




Abstract
The complement system is part of the innate immune system that comprises several small proteins activated by sequential cleavages. The majority of these complement components, such as components 3a (C3a) and C5a, are chemotactic and pro-inflammatory. However, in this study, we revealed an inhibitory role of complement component 8 gamma (C8G) in neuroinflammation. In patients with Alzheimer's disease, who exhibit strong neuroinflammation, we found higher C8G levels in brain tissue, CSF, and plasma. Our novel findings also showed that the expression level of C8G increases in the inflamed mouse brain, and that C8G is mainly localized to brain astrocytes. Experiments using recombinant C8G protein and shRNA-mediated knockdown showed that C8G inhibits glial hyperactivation, neuroinflammation, and cognitive decline in acute and chronic animal models of Alzheimer’s disease. Additionally, we identified sphingosine-1-phosphate receptor 2 (S1PR2) as a novel interaction protein of C8G and demonstrated that astrocyte-derived C8G interacts with S1PR2 to antagonize the pro-inflammatory action of S1P in microglia. Taken together, our results reveal the previously unrecognized role of C8G as a neuroinflammation inhibitor. Our findings pave the way towards therapeutic containment of neuroinflammation in Alzheimer’s disease and related neurological diseases.
Introduction
The complement system is a part of innate immunity that plays a major role in enhancing immunity to pathogens or unwanted host material. The complement system comprises a highly dynamic and orchestrated series of cascades with various factors such as soluble plasma proteins, proteases, cleavage products, cell surface receptors, and regulatory protein complexes (Ricklin et al., 2016). The activation of the complement system is achieved by two major processes: classical and alternative cascades. Following activation, complement 3 (C3) convertase (C2aC4b complex) formed from the C1 complex cleaves C3 protein into C3b and C3a in the classical pathway. C3b then generates C5 convertase (C4bC2aC3b), which cleaves the C5 component and leads to the alternative pathway. Cleavage of C3 and C5, the key proteins in the complement cascade, generates three types of effectors: anaphylatoxin (pro-inflammatory molecules C3a and C5a), opsonin (C3b and iC3b), and the membrane attack complex (MAC; C5bC6C7C8C9; alternatively termed the terminal complement complex).
Activation of the complement system has been documented in neurodegenerative diseases and other brain disorders. For example, increased CSF levels of C1q, C3d, and C4d have been found in the early stages of Alzheimer’s disease (Finehout et al., 2005; Wang et al., 2011; Daborg et al., 2012). Upregulation of the MAC (C9 or C5b-9) in the early and advanced stages of Alzheimer’s disease has also been reported (Zanjani et al., 2005; Loeffler et al., 2008). Inhibition of C5aR1 signalling reportedly suppresses cognitive loss by delaying microglial inflammatory polarization in an Alzheimer’s disease model (Hernandez et al., 2017). Moreover, the complex variation of C4 has been strongly associated with schizophrenia (Sekar et al., 2016; Donohoe et al., 2018). Accumulating evidence suggests that the complement components are not merely a potent driver of inflammation (Rahpeymai et al., 2006; Stevens et al., 2007; Coulthard et al., 2017): complement C1q, C3, and C4, for example, all participate in synaptic pruning (Stephan et al., 2012; Lui et al., 2016). In addition, sublytic C5b-9 plays a crucial role in activating the cell cycle and survival of oligodendrocytes in the CNS (Rus et al., 1996, 2001; Soane et al., 2001). These observations suggest that complement components have multiple roles in neuroinflammation and neurodegenerative diseases.
The gamma chain of complement component 8 (C8G) is one of three different subunits (alpha, beta, and gamma) of the eighth complement component (C8), which is a component of the MAC. Although the roles of C8A and C8B are directly associated with MAC formation, C8G is not essential in the assembly and activity of the MAC (Parker and Sodetz, 2002). Intriguingly, C8A, C8B, and C8G are encoded by distinct structural genes and are differentially regulated in biosynthesis and secretion (Dewald et al., 1996; Scheurer et al., 1997). The genetic loci of the human C8A and C8B genes lie in proximity on chromosome 1p, but the human C8G gene is located on chromosome 9q34.3, which harbours a cluster of lipocalin genes (Dewald et al., 1996) that encode a protein family that interacts with small bioactive molecules and specific cell-surface receptors (Flower, 1996; Logdberg and Wester, 2000). To our knowledge, limited data are available that show the inducibility and functions of C8G in healthy or diseased brains. Moreover, the role of C8G in inflammation or neuroinflammation is currently unknown.
Neuroinflammation is an innate immune process in the CNS and is thought to be a key component of Alzheimer’s disease and other neurodegenerative diseases, which are marked by a progressive deterioration in the structure and/or function of neurons (Valmikinathan and Verghese, 1966). The neuroinflammatory response is multifaceted; it is associated with the complement system, hyperactivation of glial cells, induction of inflammatory genes, and recruitment of immune cells. Physiologically, neuroinflammation plays a protective role as part of a defence system (Asai et al., 2015; Calsolaro and Edison, 2016; Spangenberg et al., 2016); however, prolonged and excessive activation of glial cells releases large amounts of cytokines, chemokines, complement factors, and reactive oxygen species. Thus, the hyperactivity of glia and subsequent detrimental neuroinflammation may trigger a vicious cycle that reduces brain resilience and increases susceptibility to neurodegenerative disease (Asai et al., 2015; Calsolaro and Edison, 2016; Spangenberg et al., 2016; Van Eldik et al., 2016). Therefore, controlling neuroinflammation is considered a potential therapeutic strategy against neurodegenerative disease.
In our previous secretomics study (Jha et al., 2018) and from unpublished data, we identified several lipocalin proteins as glia-secreted proteins. Some of these lipocalin proteins, such as lipocalin-2 (LCN2) and orosomucoid-2 (ORM2), appear to play a crucial role in modulating neuroinflammation and neurodegenerative diseases (Jin et al., 2014; Jo et al., 2017; Kim et al., 2017). These previous findings led us to question the role of C8G, another member of the lipocalin protein family, in neuroinflammation and neurodegenerative diseases such as Alzheimer’s disease. In the current study, we uncover a novel induction of C8G as well as its role during neuroinflammation in vitro in cultured cells, in vivo in animal models, and in specimens from human patients. Our results indicate that C8G is selectively expressed in reactive astrocytes of the mouse brain as well as in brain tissue of patients with Alzheimer’s disease, which has a profound neuroinflammatory component. Analysis of CSF and blood plasma of Alzheimer’s disease animal models and patients with Alzheimer’s disease indicated that C8G was a potential novel disease biomarker. Recently, a large genome-wide association study of late-onset Alzheimer’s disease indicated that 60 genes, among the 112 genes lying within Alzheimer’s disease-associated loci, encode the regulators of microglial activation and function, thereby implicating microglial activation and neuroinflammation in Alzheimer’s disease pathology (Cuyvers and Sleegers, 2016; Deming et al., 2017; Sims et al., 2017). In line with this discovery, we found that C8G has a regulatory role in inflammatory activation of microglia and attenuates disease severity in Alzheimer’s disease animal models. Finally, we showed that the anti-inflammatory role of C8G results from the suppression of pro-inflammatory sphingosine-1-phosphate receptor 2 (S1PR2) signalling in microglia.
Materials and methods
Reagents
Lipopolysaccharide (LPS) from Escherichia coli 0111:B4 was purchased from Sigma-Aldrich. Recombinant TNF-α, IL-1β, interferon (IFN)-γ, IL-10, IL-4, and IL-13 proteins were purchased from R&D Systems. Amyloid-β (Aβ; 1-42) was purchased from Bachem. Amylo-Glo® RTD™ was purchased from Biosensis. JTE-013 and sphingosine-1-phosphate (S1P) were purchased from Cayman Chemical. CYM-5478 was purchased from Aobious Inc.. Fingolimod (FTY-720) was purchased from ApexBio. Bacterially-expressed recombinant mouse C8G protein was prepared as previously described by Lee et al. (2009b). In brief, the recombinant mouse C8G protein without the signal peptide was expressed in E. coli (strain BL21) as a glutathione S-transferase (GST) fusion protein. The protein was purified using glutathione Sepharose 4B beads (GE Healthcare). GST was removed by thrombin digestion, and then C8G was purified and concentrated. SDS-PAGE with Coomassie Brilliant Blue R250 staining showed that the purity of C8G was >95%. Synthetic amyloid-β-derived diffusible ligand (ADDL) was prepared according to previously published protocols (Carriba et al., 2015). In brief, the lyophilized human sequence amyloid-β1–42 was reconstituted in ice-cold hexafluoroisopropanol (HFIP; 1,1,1,3,3,3-hexafluoro-2-propanol; Sigma-Aldrich) to a final concentration of 1 mM. The solution was subsequently incubated at room temperature until it became transparent. The HFIP was then allowed to evaporate overnight to obtain a peptide film. The films were stored at −80°C until they were used. The day before use, the films were resuspended in DMSO (Sigma-Aldrich) to a final concentration of 5 mM; they were then further diluted to 1 mM in DMEM/Ham’s F12 phenol-free medium (Sigma-Aldrich) and incubated at 4°C for 24 h. Finally, the solution was centrifuged for 10 min at 14 000g and 4°C, and the supernatant was used as an ADDL. ADDL preparation was evaluated with tricine gel SDS-PAGE and Coomassie Brilliant Blue or silver staining.
Cell culture
Neonatal astrocyte culture was prepared from mixed glial cultures as previously described (McCarthy and de Vellis, 1980) with minor modifications. In brief, whole brains from 3-day-old C57BL/6 mice were chopped and mechanically disrupted using a nylon mesh. The cells obtained were seeded in culture flasks and grown at 37°C in a 5% CO2 atmosphere in DMEM supplemented with 10% heat-inactivated FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. The culture medium was first changed after 5 days and then subsequently every 3 days. Cells were used after 14–21 days of culture. Primary astrocytes were obtained by shaking mixed glial cultures at 250 rpm overnight. To determine the degree of microglial contamination in the primary astrocyte cultures, traditional RT-PCR amplification of microglia or myeloid markers, such as ionized calcium-binding adapter molecule 1 (Iba-1), Cx3cr1, F4/80, CD11c, and CD11d, was performed (Jang et al., 2013). The purity of the astrocyte cultures was >90% as determined by glial fibrillary acidic protein (GFAP) immunocytochemistry (Jeon et al., 2010). Primary microglia were obtained by mild trypsinization from mixed glial cultures (Saura et al., 2003). Primary hippocampal neurons were prepared from E20 mice, as previously described (Beaudoin et al., 2012). In brief, mouse embryos were decapitated, and their brains were rapidly removed and placed in a culture dish containing cold PBS. Hippocampi were isolated and incubated in 0.25% trypsin-EDTA (Gibco, Thermo Fisher Scientific) in PBS for 30 min at 37°C with shaking. After neutralization of trypsin, hippocampal tissue was dissociated mechanically by gentle pipetting, and the resulting dissociated cortical cells were seeded onto plates coated with poly-d-lysine (Sigma-Aldrich) in NeurobasalTM medium (Gibco, Thermo Fisher Scientific) containing GlutaMAXTM (Gibco, Thermo Fisher Scientific), penicillin–streptomycin, nerve growth factor (Invitrogen), and B27 supplement (Gibco, Thermo Fisher Scientific). The BV-2 immortalized mouse microglial cell line, HT-22 mouse hippocampal neuron cell line (Invitrogen), HEK293 cells, and bEnd.3 mouse brain endothelial cell line (ATCC) were maintained in DMEM supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Primary human astrocytes, which were purchased from ScienCell Research Laboratories, Inc, were cultured and maintained according to the manufacturer’s instructions (i.e. in human astrocyte medium; ScienCell). All cells were cultured in mycoplasma-free conditions. The NO2 concentration in the culture medium was measured to evaluate NO production in glial cells using Griess reagent as previously described (Lee and Suk, 2007). Cell viability was measured using the MTT assay described (Lee et al., 2009b).
