





This editorial refers to ‘Impaired NF-κB signalling underlies cyclophilin D-mediated mitochondrial permeability transition pore opening in doxorubicin cardiomyopathy’, by R. Dhingra et al., pp. 1161–1174.
Anthracyclines are the most commonly used and most effective chemotherapeutic agents. Doxorubicin (Dox) was the first anthracycline introduced into the clinical arena in the 1970s and its use resulted in dramatic improvements in survival and quality of life for many cancer patients.1 Unfortunately, this success came with untoward side-effects in the form of significant cardiotoxicity. Cardiotoxic side effects occur in up to 30% of patients treated with anthracyclines, ranging from mild and reversible contractile dysfunction to severe and progressive-dilated cardiomyopathy and chronic heart failure.1 Cardiotoxic side effects are the most common reasons to discontinue anthracycline-based chemotherapy, thereby limiting the individual chances of survival.1 Therapeutic strategies to prevent cardiotoxicity in patients receiving anthracycline-based chemotherapy are urgently needed.
Over the years, intense efforts have been directed at unravelling the mechanisms responsible for Dox-induced cardiotoxicity. In the mitochondria, generation of excessive reactive oxygen species, accumulation of iron-Dox complexes and enhanced calcium influx lead to cardiomyocyte dysfunction and ultimately cell death.1,2 The heart requires tremendous amounts of ATP to maintain its mechanical work and is, therefore, highly dependent on properly functioning mitochondria.1 In addition to their role in cardiac bioenergetics, mitochondria are also essential for cardiac calcium handling, cardiac signal transduction, and the initiation and execution of cell death. Recently, evidence is accumulating that impediments in mitochondrial iron and calcium handling lead to cell death and are central to the development of Dox-induced cardiotoxicity.1,2 The mechanisms underlying these impediments are poorly described but could offer therapeutic targets to prevent Dox-induced cardiotoxicity.
The article by Dhingra et al. is part of this continued effort by their and other research groups to unravel mitochondrial pathways that control cell death in cardiomyocytes. While NF-κB is often considered to be a villain in cardiac physiology, the authors have previously demonstrated that activation of NF-κB can suppress cardiomyocyte necrosis during myocardial ischaemia/reperfusion injury. Cardio-protection offered by NF-κB is at least partially mediated by NF-κB-mediated transcriptional repression of Bcl-2/adenovirus E1B 19Kda protein-interacting protein 3 (Bnip3).3 Bnip3 is a member of the Bcl-2 protein family which control the permeability of the outer mitochondrial membrane. Depending on the cellular context and post-translational modifications to Bnip3, expression of Bnip3 can promote cell death or foster cell survival. In the context of Dox-induced cardiomyopathy, transcriptional activation of Bnip3 has been implicated in the promotion of mitochondrial permeability transition pore (mPTP) opening leading to cardiomyocyte necrosis.4 The mechanisms responsible for Dox-induced Bnip3 transcription are only partially understood, and the mode by which Bnip3 provokes mPTP opening is unknown. Based on their previous work, the authors hypothesized that inhibition of NF-κB and subsequent de-repression of Bnip3 could be responsible for Dox-induced mPTP formation and cell death. This hypothesis was tested in a series of experiments in Dox-treated mice and neonatal rat ventricular cardiomyocytes (NRVCs).5
First, it was confirmed that Dox reduces NF-κB activity in cardiomyocytes, associated with increased Bnip3 expression, calcium-induced mitochondrial swelling and mPTP formation. The importance of mPTP formation in Dox-induced cell death was also established through knock down or inhibition of Cyclophilin D (CyP-D), a well-established component of the mPTP. Next, the general association between the suppression NF-κB and mPTP formation was established through gain and loss of function experiments with NF-κB and CyP-D. Furthermore, overexpression of IKKβ was sufficient to reverse mPTP formation and cell death, indicating that suppression of NF-κB is required for Dox-induced cardiac injury. The final and perhaps most significant discovery was that both Bnip3-mediated mPTP formation and Bnip3-mediated cell death were found to be contingent upon the association between Bnip3 and CyP-D. Hence, this study integrates a number of previous independent observations to establish a novel signalling pathway in which suppression of NF-κB sets the stage for Bnip3-mediated mPTP formation and cell death. The study also provides reassuring proof that we may 1 day be able to prevent or reverse anthracycline-induced cardiomyopathy. Finally, the study leaves us with promising nodal points for intervention including stimulation of NF-κB activity, suppression of Bnip3 transcription or prevention or disruption of the Bnip3-Cyp-D protein complex.3–5
