate their causal contribution to AF, along with the underlying molecular mechanisms, particularly in structurally remodelled hearts.
Conflict of interest: D.D. is member of Scientific Advisory Boards of Omeicos Therapeutics GmbH and Acesion Pharma. S.C.D. reports no conflicts of interest.
Funding
The author’s research is supported by NIH (R01HL136389, R01HL131517, and R01HL089598 to D.D.; R01HL104025 and R01HL106592 to S.C.D.); German Research Foundation (DFG, Do 769/4-1 to D.D.); and EU (large-scale integrative project MEASTRIA, No. 965286 to D.D.).
The opinions expressed in this article are not necessarily those of the Editors of Cardiovascular Research or of the European Society of Cardiology.

{kind=link}