Mice and animal care procedures
Male C57BL/6 mice (8–9 weeks of age) were purchased from Samtako Bio). The 5xFAD mice (3–8 months of age), which overexpress the K670N/M671L (Swedish mutation), I716V (Florida mutation), and V717I (London mutation) mutations in human amyloid precursor protein (isoform 695), as well as the M146L and L286V mutations in human presenilin-1, were kindly provided by Korea Brain Research Institute (Daegu, Korea). All mice were housed in groups of three to five per cage under specific pathogen-free conditions and a 12-h light/dark cycle.
Mouse models of neuroinflammation and Alzheimer’s disease
Systemic LPS injection was performed as previously described by (Qin et al., 2007). Mice were subjected to intraperitoneal injection with vehicle (same volume of PBS) or LPS (5 mg/kg). For intracerebroventricular injection of C8G and LPS, mice were first anaesthetized using isoflurane and then restrained on a stereotaxic device (Stoelting Co.). Recombinant C8G protein (1 μg/ml, 3 μl) or LPS (10 ng per mouse, 3 μl) was then slowly injected (0.2 μl/min) into the left lateral ventricle using a 5-μl Hamilton syringe and microinjector (Panlab, Harvard Apparatus). For a non-genetic mouse model of Alzheimer’s disease, an intracerebroventricular injection of ADDL (1 μM in 7.5 μl of PBS) was used. The coordinates used for all of intracerebroventricular injections were −1.0 mm lateral, −0.3 mm posterior, and 1.5 mm ventral to the bregma (Freir et al., 2011; Rostami et al., 2017). When necessary, animals were sacrificed under CO2-induced anaesthesia.
Surgical procedure for cannula implantation and C8G infusion
A long-term intracerebroventricular cannula (Brain Infusion Kit 3; Alzet) was implanted into the lateral ventricle of the brain of the mice under isoflurane anaesthesia. The cannula was stereotaxically positioned in the lateral ventricle with the coordinates: −1.0 mm lateral, −0.3 mm posterior, and 1.5 mm ventral to the bregma. Once positioned, it was secured in place using dental cement. Mice were allowed a 14-day recovery period from surgery and brain wound injury; subsequently, a mini-osmotic pump (Alzet 1007D) for infusing either vehicle (PBS) or C8G (1 μg/ml at a rate of 0.5 μl/h) was implanted subcutaneously and connected to the cannula by a polyethylene tube catheter, which was introduced under the dorsal skin of the animal.
Clinical samples
Twelve autopsy cases were examined from Boston University’s Alzheimer’s Disease Center (BUADC) including individuals with and without cognitive impairment who had undergone annual cognitive evaluations using the National Alzheimer’s Disease Coordinating Center (NACC) Uniform Data Set (UDS) protocol (Beekly et al., 2007). Neuropathological processing of control and Alzheimer’s disease human brain samples was performed according to the procedures previously established by the BUADC and Chronic Traumatic Encephalopathy (CTE) Center (Vonsattel et al., 2008; Mez et al., 2015). Institutional review board approval for ethical permission was obtained through the BUADC and CTE Center. This study was reviewed by the Boston University School of Medicine Institutional Review Board (Protocol H-28974) and was approved as exempt because the study involved only tissue collected from post-mortem individuals that are not classified as human subjects. Nevertheless, next of kin provided informed consent for participation and brain donation. The study was performed in accordance with the institutional regulatory guidelines and principles of human subject protection in the Declaration of Helsinki. Detailed information about the brain tissues is described in Supplementary Table 1. In all cases in which Alzheimer’s disease was diagnosed at autopsy, Alzheimer’s disease was also stated as the cause of death. Human CSF and plasma were collected from patients who visited the Dementia Clinic of Kyungpook National University Hospital (Daegu, South Korea). All participant characteristics are summarized in Supplementary Table 2.
Histology
Mice were anaesthetized with ether and transcardially perfused with 4% paraformaldehyde in PBS, and their extracted brains were post-fixed and cryoprotected with 30% sucrose solution for 3 days. The fixed brains were then embedded in optimal cutting temperature compound (Tissue-Tek; Sakura Fine-Tek) and cut into 20-μm thick sections on a cryostat. For immunofluorescence analysis of animal tissues, frozen brain sections were permeabilized in 0.1% TritonTM X-100 and blocked using 1% bovine serum albumin (BSA) and 5% normal donkey serum for 1 h at room temperature. Brain sections were incubated with the following primary antibodies at 4°C overnight: anti-C8G, anti-Iba-1, and anti-GFAP antibodies. This was followed by incubation for 1 h at room temperature with fluorescence-conjugated secondary antibodies. Finally, the sections were mounted and counterstained using DAPI-containing gelatin. Amyloid-β plaques were stained with 1× Amylo-Glo® RTD™ (1:100) or Thioflavin-S solution. Briefly, for Amylo-Glo fluorescence labelling, brain tissues were prewarmed at 60°C before being transferred into 70% ethanol for 5 min and then rinsed in distilled water. The slides were subsequently incubated for 10 min in the 1× staining solution. After being rinsed with 0.9% saline and distilled water, the sections were mounted with gelatin solution. Amyloid plaques were visualized by UV illumination. For Thioflavin-S staining, tissue sections were rinsed in distilled water and incubated with 1% aqueous Thioflavin-S for 8 min at room temperature. After rinsing with distilled water, the sections were mounted with gelatin solution. To visualize degenerative neurons, the following process was first followed: the slides were immersed in a solution containing 1% NaOH in 80% ethanol for 5 min, rinsed first for 2 min in 70% ethanol and then for 2 min in distilled water, incubated in a 0.06% potassium permanganate solution for 10 min, rinsed with distilled water for 2 min, incubated in Fluoro-Jade® C (FJC) staining solution for 10 min, and finally washed and then air-dried on a slide warmer at 50°C for 30 min. After clearing the slides with xylene and adding a coverslip, the sections were examined under an epifluorescence microscope. Amyloid plaques and degenerative neurons within the region of interest were semi-automatically counted using the ‘Analyze particles’ function of FIJI/ImageJ software (NIH).
For immunohistochemistry using 3,3′-diaminobenzidine (DAB) staining, peroxidase activity was depleted by incubation with hydrogen peroxide before staining. After blocking with 5% normal donkey serum and 0.1% BSA, sections were incubated with anti-GFAP or anti-Iba-1 antibodies at 4°C overnight. After washing, a Vectastain Elite ABC kit (Vector Laboratories) was used according to the manufacturer's instructions in order to reveal the distribution of primary antibodies. After washing three times with PBS for 15 min, sections were incubated with DAB chromogen solution (Thermo Fisher Scientific) at room temperature and mounted. All antibodies and experimental information are listed in Supplementary Table 3.
The number of microglia or astrocytes was analysed according to immunohistological intensity using FIJI/ImageJ software (NIH) as previously described (Wada et al., 2006). Briefly, five squares (300 × 300 pixels each) were randomly placed in the hippocampal or cortical areas of each mouse image. The composite images obtained from each animal were binary thresholded at 50% of the background level, and the particles were converted to a subthreshold image area with a size <300 and >5 pixels, which was judged as either GFAP- or Iba-1-positive cells. Binary images among the groups were adjusted using the same threshold, and cells were recognized and quantified by counting pixels. The cell number per animal was calculated from the average of five squares and is presented as an area (mm2).
Co-localization of specific protein markers was calculated using Pearson's correlation coefficients, as previously described (Dunn et al., 2011). We obtained a Pearson’s coefficient value from the five images of each mouse using FIJI/ImageJ software and the plugin ‘Colocalization Threshold.’ The mean value of the five images was considered to be the final Pearson’s coefficient value for a given mouse. Using this value, the final graph was generated.
Double staining immunohistochemistry for human post-mortem brain tissue
C8G staining
Paraffin-embedded tissues were sectioned in a coronal plane at 10–20 μm as previously described (Lee et al., 2018). The tissue sections were rehydrated, blocked with blocking solution (1% H2O2), and incubated with rabbit polyclonal antibody against C8G for 24 h. After washing three times, the slides were processed with biotinylated anti-rabbit IgG and a Vector ABC Kit (Vector Laboratories, Inc.). The immunoreactive signals were developed with DAB chromogen (Thermo Fisher Scientific).
GFAP staining
Endogenous alkaline phosphatase was blocked using 3% hydrogen peroxide in TBS. Sections were blocked with 2.5% normal horse serum (Vector Laboratories) before incubation for 24 h with a mouse monoclonal antibody to GFAP. After washing, sections were incubated with ImmPRESS-AP anti-mouse IgG (alkaline phosphatase) polymer detection reagent (Vector Laboratories) for 30 min at room temperature. Colours were developed with a Vector Red alkaline phosphatase substrate kit (Vector Laboratories). Slides were subsequently counterstained with haematoxylin (Vector Laboratories), processed back to xylene through an increasing ethanol gradient [70%, 80%, and 95% (once each) and 100% (twice)], and then mounted. The images were analysed under a bright field microscope. After double immunohistochemistry staining, slides were placed in 1× Amylo-Glo® RTD™ (1:100) staining solution for 10 min at room temperature without shaking. After staining, sections were washed in PBS three times for 5 min each wash, briefly rinsed in Milli-Q® DI water, and then mounted. The images were analysed under a fluorescence microscope. All antibodies and experimental information are listed in Supplementary Table 3.
Image analysis of human immunohistochemistry sections
Semi-quantitative assessment of immunohistochemistry signals and co-localization pattern was performed using CellSens Imaging Software (Olympus Korea Co.). Co-localized areas (regions of interest) in merged images from two of the overlapping chromogenic dyes were selected by drawing a transverse line. The signal intensity of region of interest on the selected line was automatically calculated by the software module and transferred to a Microsoft Excel file to generate statistics and representative graphs.