The study by Dhingra et al. provides new insights that are welcomed in the cardio-oncology field. The finding that Dox-activated signalling pathways regulate mPTP formation is in accordance with other studies and suggests that other regulators of mPTP formation could prevent anthracycline-associated cardiomyopathy as well.6,7 The contribution of mPTP formation to Dox-mediated cardiac injury is not well described and mostly involves experiments in cultured cardiomyocytes or isolated mitochondria.8 The temporal patterns of mPTP-mediated cell death after Dox administration are, therefore, unknown but are probably short-lived. This does not necessarily represent a therapeutic limitation, as sustained activation of NF-κB could promote tumour growth and cause cardiac inflammation.9
Another therapeutic avenue to overcome Dox-induced mitochondrial injury could lie in targeting mitochondrial dynamics. For instance, Abdullah et al.10 have shown that dysregulated mitochondrial dynamics and impaired mitochondrial respiration play a prominent role in Dox-induced cardiotoxicity. Inhibition of mitophagy promotes apoptosis and mitochondrial dysfunction after Dox treatment, whereas stimulation of mitophagy may have protective effects.11 Stimulating mitochondrial biogenesis also appears to be protective in Dox-induced cardiotoxicity. As up-regulation of PGC1-alpha and its downstream factors including tFAM and NRF-1/2 reduce Dox-induced cardiotoxicity.12,13 Of note, this protection was accompanied by the inhibition of NF-κB, reinforcing the notion that the protection offered by NF-κB may be temporary in vivo.13 Interestingly, exercise training is known to stimulate mitochondrial biogenesis and also stimulates mitophagy in rats treated with Dox.8,14,15 Therefore, exercise may also offer protection against Dox-induced cardiotoxicity by enhancing mitochondrial quality control.
Taken together, the study by Dhingra et al. reveals a unique and novel signalling pathway responsible for Dox-induced mPTP formation and cell death, which offers several nodal points for intervention. The translational potential obviously is somewhat limited because the study predominantly focused on the use of NRVCs. Future studies are, therefore, required to establish the true relevance in vivo. Furthermore, it is likely that Dox-induced mPTP formation is short-lived. Interventions focused on improving mitochondrial quality control could, therefore, offer the second line of defense after Dox administration. Mitochondrial targeted therapy against Dox-induced cardiomyopathy may, therefore, require a staged approach (summarized in Figure 1). Therapies that stimulate NF-κB, inhibit Bnip3, or disrupt the association with Cyp-D, could offer protection early after Dox-infusion. In the later stages, therapies directed at restoring mitochondrial fitness may be more forthcoming. This may include measures as simple as implementing an exercise regimen. Concludingly, these findings provide the evidence to develop novel therapeutic interventions to prevent or reverse Dox-induced cardiotoxicity.
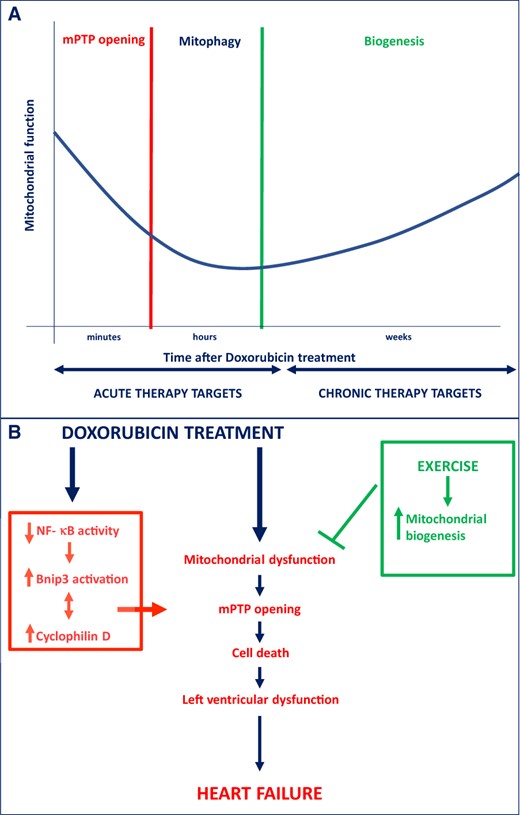
(A) Staged approach of potential mitochondrial targets to attenuate Dox-induced cardiotoxicity, ranging from acute to more chronic stages after Dox administration. (B) Underlying mitochondrial and cell death mechanisms leading to Dox-induced cardiotoxicity. Bnip3, Bcl-2/19 kDa interacting protein 3; mPTP, mitochondrial permeability transition pore; NF-κB, nuclear factor-κB.
Conflict of interest: The UMCG, which employs the authors, has received research grants and/or fees from AstraZeneca, Abbott, Bristol-Myers Squibb, Novartis, Novo Nordisk, and Roche. Dr de Boer received speaker fees from Abbott, AstraZeneca, Novartis, and Roche. The other authors do not have any conflict of interest to declare.
The opinions expressed in this article are not necessarily those of the Editors of Cardiovascular Research or of the European Society of Cardiology.

{kind=link}