Traditional or real-time reverse transcription-PCR
Total RNA was isolated from cells, hippocampal tissue, or whole brains using QIAzol reagent (Qiagen) following the manufacturer’s instructions. Reverse transcription and PCR amplification were performed using a C1000 Touch thermal cycler (Bio-Rad) with specific primer sets. GAPDH was used as an internal control. The nucleotide sequences of the primers were based on published cDNA sequences. For real-time PCR, 100 μg of RNA was used for cDNA preparation with an iScriptTM cDNA Synthesis Kit (Bio-Rad) according to the manufacturer’s protocol. The cDNA from each sample was amplified by real-time PCR using iQTM SYBR® Green Supermix (Bio-Rad). RNA quantities were normalized using GAPDH mRNA as a reference. PCR cycling conditions were as follows: denaturation for 3 min at 95°C; 40 cycles of amplification for 15 s at 95°C, 15 s at 60°C, and 20 s at 70°C; followed by 30 s at 72°C. For melting curve analysis, data were collected from 33 cycles (6 s each) with the temperature increased from 60°C to 92°C (set point temperature increased after cycle 2 by 1°C). All primers used are listed in Supplementary Table 4.
ELISA
To assess the level of C8G in the CSF and plasma of Alzheimer’s disease model mice, samples were first diluted in PBS. For in-house ELISA, the diluted samples (antigens) were coated on a PVC microtitre plate and maintained overnight at 4°C. After blocking with 1% BSA in PBS, samples were incubated overnight with anti-C8G primary antibody in a 96-well plate at 4°C. Anti-rabbit HRP-linked secondary antibody (1:500) and 3,3′,5,5′-tetramethylbenzidine (TMB) substrate reagent (R&D Systems) were used to quantify C8G protein. A Mouse C8G ELISA Kit (Mybiosource) was also used according to the manufacturer’s instructions. The absorbance was read at 450 and 540 nm using a microplate reader (Molecular Devices). Recombinant C8G was used as the standard. To determine levels of human C8G, the plasma (1:10 000) and CSF (1:10) were first diluted. A Human C8G ELISA Kit (Cloud-Clone Corp.) was used for the assay according to manufacturer’s instructions. The levels of TNF-α and S1P in the culture medium were measured with a Mouse TNF-α Sandwich ELISA Kit (R&D Systems) and an S1P ELISA Kit (Echelon Biosciences), respectively.
Immunoblot and immunoprecipitation
Samples were lysed using radioimmunoprecipitation assay (RIPA) lysis buffer (Thermo Scientific) with a protease and phosphatase inhibitor mixture (Roche Holding AG, Basel, Switzerland) and then centrifuged at 4°C and 14 000g for 20 min. The supernatant was transferred to a new tube, and the concentration was determined using a BCA kit (Thermo Scientific). The same amount of protein was added to a polyacrylamide gel (10%–12%) for each sample. Proteins separated by SDS-PAGE were electro-transferred to PVDF membranes (Bio-Rad), and the membranes were blocked using 5% skimmed milk in PBS with 0.25% Tween-20 (PBST) and then incubated at 4°C overnight with anti-C8G or β-actin antibodies. After washing, the membranes were bound to HRP-conjugated secondary antibodies and incubated at room temperature for 1 h. The blots were developed using enhanced chemiluminescence (ECL) western blotting detection reagent (Thermo Fisher Scientific) and analysed with the MicroChemi system (DNR Bio-imaging Systems). All antibodies used and experimental information are listed in Supplementary Table 3.
Construction of recombinant adeno-associated virus vectors containing hybrid AAV serotype C8G shRNA
For in vivo gene silencing, the validated mouse shRNA sequence for C8G [target sequence: ggaagctgtctgtgaaactat; NM_027062 (706–726 nt)] was cloned into the pSicoR vector using HpaI/XhoI sites (Addgene) and subcloned into the pAAV-MCS vector (Stratagene) using MluI/BglII sites. The high-titre recombinant adeno-associated virus (rAAV) vectors were produced in HEK293TN cells using a helper virus-free system. In brief, the rAAV vectors were produced after co-transfecting with equimolar amounts of a rep/cap/helper plasmid. After incubation for 72 h, the cells were lysed, treated with benzonase (Sigma-Aldrich), and further purified using HiTrap heparin columns (GE Healthcare). Amicon ultra-15 centrifugal filter units (Millipore) were used to increase the concentration to the final volume.
Animal behaviour tests
A sucrose preference test was performed as previously described using mice with free access to both water and sucrose solutions (Henry et al., 2008), which began after 2 days of treatment. One water bottle filled with 1% sucrose solution and another filled with water were placed in the cage for 24 h. The consumption from each bottle was measured daily and sucrose preference was expressed as follows: (Δ weight sucrose)/(Δ weight sucrose + Δ weight water) × 100.
A step-through passive avoidance test was performed as previously described by Karlen et al. (2009) with minor modifications. In brief, animals were placed in the light compartment of a two-compartment box (one light, one dark; San Diego Instruments) with the door to the dark compartment closed. Following 30 s of exploration, the door to the dark compartment was opened. When a mouse entered the dark compartment, the door was closed and it received a single electric shock (0.5 mA, 3 s). Retention tests were conducted after 24, 48, and 72 h, and the step-through latency for the animal entering the dark compartment with all four paws was recorded with no further shock administered. A maximum latency of 300 s was scored if the animal did not enter the dark compartment. All recording and operation were conducted using the manufacturer’s software (Gemini Systems).
A Y-maze is a horizontal maze with three arms (40-cm long, 3-cm wide, and 12-cm high). For the Y-maze test, the animals were initially placed in the centre of the maze, and the order (i.e. ABCCAB) and number of arm entries was manually recorded for 7 min for each animal. Voluntary shifts were defined as trials with entries into all three arms in sequence (i.e. ABC, CAB, or BCA, but not BAB). The maze was thoroughly cleaned with water after each animal was tested in order to remove residual odour. The ratio of alternatives was calculated according to the following equation: % alternation = [(number of alternations)/(total arm entries)] × 100. The total number of arm entries was used as an indicator of locomotor activity. All recording and calculation were automatically obtained using SMART video tracking software (version 3.0, Harvard Apparatus, Holliston, MA).
To conduct a forced swim test, mice were placed individually in a vertical acrylic cylinder (height: 60 cm; diameter: 20 cm) filled with tap water at 26°C to a depth that made it impossible for the mice to touch the bottom with their hind paws (i.e. 20 cm). Animals were removed from the water after 6 min and dried quickly before being returned to their cages. Each session was video-recorded and analysed blindly. Three different behaviours were considered during analysis according to Porsolt’s criteria (Can et al., 2012): (i) immobility: mice were judged as immobile when they floated passively, making only small movements to keep their nose above the surface; (ii) climbing (or thrashing): upward-directed movements with the forepaws, in and out of the water, along the side of the swim chamber; and (iii) swimming: active movements (usually horizontal) more than necessary to merely maintain their head above the water. Diving and face shaking behaviours were not scored. The time spent immobile was measured.
To conduct a Barnes maze task, mice were individually placed for 10 s in a 10-cm high cylindrical black start chamber in the middle of a maze, which constituted a white circular platform (90 cm in diameter) with 20 equally spaced holes around the perimeter. Before measurements were taken, mice were gently guided to enter the escape hole by hand after removing the start chamber. Once mice entered the hole, the target hole was covered and mice were allowed to stay inside it for 2 min. When the mice could find the escape hole within 300 s (the habituation phase), we began recording. In the training phase, mice received two trials per day, with an interval of 15 min between trials for a total of 5 days. From the start point, the time taken to enter the escape hole was measured as the latency of escape. On Day 6, we performed the probe test. Mice were camera-recorded for 60 s per trial. The maze was divided into four quadrants, and the time spent in each quadrant was measured.
Construction of plasmids
The cDNAs encoding full-length mouse C8G (GenBank accession number: NM_027062) and mouse S1PR2 (GenBank accession number: NM_010333) were obtained using a RT-PCR-based Gateway cloning method (Invitrogen). C8G without a signal peptide (C8G-ΔSP) and S1PR2 mutants, such as N-terminus-deleted S1PR2 (S1PR2-ΔN) and extracellular loops-deleted S1PR2 (S1PR2-ΔECL), were generated using full-length mouse C8g or S1pr2 cDNA as a template with an EZchangeTM Site-directed Mutagenesis Kit (Enzynomics). The constructs were subcloned into several vectors, including pDEST-HA-C and pDEST-GFP-C, by Gateway cloning (Invitrogen). The C8G and S1PR2 mutants were co-expressed in HEK293 cells.
Protein-protein interaction assay
GST-C8G pull-down assay
To precipitate extracellular or intracellular proteins which bind to C8G, we conducted a pull-down assay using GST-fused C8G protein (GST-C8G) and two different methods in BV-2 microglial cells: (i) GST–C8G incubation with total cell lysates (Assay 1); or (ii) GST-C8G incubation with intact cells to examine the role of cell surface S1PR2 (Assay 2). For GST-C8G pull-down assay 1, BV-2 microglial cell lysates were obtained via sonication on ice for 10 s and by centrifugation at 4°C and 14 000g for 20 min in RIPA buffer with a protease and phosphatase inhibitor mixture (Roche Holding AG). The lysates were incubated with recombinant GST-C8G (10 μg/ml) at 37°C for 1 h, gently mixed with glutathione (GSH) beads, and incubated at 4°C for overnight. The beads were separated by centrifugation at 8000g and 4°C for 5 min. The beads were then washed five times with PBS. The proteins bound with GST-C8G were separated using SDS-PAGE and transferred to a PVDF membrane for immunoblotting. GST protein was used as a negative control. For GST-C8G pull-down assay 2, intact BV-2 microglial cells were incubated with extracellular GST–C8G (10 μg/ml) for 2 h, then unbound GST-C8G proteins were removed by washing with PBS and bound proteins were cross-linked by using 4% PFA for 1 min. The cells were lysed in lysis buffer (Thermo) and centrifuged at 13 000g and 4°C for 30 min. The supernatant was gently mixed with glutathione (GSH) beads and incubated at 4°C for overnight. The beads were separated by centrifugation at 8000g and 4°C for 5 min. The beads were then washed five times with PBS. The proteins bound with GST-C8G were separated using SDS-PAGE and transferred to a PVDF membrane for immunoblotting. GST protein was used as a negative control. All antibodies used and experimental information are listed in Supplementary Table 3.
Co-immunoprecipitation
For the co-immunoprecipitation experiment, transfected HEK293T cells were lysed in lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5-mM EDTA, 1 mM PMSF, 1% NP-40, 1% SDS, and 0.25% sodium deoxycholate) containing a protease-inhibitor cocktail (Roche) for 30 min at 4°C. This was followed by centrifugation at 13 000g and 4°C for 30 min. Protein supernatants were incubated overnight at 4°C with 15 μg of anti-GFP antibody (B-2; Santa Cruz Biotechnology). Protein A/G PLUS-agarose (Santa Cruz Biotechnology) was then added for 1 h before the samples were washed three times with TBST. For immunoblotting, protein samples were separated by 10% SDS-PAGE, and the separated proteins were transferred to PVDF membranes. The blots were incubated overnight at 4°C with rat anti-HA antibody (3F10; Roche Applied Science, 1:1000) and mouse anti-GFP antibody (B-2; Santa Cruz Biotechnology, 1:1000). Blots were then washed and incubated with horseradish peroxidase-conjugated anti-rat or anti-mouse IgG, followed by washing and detection of immunoreactivity with enhanced chemiluminescence (34095; Thermo Scientific).
Endogenous protein interaction assay: tissue co-immunoprecipitation
For the endogenous protein-protein interaction assay, brain tissue from LPS-injected mice was rapidly lysed in RIPA buffer supplemented with a protease and phosphatase inhibitor mixture (Roche Holding AG). Following lysis for 30 min, samples were spun at 14,000g for 10 min at 4°C, and the supernatant was transferred to a clean tube. Brain lysate was incubated overnight at 4°C with either anti-C8G or anti-S1PR2 antibodies. Sepharose beads (GE Healthcare) were washed and added to tubes containing the lysed protein antibody and incubated overnight at 4°C. Following incubation, the beads were washed and subjected to SDS-PAGE and immunoblotting.
Assessment of RhoA activation
RhoA activation assays were performed using a kit (STA-403A; Cell Biolabs). Following various treatments, cells with 80%–90% confluence were lysed with a lysis buffer (a kit component) for 15 min on ice. The lysate was centrifuged for 10 min at 14 000g and 4°C. The supernatant was then incubated with 40 μl of Rho-binding domain (RBD) beads for 1 h on ice. Subsequently, the beads were centrifuged for 20 s at 14 000g and 4°C; the supernatant was then discarded and 0.5 ml of lysis buffer was added to the beads. This step was repeated three times. An equal volume (40 μl) of 2× reducing SDS-PAGE sample buffer was added to the bead solution, which was then boiled at 95°C for 5 min. After centrifugation for 30 s at 14 000g and 4°C, 20 μl of the sample was used for immunoblotting with an anti-RhoA antibody (1:200, another kit component).
Statistics
Statistical analyses were performed with Prism software version 8.0 (GraphPad Software, La Jolla, CA). Values are expressed herein as means ± standard error of the mean (SEM). Statistical analysis to compare the mean values for multiple groups was performed using a one-way or two-way ANOVA with correction for multiple comparisons. Comparison of two groups was performed using either Welch’s t-test or unpaired non-parametric Mann-Whitney U-test, based on the identical interval and continuity, independence, normality, and equal variance.
Study approval
The current study was approved by the appropriate local ethics committee. Informed consent was procured from all human participants or their caregivers who donated their tissue. All animal behaviour tests were approved by Kyungpook National University Animal Care Committee (KNU 2015-0010), and care was taken to minimize animal suffering.
Data availability
The data generated or analysed during the current study are available from the corresponding author on reasonable request.
Results
C8G expression is upregulated in Alzheimer’s disease brain tissue, CSF, and plasma
In our first series of experiments, we investigated the expression of C8G in the brains of 5xFAD mice (8-months-old). The expression of C8G was mainly co-localized with GFAP-positive reactive astrocytes (Fig. 1A) but not in Iba-1-positive microglia in the brain (Supplementary Fig. 1A), and it was upregulated (Fig. 1B) in 5xFAD mice. Such a marked increase in the expression was not observed for C8A or C8B: only a modest upregulation of C8A was detected in 5xFAD mice. C8G expression was pronounced in the reactive astrocytes surrounding amyloid plaques but not in microglia. Upregulation of C8G expression was similarly observed in ADDL-injected mice (Supplementary Fig. 2A–C).
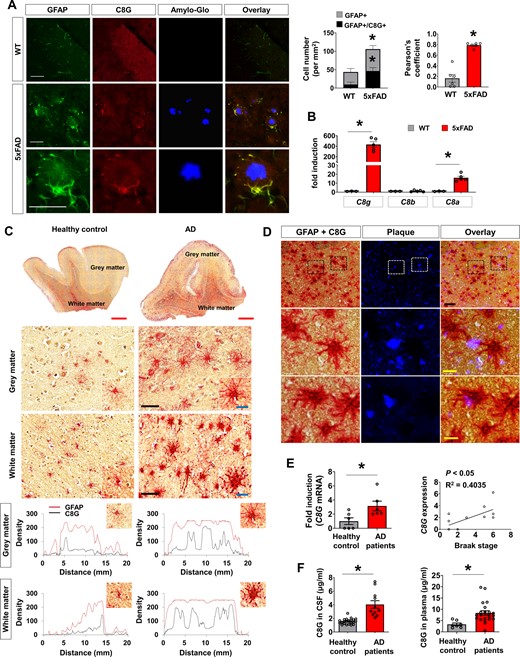
C8G expression in Alzheimer's disease mice and patients. (A) Immunofluorescence analysis of C8G in the hippocampus of wild-type (WT) and 5xFAD mice. Amyloid plaques were stained with Amylo-Glo® fluorescent tracer. Scale = 100 μm. The right panel shows normalized counts of GFAP-positive cells and co-localized cells per unit area in the hippocampus (n = 6) and averages of Pearson’s coefficient (n = 6). (B) C8 subtype mRNA level in the brain of control (Con) and 5xFAD mice (n = 5 per group). (C) Human C8G immunoreactivity in GFAP-positive astrocytes of grey matter and white matter of healthy controls and patients with Alzheimer’s disease (AD) (red: GFAP; black dots: C8G). C8G was detected using anti-C8G antibody and DAB chromogen. GFAP was developed using anti-GFAP antibody and a red alkaline phosphatase substrate kit. Red scale bar = 2 mm; black scale bar = 80 μm; and blue scale bar (inset) = 20 μm. Image analysis (green): C8G signal (black line) was elevated in GFAP-positive astrocytes (red line) in the cortex of patients. Green scale bar = 20 μm. (D) Immunoreactivity of human C8G/GFAP-positive astrocytes surrounding amyloid-β plaque (red: GFAP; black dots: C8G; blue: amyloid-β plaque). Double staining of C8G and GFAP was performed as described in C, and amyloid-β plaque was stained by Amylo-Glo tracer. Scale bar = 20 μm. (E) C8G mRNA level in the brain of healthy controls and patients with Alzheimer’s disease (both n = 6). C8G mRNA measured by real-time PCR. Right: Correlation between Braak stage and C8G expression. (F) Human C8G protein level in the CSF and plasma of healthy controls and patients with Alzheimer’s disease. Data are mean ± SEM. *P < 0.05, unpaired non-parametric test (Mann-Whitney) or Welch’s t-test.
Furthermore, we evaluated the expression of C8G in the brain tissue of Alzheimer’s disease patients. As in animal Alzheimer’s disease models, significant C8G expression was detected in reactive astrocytes around amyloid plaques (Fig. 1C and D) and was upregulated in the brains of Alzheimer’s disease patients (Fig. 1E). The upregulation of C8G mRNA (P = 0.0238) was strongly correlated with the progression through the Braak stages (Braak et al., 2006) (P < 0.05, R2 = 0.4035; Fig. 1E), which are defined as follows: Stage I–II: either mild or severe alteration of the transentorhinal layer Pre-alpha; Stage III–IV: a conspicuous effect on the layer Pre-alpha in both the transentorhinal region and the actual entorhinal cortex, as well as mild involvement of the first Ammon's horn sector; Stage V–VI: the destruction of virtually all isocortical association areas. To test whether C8G was detectable in body fluids, we measured C8G levels in the CSF and blood plasma of mouse Alzheimer’s disease models and human subjects. C8G protein levels were significantly higher in the plasma and CSF of mouse Alzheimer’s disease models (Supplementary Fig. 2D and E) and those of patients with Alzheimer’s disease (Fig. 1F) relative to healthy controls.
C8G is upregulated in inflamed brains and is mainly localized to astrocytes
To determine the link between upregulation of C8G and the neuroinflammatory condition, we assessed the expression of C8G in the inflamed mouse brain. Intraperitoneal LPS-injected mice were sacrificed 3 days after injection. Immunofluorescence imaging data from these mice showed that C8G expression was significantly increased in the hippocampal region of the brain following systemic LPS administration (Fig. 2A and Supplementary Fig. 3A). This preferential induction of C8G in hippocampal astrocytes might be due to hippocampal susceptibility to acute neuroinflammatory triggers; for example, the hippocampus has a higher density of interleukin-1 receptors than other brain regions (Parnet et al., 2002). The upregulated C8G expression was mainly detected in reactive astrocytes (Fig. 2A and Supplementary Fig. 1B). Significant upregulation of C8G in whole-brain samples was observed at 24 h after LPS intraperitoneal administration; this peaked at 72 h at the protein and mRNA levels and diminished thereafter (Fig. 2B and C). When compared with pro-inflammatory cytokines (IL-1β, IL-6, and TNF-α) or the acute phase (AP) protein LCN2, C8G expression was upregulated and sustained during later phases of neuroinflammation (Fig. 2C).
![C8G induction in astrocytes under neuroinflammation. (A) Immunofluorescence analysis of C8G in the hippocampus. Mice injected with LPS (5 mg/kg) intraperitoneally (i.p.). Scale bar = 25 μm. Right: The number of GFAP-positive cells and co-localized cells per unit area in the hippocampus (n = 6) as well as averages of Pearson’s coefficients (n = 6). (B) Time course of C8G protein expression in the inflamed brain. C57BL/6 mice (n = 3) injected with LPS (5 mg/kg) i.p. (C) Comparison of mRNA expression of C8G, pro-inflammatory cytokines, and lipocalin-2 (Lcn2) in the LPS-injected brain of mice (n = 3). (D) C8G expression in different cell types of the CNS. Microglia, astrocytes, and bEnd.3 mouse brain endothelial cells (n = 3 each) were incubated with LPS (1 μg/ml) alone or with L/I [LPS (1 μg/ml) and IFN-γ (50 units/ml)] as indicated. Primary cultured hippocampal neurons were incubated with conditioned medium of mixed glial cells stimulated with L/I. After a 6 h treatment, C8G level was measured with traditional RT-PCR. (E) C8G induction by inflammatory stimuli in mouse primary cultured astrocytes (n = 3). Cells were incubated with L/I, recombinant mouse TNF-α (10 ng/ml), IL-1β (10 ng/ml), IL-6 (10 ng/ml), IFN-γ (50 units/ml), IL-10 (10 ng/ml), IL-4 (10 ng/ml), or IL-13 (10 ng/ml) for 6 h. Mouse liver was used as a positive control. (F) C8G mRNA induction by inflammatory stimuli in human astrocytes (n = 3). Astrocytes were incubated with recombinant human TNF-α, IL-1β, IL-6, IFN-γ, IL-10, IL-4, or IL-13 protein or their combination (Mix: TNF-α + IL-1β + IFN-γ) with the same concentration as mouse astrocytes for 6 h. HepG2 cells were used as a positive control. The assay (n = 3; done in triplicate) was repeated at least two times (inter-assay coefficients of variation were < 20%). Data are mean ± SEM. *P < 0.05 versus control (Con). Unpaired non-parametric test in A (Mann-Whitney) and D (Welch’s t-test); Dunnett's multiple comparison post hoc tests after one-way ANOVA in B, C, E and F.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/brain/144/2/10.1093_brain_awaa425/2/m_awaa425f2.jpeg?Expires=1747880363&Signature=129Byf-9ZZTsvvHAUVIfli7UOQD7dlDSbLwF0zX4rBacxJ3I0LhDhJpmw76cqi1bvStiaNUKqx2NdClVbssVjufaeogDMqEqrabrTUH~1z89vSesI9MYLVD7OYAsrrAAY8bDFVwkmANnHi6mCULeYIzn8N61igGPvDRWB8YJVzMo~CrgP-MZ5UzeerTnHL9teasLHGeD7CcstIHs-RtoI2bSlxKBUsdkO4q0TYs~vpGjQfaY68a0Wfz0qBczxxakEN5p-Ir9XYBng-bhLNwfZpSPM~pkNIPKlW6sNv9i-pWqzCF2QTPAhlPvBHl~WDt2uNquD0z88ju4vjRinb-CDQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
C8G induction in astrocytes under neuroinflammation. (A) Immunofluorescence analysis of C8G in the hippocampus. Mice injected with LPS (5 mg/kg) intraperitoneally (i.p.). Scale bar = 25 μm. Right: The number of GFAP-positive cells and co-localized cells per unit area in the hippocampus (n = 6) as well as averages of Pearson’s coefficients (n = 6). (B) Time course of C8G protein expression in the inflamed brain. C57BL/6 mice (n = 3) injected with LPS (5 mg/kg) i.p. (C) Comparison of mRNA expression of C8G, pro-inflammatory cytokines, and lipocalin-2 (Lcn2) in the LPS-injected brain of mice (n = 3). (D) C8G expression in different cell types of the CNS. Microglia, astrocytes, and bEnd.3 mouse brain endothelial cells (n = 3 each) were incubated with LPS (1 μg/ml) alone or with L/I [LPS (1 μg/ml) and IFN-γ (50 units/ml)] as indicated. Primary cultured hippocampal neurons were incubated with conditioned medium of mixed glial cells stimulated with L/I. After a 6 h treatment, C8G level was measured with traditional RT-PCR. (E) C8G induction by inflammatory stimuli in mouse primary cultured astrocytes (n = 3). Cells were incubated with L/I, recombinant mouse TNF-α (10 ng/ml), IL-1β (10 ng/ml), IL-6 (10 ng/ml), IFN-γ (50 units/ml), IL-10 (10 ng/ml), IL-4 (10 ng/ml), or IL-13 (10 ng/ml) for 6 h. Mouse liver was used as a positive control. (F) C8G mRNA induction by inflammatory stimuli in human astrocytes (n = 3). Astrocytes were incubated with recombinant human TNF-α, IL-1β, IL-6, IFN-γ, IL-10, IL-4, or IL-13 protein or their combination (Mix: TNF-α + IL-1β + IFN-γ) with the same concentration as mouse astrocytes for 6 h. HepG2 cells were used as a positive control. The assay (n = 3; done in triplicate) was repeated at least two times (inter-assay coefficients of variation were < 20%). Data are mean ± SEM. *P < 0.05 versus control (Con). Unpaired non-parametric test in A (Mann-Whitney) and D (Welch’s t-test); Dunnett's multiple comparison post hoc tests after one-way ANOVA in B, C, E and F.
Using different types of cultured CNS cells, we found that significant C8G mRNA expression was mainly detected in astrocytes after LPS/IFN-γ treatment (Fig. 2D). Considering the temporal kinetics of C8G induction in the inflamed brain and the expression profile of C8G in cultured cells, we speculate that pro-inflammatory cytokines induce C8G expression in astrocytes during neuroinflammation. In primary cultured mouse and human astrocytes, treatment with LPS/IFN-γ, IL-1β, IL-6, and IFN-γ significantly induced C8G expression; however, anti-inflammatory cytokines, such as IL-10, IL-4, and IL-13, did not produce such effects (Fig. 2E and F).
We also used an LPS intracerebroventricular model of neuroinflammation to investigate C8G induction further. A similar pattern of C8G induction was observed in the LPS intracerebroventricular model (Supplementary Fig. 3B). To determine the direct effect of pro-inflammatory cytokines on C8G induction, we blocked TNF-α, IL-1β, or IL-6 using neutralizing antibodies in the astrocyte cultures following stimulation with LPS/IFN-γ, and then assessed C8G induction (Supplementary Fig. 3C). Data revealed that C8G induction was significantly reduced by neutralizing IL-1β and IL-6, but not TNF-α. These results indicate that C8G is potentially inducible by IL-1β and IL-6 in neuroinflammatory conditions.
C8G attenuates neuroinflammation and memory impairment in 5xFAD mice
To determine the potential anti-inflammatory and neuroprotective effects of C8G in a chronic Alzheimer’s disease model, we delivered recombinant C8G protein into the brains of 5xFAD mice using mini-osmotic pumps (Fig. 3A). To minimize brain injury during the experiment, a guide cannula was implanted 14 days before C8G injection. C8G protein was slowly infused into 5xFAD mice (intracerebroventricularly) during the experimental period (0.5 µl/h). The increased number (cortex: P = 0.0202; hippocampus: P = 0.0042) and altered morphology (cortex: P = 0.0058; hippocampus: P = 0.0166) of microglia in the cortex and hippocampus of 5xFAD mice were significantly reversed by intracerebroventricular C8G infusion (Fig. 3B–D and Supplementary Fig. 4). In addition, the elevation of pro-inflammatory cytokines [TNF-α (P = 0.0312) and IL-1β (P < 0.0004)] in 5xFAD mice was significantly attenuated by intracerebroventricular C8G infusion (Fig. 3E). In terms of astrocytes, we did not observe clear differences between 5xFAD and C8G-infused 5xFAD mice (Supplementary Fig. 4). This implies that C8G infusion failed to reduce astrogliosis in the 5xFAD mice.
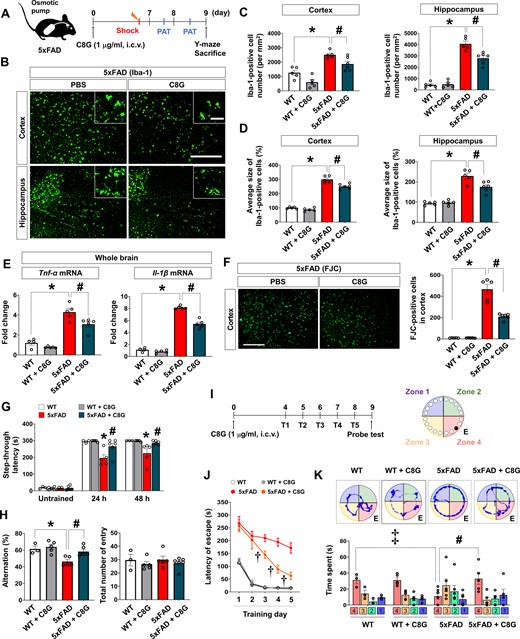
C8G attenuates neuroinflammation and behavioural deficits in 5xFAD mice. (A) Scheme of the experiment. 5xFAD mice and WT littermates were used (8-months-old, male). A guide cannula was implanted 14 days before vehicle or C8G protein (1 μg/ml) infusion into the left lateral ventricle using an osmotic pump (0.5 μl/h infusion rate). Number of mice: wild-type (WT; n = 5), WT + C8G (n = 5), 5xFAD (n = 5), and 5xFAD + C8G (n = 6). (B) Immunohistochemistry for Iba-1. Scale bar = 400 μm (inset = 50 μm). (C) Quantification of microglia. (D) Quantification of microglia morphology. (E) Expression of pro-inflammatory cytokines (TNF-α and IL-1β) in the brain. (F) Degenerative cells in the brain of vehicle- or C8G-infused 5xFAD mice. Degenerative cells were labelled by Fluoro-Jade® C (FJC) staining. Scale bar = 400 μm. (G) Passive avoidance test (PAT). Memory retention was recorded as the latency of the mice in entering the dark compartment after a learning session (electric shock, 2.5 mA). (H) Y-maze spontaneous alternation test. (I) Scheme of the experiment for the Barnes maze task. Number of mice: WT (n = 3), WT + C8G (n = 5), 5xFAD (n = 6), and 5xFAD + C8G (n = 5). Mice were given two trials a day for 5 days. The probe test was performed 24 h after the final training session. (J) Latency time in training session. (K) Time spent in quadrants during the probe test. All data are mean ± SEM. *P < 0.05; #P < 0.05; ‡P < 0.05, Welch’s t-test in C–G (within the time point), H, and K; †P < 0.05, two-way ANOVA with Bonferroni post hoc test in J. White bars = WT; grey = WT + C8G; red = 5xFAD; dark green = 5xFAD + C8G. n.s. = not significant.
To examine whether C8G protects from neurodegeneration, we conducted Fluoro-Jade® C staining of the brain tissue slides from 5xFAD mice with or without C8G infusion. The number of Fluoro-Jade® C -stained cells was significantly increased in the cortex and hippocampal regions of 5xFAD mice (Fig. 3F and Supplementary Fig. 5A). Conversely, the number of Fluoro-Jade® C -stained degenerating cells was significantly reduced in the brains of C8G-infused 5xFAD mice (Fig. 3F, P = 0.0041). However, C8G did not affect the number of amyloid beta plaques in the brain of 5xFAD mice (Supplementary Fig. 5B).
To determine whether impaired spatial memory could be reversed by C8G administration, 5xFAD mice were subjected to a passive avoidance test. The step-through latency after electric shock was noticeably shorter in 5xFAD mice than in control mice (Fig. 3G; 24 h: P = 0.0169; 48 h: P = 0.0173). However, this reduction in latency was reversed in C8G-infused 5xFAD mice. In a Y-maze test, the impaired spatial memory in 5xFAD mice was alleviated by C8G infusion (Fig. 3H;P = 0.008). In a Barnes maze test (Fig. 3I), the latency in entering the escape box during the training sessions was significantly different between 5xFAD mice and C8G-infused 5xFAD mice [Fig. 3J; 3 days (P = 0.0023); 4 days (P < 0.0001); 5 days (P = 0.0002)]. Compared with vehicle-treated 5xFAD mice, C8G-infused 5xFAD mice showed a significant decrease in the escape latency from Days 3 to 5, indicating that memory improvement was meditated by C8G. Similarly, during the probe-trial session, C8G infusion significantly increased the time spent in the target quadrant in 5xFAD mice, which supports C8G-mediated memory improvement (Fig. 3K;P = 0.0144).
Knockdown of C8G exacerbates the pathology of Alzheimer’s disease by enhancing neuroinflammation
To elucidate the role of C8G in Alzheimer’s disease pathogenesis and neuroinflammation, we examined the effects of C8G knockdown on 3-month-old 5xFAD mice. For this investigation, AAV-shRNA of C8G (109 TU/ml, 1.5 μl) was delivered through intracerebroventricular injection to locally reduce C8G gene expression. AAV-shRNA scramble of C8G was used as a control (Fig. 4A). Virus expression was broadly observed via an mCherry signal (Supplementary Fig. 6), and successful knockdown of C8G expression was confirmed at 7 days after virus injection (Fig. 4B). The infection rate and knockdown efficacy were 67.7% and 63.3%, respectively. We also conducted Fluoro-Jade® C staining, which labels degenerating cells (Schmued et al., 2005; Brown and Sawchenko, 2007; Chidlow et al., 2009), of the brain tissue slides from 5xFAD mice infected with shRNA scramble (Scr) or shRNA C8G (shC8G) (Fig.4C). Data revealed that the number of Fluoro-Jade® C-stained cells and amyloid-β plaques in the cortex and hippocampal regions of shC8G-injected 5xFAD mice was significantly higher than in these regions in scramble-injected 5xFAD mice (Fig. 4C). To investigate the role of C8G on neuroinflammation in 5xFAD mice, we compared glial activation between the two groups. Immunohistochemistry data of GFAP and Iba-1 revealed a striking increase in the number of astrocytes and microglia as well as hypertrophic microglia by knockdown of C8G (Fig. 4D). In addition, we also found that the expression of pro-inflammatory cytokines (TNF-α and IL-1β) were elevated by C8G knockdown (Fig. 4E). As described above, injection of recombinant C8G did not alter the number of astrocytes or amyloid plaques in 5xFAD mice (Supplementary Fig. 4 and 5). We cannot precisely explain this discrepancy; however, differences in the age of animals used or the extent of disease progression might be potential reasons (3 months of age in the C8G knockdown experiments; 8 months of age in the recombinant C8G experiments).
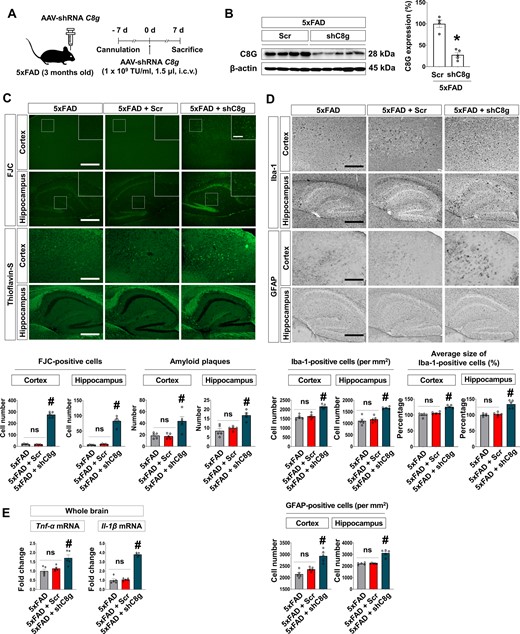
Knockdown of C8G exacerbates the pathology of Alzheimer’s disease by enhancing neuroinflammation. (A) Scheme of the experiment. 5xFAD mice (3 months of age) were given an intracerebroventricular (i.c.v.) injection of AAV-shRNA Scramble (Scr) or C8g (shC8g) at 7 days using a stereotaxic device and microinjector. (B) Knockdown of C8g in the mouse brain. Immunoblot data show the decreased C8G expression. (C) Degenerative cells and amyloid-β plaques in the brain of Scr- or shC8g-infused 5xFAD mice. Degenerative cells or amyloid plaques were labelled by Fluoro-Jade® C (FJC) or Thioflavin-S staining. Scale bar = 400 μm. (D) Immunohistochemistry for Iba-1 and GFAP. Scale bar = 400 μm. (E) Expression of pro-inflammatory cytokines (TNF-α and IL-1β) in the brain of mice (n = 5 each). Knockdown of C8g elevated the expression of pro-inflammatory cytokines in the brain of 5xFAD mice. Data are mean ± SEM. *P < 0.05 versus Scrambled, non-parametric test (Mann-Whitney) in B; #P < 0.05, Tukey’s multiple comparison post hoc test after one-way ANOVA in C–E. n.s. = not significant.
Knockdown of C8G exacerbates LPS-induced neuroinflammation
To demonstrate the direct role of C8G in the neuroinflammatory brain condition, we confirmed the effect of C8G knockdown in the LPS-induced inflamed brain (Supplementary Fig. 7). Since an intracerebroventricular injection of LPS can evoke rapid and aggressive glial activation (Zujovic et al., 2001; Lund et al., 2006; Beynon and Walker, 2012), we considered this neuroinflammation model unsuitable for our purpose, which was to examine the potential accelerating effects of C8G knockdown on neuroinflammation. Therefore, we used the systemic LPS injection model, which induces a slowly progressing, modest neuroinflammation (Chung et al., 2010; Biesmans et al., 2013; Zhao et al., 2019). Our data revealed that this knockdown significantly aggravated LPS-induced astrogliosis (P = 0.0079) and microgliosis (P = 0.0317) and promoted the alteration of microglial morphology (P = 0.0317) (Supplementary Fig. 7C). A trend towards microglia activation triggered by C8G knockdown was observed in untreated mice; this may have been due to brain injury caused by guide cannula injection in control mice. In addition, we found that C8G enhanced the expression of pro-inflammatory cytokines [TNF-α (P = 0.0079) and IL-1β (P = 0.0079)] (Supplementary Fig. 7D).
C8G suppresses inflammatory activation of microglia
To determine the potential role of C8G in microglial activation and neuroinflammation, recombinant C8G protein was applied to microglial cells stimulated with LPS. The results indicated a significant inhibitory effect of C8G on LPS-induced NO production in the BV-2 mouse microglial cell line (Fig. 5A, left; P < 0.001 versus LPS-treated group) and primary microglial cells (Fig. 5A, right; P < 0.01 versus LPS-treated group).
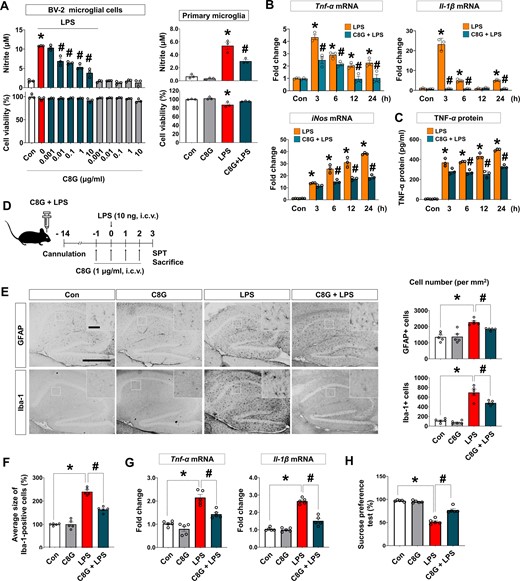
Recombinant C8G protein inhibits LPS-induced microglial activation. (A) C8G inhibited LPS-induced NO production in BV-2 microglial cell lines and primary microglia. BV-2 cells were stimulated with LPS (100 ng/ml) for 24 h after a 2 h pretreatment with different concentrations of C8G (n = 3 each). (B) Recombinant C8G protein inhibits the levels of pro-inflammatory cytokines and NO synthase 2 (Nos2), which were upregulated by LPS in microglial cells. After a 2 h C8G pretreatment (1 μg/ml), BV-2 microglia were exposed to LPS (100 ng/ml) for 6 h (n = 3 each). (C) Recombinant C8G protein inhibits TNF-α production in BV-2 cells. After a 2 h C8G pretreatment (1 μg/ml), BV-2 microglia were exposed to LPS (100 ng/ml) for 24 h. The TNF-α protein level in cultured medium of BV-2 cells was assessed using sandwich ELISA (n = 3 each). The assay (n = 3) was repeated at least twice (inter-assay coefficients of variation were < 20%). (D) A schematic of the experiment with an intracerebroventricular LPS-induced neuroinflammation model. C57BL/6 mice (10 weeks old) were administered vehicle (PBS), C8G (1 μg/ml, 3 μl), and/or LPS (10 ng per brain, 3 μl) using a stereotaxic device and microinjector. (E) Immunohistochemistry of hippocampal gliosis. Reactive astrocytes and microglia in the hippocampus were immunostained with anti-GFAP and anti-Iba-1 at 3 days after LPS injection. Scale bar = 400 μm (inset = 50 μm). Quantified graphs of gliosis are shown in the right panels (n = 5 each). (F) Quantification of activated microglia morphology (n = 5 each). (G) Expression of pro-inflammatory cytokines (TNF-α and IL-1β) in the brains of mice (n = 5 per treatment). (H) Sucrose preference test (n = 5 per treatment). Data are shown as mean ± SEM. *P < 0.05 versus control (Con); #P < 0.05 versus LPS, unpaired non-parametric test (Mann-Whitney) or Welch’s t-test. White bars = control; grey = C8G; red = LPS; dark green = C8G + LPS.
We also examined whether C8G modulates the production of pro-inflammatory cytokines in microglia. The mRNA expression of LPS-induced pro-inflammatory cytokines (TNF-α and IL-1β) and inducible nitric oxidase (iNOS) was clearly inhibited by C8G in BV-2 cells (Fig. 5B and Supplementary Fig. 8A). Additionally, elevated TNF-α protein secretion by LPS treatment in BV-2 cells was inhibited by the addition of C8G (Fig. 5C). However, C8G did not affect LPS/IFN-γ-induced NO production in cultured astrocytes (Supplementary Fig. 8B), suggesting that astrocyte-derived C8G may act on microglia but not on astrocytes.
To test the anti-inflammatory effect of C8G in vivo, the severity of neuroinflammation and depression-like behaviour (loss of sucrose preference) were assessed following LPS and recombinant C8G protein administration (Fig. 5D). C8G injection significantly reduced the LPS-induced increase in the cell number of GFAP-positive astrocytes (Fig. 5E;P = 0.0079) and Iba-1-positive microglia (Fig. 5E;P = 0.0259). C8G injection also altered LPS-induced changes in microglial morphology (Fig. 5F;P < 0.001). Moreover, LPS-induced mRNA expression of pro-inflammatory cytokines [Fig. 5G;Tnfa (TNF-α; P = 0.0079) and Il1b (IL-1β; P < 0.0079)] and loss of sucrose preference (Fig. 5H;P = 0.0003) were clearly alleviated in C8G-injected mice.
C8G suppresses oligomeric amyloid-β-induced microglial activation and confers neuroprotection
Unlike other complement components, C8G is a member of the lipocalin protein family and has a core beta-barrel structure forming a calyx with a distinct binding pocket for small molecules or ligands (Lovelace et al., 2008). Therefore, we explored whether C8G affects amyloid-β neurotoxicity. For this, we generated ADDLs (Supplementary Fig. 9A) and explored the effects of C8G on ADDL neurotoxicity using the HT-22 mouse hippocampal neuronal cell line. Consequently, we found that C8G treatment did not significantly influence the direct neurotoxicity of ADDL (Supplementary Fig. 9B).
Because ADDL is not only a neurotoxin but also a strong stimulator of microglial activation (Ii et al., 1996; Pan et al., 2009), we also investigated whether C8G indirectly reduces ADDL neurotoxicity by modulating ADDL-induced microglial activation. Activated microglial cells release neurotoxic substances or pro-inflammatory cytokines; thus, we designed a non-contact co-culture system (Fig. 6A). We collected the conditioned medium from BV-2 microglial cells incubated with ADDL for 24 h with and without pretreatment with C8G. The conditioned medium was then added to HT-22 neurons and cultured for 24 h. C8G pretreatment almost completely abolished the neurotoxicity of ADDL-treated microglial conditioned medium (Fig. 6A;P < 0.0001). Similar results were obtained with LPS-stimulated microglial conditioned medium (Fig. 6A;P < 0.0001). These results imply that C8G protects neuronal cells against ADDL-induced microglial toxicity.
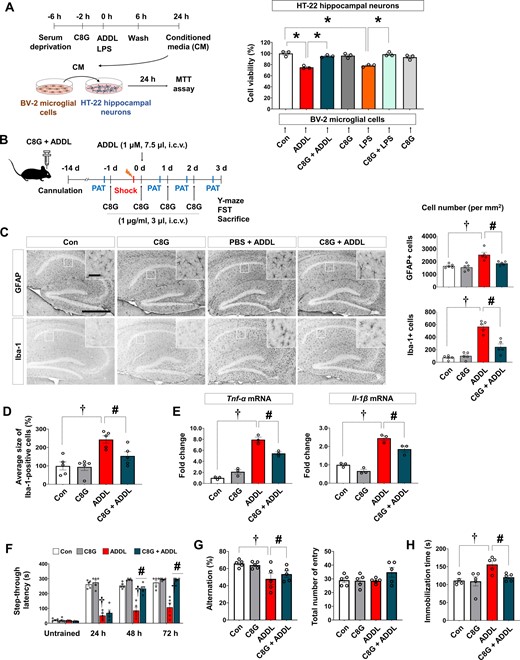
C8G attenuates neuroinflammation and behavioural deficits in an ADDL-induced Alzheimer's disease model. (A) Effect of C8G on microglia-mediated ADDL-induced neurotoxicity. BV-2 microglial cells were stimulated by ADDL (2 μM) or LPS (100 ng/ml) for 6 h with/without recombinant C8G protein preincubation (1 μg/ml) for 2 h. Cells were washed with PBS, and new serum-free medium was applied. Conditioned medium (CM) was harvested after 18 h. HT-22 hippocampal neuronal cells were cultured with conditioned medium with/without recombinant C8G protein preincubation for 24 h. Cell viability was measured using the MTT assay (n = 3). (B) Scheme of the experiment including the ADDL-induced Alzheimer’s disease model. C57BL/6 mice (10 weeks old, n = 6 per group) were administered with a vehicle (0.1% DMSO diluted in PBS), ADDL (1 μM, 7.5 μl, i.c.v.), ADDL + C8G (1 μg/ml, 3 μl, i.c.v.), and C8G using a stereotaxic device and microinjector. (C) Immunohistochemistry of hippocampal gliosis. Scale bar = 400 μm (inset = 50 μm). Quantification of gliosis is shown in the right panel. (D) Quantification of activated microglia morphology (n = 5 each). (E) Expression of pro-inflammatory cytokines (TNF-α and IL-1β) in the brain of the mice (n = 3 each). (F) Passive avoidance test (PAT). Memory retention recorded as the latency of the mice in entering the dark compartment after a learning session (electric shock, 2.5 mA). (G) Y-maze spontaneous alternation test. (H) Forced swim test (FST). Depression-like behaviour was defined as the latency in immobilization in the FST. Data are shown as mean ± SEM. †P < 0.05; #P < 0.05 versus ADDL, unpaired non-parametric test (Mann-Whitney) or Welch’s t-test. White bars = control; grey = C8G; red = ADDL; dark green = C8G + ADDL.
As C8G mitigated LPS-induced microglial activation and neuroinflammation in vitro and in vivo, we also evaluated the anti-inflammatory effects of C8G in the ADDL-injected mouse brain. C8G was administered daily for 4 days through an implanted guide cannula starting the day before intracerebroventricular ADDL injection (Fig. 6B). Reactive astrogliosis and microgliosis were observed in the hippocampus of ADDL-injected mouse brains but not in the control groups, which were injected with PBS, C8G, and reverse ADDL (Fig. 6C and Supplementary Fig. 9C). However, the administration of recombinant C8G protein before ADDL injection significantly reduced astrogliosis (Fig. 6C;P = 0.0228) and microgliosis (Fig. 6C;P < 0.001), and it suppressed alteration in microglial morphology (Fig. 6D;P = 0.0207). In addition, C8G mitigated the pro-inflammatory cytokines [TNF-α (P = 0.0154) and IL-1β (P = 0.0486)] induced by ADDL (Fig. 6E). To determine whether C8G had neuroprotective effects in ADDL-injected brain, we estimated neuronal cell death in the hippocampal region (Supplementary Fig. 9D). Our data revealed that neuronal density in the hippocampal CA3 region was significantly reduced by ADDL injection compared with vehicle injection, and this decrease was attenuated by C8G injection.
In addition, we also explored whether C8G attenuates ADDL-induced acute memory impairment. Mice were subjected to the passive avoidance test (hippocampus-dependent contextual learning and memory test; Fig. 6F) and Y-maze spontaneous alternation test (spatial learning and memory test; Fig. 6G). In the passive avoidance test, all groups showed a similar initial latency with respect to the time that mice took to move into the dark compartment of the box without any stimulation. After an electric shock, the step-through latency was greatly increased, indicating that memory retention was well formed by electric stimulation. In ADDL-injected mice, memory retention was significantly impaired at 24 h, 48 h, and 72 h after the shock. However, mice receiving both C8G and ADDL showed better memory retention from 48 h than ADDL-injected mice (Fig. 6F). The Y-maze test was performed 1 h after the last session of the passive avoidance test. Our data for this test showed that the impaired spatial memory caused by ADDL injection was alleviated by C8G preinjection (Fig. 6G;P = 0.018). The total number of entries did not differ among the groups. Additionally, we assessed the effect of C8G on ADDL-induced depression-like behaviour (in a forced swim test). A significant increase in immobility observed in ADDL-injected mice was alleviated by C8G injection (Fig. 6H;P = 0.0161). Behavioural deficiencies were not detected in the mice injected with PBS or reverse ADDL (Supplementary Fig. 9C).
C8G protein interacts with S1PR2 in microglia
To uncover the molecular mechanisms underlying the modulation effects of C8G, we searched for binding partners of C8G. First, we utilized BioPlex 2.0 (Biophysical Interactions of ORFeome-derived complexes), a database that uses robust affinity purification-mass spectrometry data to elucidate protein interaction networks and co-complexes. Among several candidates of novel interaction partners of C8G, we focused on S1PR2, which is known to play a critical role in vascular inflammation (Zhang et al., 2013; Kim et al., 2015); the S1P signalling pathway is closely associated with inflammatory diseases and Alzheimer’s disease (Maceyka et al., 2012).
To confirm interactions between C8G and S1PR2 in microglia, we performed a pull-down assay using GST-fused C8G protein (GST-C8G) and two different methods in BV-2 microglial cells (Fig. 7A): (i) GST-C8G incubation with total cell lysates (Assay 1); or (ii) GST-C8G incubation with intact cells to examine the role of cell surface S1PR2 (Assay 2). Endogenous C8G protein expression was not detected in BV-2 cells. The results from both assays showed specific interactions between C8G and S1PR2 (Fig. 7A). To identify the C8G-binding domain of S1PR2, we generated two HA-tagged deletion derivatives of full-length S1PR2 (mS1PR2 FL): N-terminus region deletion (mS1PR2 ΔN; amino acid 1–34) and extracellular loop deletion (mS1PR2 ΔECL; amino acid 96–108, 174–189, and 268–271) (Fig. 7B). In the co-immunoprecipitation assay, C8G was able to co-immunoprecipitate with mS1PR2 ΔECL derivatives but not with mS1PR2 ΔN. This indicates that C8G binds to the N-terminus region of S1PR2. Additionally, we confirmed the interaction between endogenous C8G and S1PR2 proteins in the brain tissue (Fig. 7C).
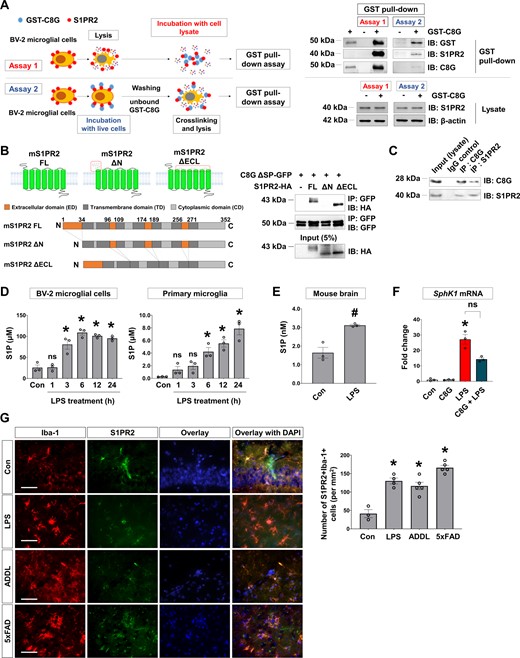
S1PR2 is a novel interaction partner of C8G. (A) A schematic of two different methods applied for the GST pull-down assay using recombinant GST-C8G (10 μg/ml) in BV-2 microglial cell lysates (Assay 1) or following extracellular GST-C8G (10 μg/ml) application to intact BV-2 microglial cells for 2 h (Assay 2). Recombinant GST-C8G protein was used as a positive control. S1PR2 in lysate and β-actin were used as loading controls. (B) Schematics of full-length (mS1PR2-FL), N-terminus-deleted mutant (mS1PR2-ΔN), and extracellular loops-deleted mutant (mS1PR2-ΔECL), and the co-immunoprecipitation assays in which these constructs were used. C8G without a signal peptide (ΔSP-C8G) and S1PR2 mutants were co-expressed in HEK293 cells. The whole-cell lysate of HEK293 expressing the mutants was immunoprecipitated (IP) with anti-GFP antibody and then analysed by immunoblotting with anti-GFP or anti-HA antibody; 5% of the total lysate was used as an input control for immunoprecipitation. (C) Co-immunoprecipitation of endogenous proteins in the systemic LPS-induced inflammatory brain. Mouse whole-brain lysate was immunoprecipitated with anti-C8G or anti-S1PR2 antibody and then immunoblotted with the antibodies indicated. (D) Levels of S1P in BV-2 microglial cells and primary microglia stimulated with LPS (100 ng/ml) (n = 3 per group). The level of S1P was assessed using sandwich ELISA. (E) Level of S1P in the inflamed brain (n = 3 per group). (F) Sphk1 mRNA level in the inflamed brain following systemic LPS injection. C57BL/6 mice (10 weeks old) were administered with vehicle (PBS), C8G (1 μg/ml, 3 μl, i.c.v.), and/or LPS (5 mg/kg, i.p.) before Sphk1 mRNA measurement in the brain. (G) Immunohistochemistry of S1PR2 in LPS-injected (5 mg/kg. i.p.) and Alzheimer’s disease mouse models. Scale bar = 50 μm. Right: Quantification of S1PR2 immunoreactivity in the brain of neuroinflammation and Alzheimer’s disease mice (n = 3 per group). Data are mean ± SEM. *P < 0.05 versus control (Con), Dunnett’s multiple comparison post hoc test after one-way ANOVA; #P < 0.05 versus PBS, Welch’s t-test.
As we hypothesized that C8G-S1PR2 interaction inhibits pro-inflammatory S1P signalling in microglia, we also addressed the biological functions of the C8G-S1PR2 interaction by assessing the expression of S1P and S1PR2. When we evaluated S1P levels following LPS exposure using ELISA, S1P levels were upregulated both in vitro (BV-2 microglial cells and primary microglia cultures; Fig. 7D) and in vivo (in the inflamed brain; Fig. 7E). The expression of sphingosine kinase 1 (SphK1), a lipid kinase that catalyses the conversion of sphingosine to S1P, was also increased in the brain of LPS-injected mice (Fig. 7F;P = 0.0331 versus control); however, C8G did not affect Sphk1 expression. S1PR2 was mainly located in microglia and was upregulated in the brain of LPS-injected and Alzheimer’s disease mice (P < 0.0001) (Fig. 7G and Supplementary Fig. 10A and B). Collectively, our data suggest that pro-inflammatory S1P and microglial S1PR2 are upregulated under neuroinflammatory conditions.
Antagonistic role of C8G towards S1PR2 is neuroprotective in Alzheimer’s disease
We also compared the effect of C8G with that of the S1PR2 antagonist JTE-013 on S1P-induced NO production in microglia (Supplementary Fig. 11A). Both JTE-013 and C8G had a suppressive effect on the S1P-stimulated production of NO in microglia. C8G treatment decreased NO production induced by the S1PR2 agonist CYM-5478 (Supplementary Fig. 11B). In addition, we explored the role of C8G in the RhoA/Rho kinase (ROCK) pathway, which is crucial in microglial activation for migration, phagocytosis, and release of cytokines and chemokines (Barcia et al., 2012; Yan et al., 2012; Borrajo et al., 2014; Roser et al., 2017). RhoA activity was significantly increased by LPS and S1PR2 agonists (S1P and CYM-5478), and this was reversed by C8G pretreatment (Supplementary Fig. 11C). Finally, LPS- or S1PR2-agonist-induced NF-κB activation was inhibited by C8G pretreatment (Supplementary Fig. 11D). These pharmacological experiments indicate that C8G inhibits proinflammatory microglial activation via the specific antagonistic actions on S1PR2 (Supplementary Fig. 11E).
To extend our in vitro assessments, we investigated whether C8G treatment mimics the effect of the S1PR2 antagonist and whether the antagonistic role of C8G towards S1PR2 receptor confers a neuroprotective effect in vivo. For this investigation, we compared the neuroinflammatory status between 5xFAD mice injected with C8G and those injected with the S1PR2 antagonist (JTE-013). In addition, we examined the competitive antagonistic effect of C8G in 5xFAD mice injected with the S1PR2 agonist (CYM-5478) (Fig. 8A). Vehicle, C8G, JTE-013, CYM-5478, or C8G + CYM-5478 were delivered by an osmotic pump for 7 days. Expression of pro-inflammatory cytokines (TNF-α and IL-1β), the number of reactive astrocytes and microglia, and alteration in microglial morphology in the 5xFAD mice injected with C8G or JTE-013 were significantly reduced relative to these measures in vehicle-injected 5xFAD mice (Fig. 8B and C). In addition, mice were subjected to the passive avoidance test (Fig. 8D) and Y-maze spontaneous alternation test (Fig. 8E). In the passive avoidance test, all groups showed a similar initial latency. After the electric shock, memory retention was significantly impaired at 24, 48, and 72 h post-shock in vehicle- and CYM-5478-infused 5xFAD mice. However, mice receiving both C8G and JTE-013 showed better memory retention than vehicle-injected 5xFAD mice (Fig. 8D). In addition, C8G infusion clearly rescued the impairment of memory retention in CYM-infused 5xFAD mice. In the Y-maze test, the impaired spatial memory in the vehicle-infused 5xFAD mice was alleviated by C8G or JTE-013 infusion. Additionally, C8G improved the spatial memory that was previously reduced by CYM-5478 infusion in 5xFAD mice (Fig. 8E).
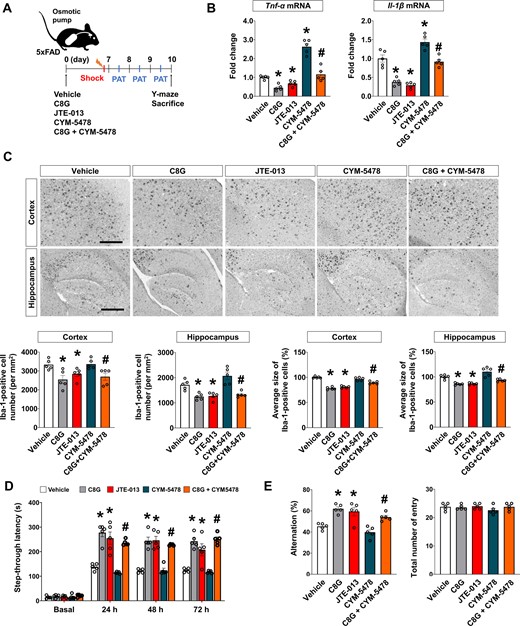
Antagonistic role of C8G to S1PR2 confers a neuroprotective effect via modulation of neuroinflammation in Alzheimer's disease. (A) Scheme of the experiment. A guide cannula was implanted 14 days before infusion of the vehicle (0.1% DMSO diluted in PBS), C8G (1 μg/ml), JTE-013 (S1PR2 antagonist; 1 μM), CYM-5478 (S1PR2 agonist; 1 μM), or C8G + CYM-5478 into the left lateral ventricle using an osmotic pump (0.5 μl/h infusion rate). Five mice were used per group. (B) Expression of pro-inflammatory cytokines (TNF-α and IL-1β) in the brain of the mice. (C) Immunohistochemistry for Iba-1. Scale bar = 400 μm. Bottom panels show quantification of microglia and microglial morphology. (D) Passive avoidance test (PAT). Memory retention recorded as the latency of the mice in entering the dark compartment after a learning session (electric shock, 2.5 mA). (E) Y-maze spontaneous alternation test. Data are mean ± SEM. *P < 0.05 versus vehicle, Dunnett's multiple comparison post hoc tests after one-way ANOVA; #P < 0.05 versus CYM-5478, Welch’s t-test.
Discussion
C8G induction may be a part of the acute phase response in the CNS
The present study revealed that astrocytic C8G is upregulated in the inflamed brain and has an induction pattern that is independent of the complement system. Our results also indicate that C8G is an inhibitor of neuroinflammation as it plays a role in the modulation of microglial activation. Unexpectedly, however, C8G expression levels were higher in the brain and fluids (CSF and plasma) of patients with Alzheimer’s disease than in those of control subjects. Our in vitro experiments showed that C8G was upregulated in astrocytes following stimulation with pro-inflammatory cytokines (mainly IL-1β and IL-6; Fig. 2E and F). These cytokines are major reactants of the acute phase response (APR), which can be defined as an orchestrated response to infection, tissue injury, inflammation, neoplastic growth, and other immunological disorders (Castell et al., 1989; Kramer et al., 2008).
A prominent feature of the APR is the induction of AP proteins, such as complement components, antiproteases, clotting factors, and pentraxins. Such AP proteins include serum amyloid-A (SAA), serum amyloid-P (SAP), and C-reactive protein (CRP) (Pannen and Robotham, 1995). The APR largely functions in restoring homeostasis via actions such as pathogen neutralization, proteolysis, opsonization, debris clearance, and attenuation of local inflammatory responses in order to minimize injury to healthy tissues. The APR has also been found in the CNS, in which microglia, astrocytes, and the choroid plexus mainly participate in the APR. Various AP proteins, such as α1-anti-chymotrypsin (ACT), α-anti-trypsin, SAP, CRP, ceruloplasmin, pentraxin-3, and complement components, have been documented to be secreted by glial cells (McGeer and Rogers, 1992). Upregulation of astrocytic α1-ACT contributes to amyloid deposits (Tyagi et al., 2013), and astrocyte-derived pentraxin-3 limits macrophage-mediated phagocytosis in the Alzheimer’s disease brain (Ko et al., 2012). Although the mechanisms of the brain APR have not been fully elucidated, upregulation of AP proteins in the CNS may be involved in the pathogenic mechanisms of neuroinflammatory or neurodegenerative diseases (Clark et al., 1998; Ko et al., 2012). In the present study, C8G showed a delayed pattern of expression that seems to follow the induction of AP reactants in the LPS-induced neuroinflammation mouse model (Fig. 2C and Supplementary Fig. 3B). Additionally, in our study, C8G was found at higher levels in the brain tissue of three different types of Alzheimer’s disease mouse models [oligomeric amyloid-β toxicity (intracerebroventricular ADDL) and transgenic amyloid-β deposit (5xFAD) models] as well as in the brain and fluids of patients with Alzheimer’s disease (Fig. 1E and F, and Supplementary Fig. 2D and E). Therefore, the secretion of C8G from reactive astrocytes may be a part of a delayed APR to regain brain homeostasis in response to acute inflammation.
Crosstalk between astrocytes and microglia via C8G
Intercellular crosstalk in the CNS is important for initiating and regulating neuroinflammation (Liu et al., 2011). For instance, microglial C1q, TNF-α, and IL-1β prime cytotoxic astrocytes (Liddelow et al., 2017) and microglia-derived transforming growth factor (TGF)-α and VEGF-B regulate astrocyte pathogenic activities (Rothhammer et al., 2018). Microglial IL-1 leads to either astrogliosis (John et al., 2004) or astrocytic production of TGF-β (da Cunha et al., 1993), which in turn attenuates microglial activation (Norden et al., 2014). Importantly, interpretation and understanding of interglial crosstalk may provide a therapeutic strategy for the modulation of neuroinflammation. Typically, initial microglial activation is followed by astrocytic activation. This sequential activation of glia has been reported in various neurodegenerative animal models (Matsumoto et al., 1992; Colburn et al., 1997; Frautschy et al., 1998; Norden et al., 2016). Similarly, C8G-inducing signals may originate from stimulated microglia, which are on the front line in a pathological brain. The resident microglia are activated by pathological stimuli in the early neuroinflammatory phase. The activated microglia produce pro-inflammatory cytokines, such as IL-1β and IL-6, to induce astrocytic C8G expression, and the released astrocytic C8G may then downregulate microglia hyperactivation. Our findings support the notion that C8G mediates a novel means of communication between astrocytes and microglia to suppress CNS inflammation.
C8G as a biomarker for neuroinflammation and Alzheimer’s disease
We observed significant upregulation of C8G compared with the other C8 subunits, with C8A not being detected and C8B showing reduced levels in reactive astrocytes (Fig. 2E and F). Typically, C8G and C8A subunits are induced faster than C8B; they are assembled together intracellularly, after which the rigid C8A-C8G heterodimer binds to C8B non-covalently in the extracellular space (Ng and Sodetz, 1987). Our data imply that the astrocytic C8G may have a distinct induction mechanism and may act in its free form in the CNS. Cys40 of human C8G is involved in the C8A-binding activity, forming a disulphide bond between C8A and C8G (Brickner and Sodetz, 1985). This residue may remain exposed in the free form of C8G and may be a useful target for the design of agents that recognize brain-specific C8G. Moreover, C8G may be a diagnostic biomarker for Alzheimer’s disease, as well as other neurodegenerative diseases.
Microglia-inhibiting C8G may be neuroprotective in Alzheimer’s disease
Our results suggest that the microglial modulation caused by astrocytic C8G in neuroinflammation could confer a beneficial outcome in Alzheimer’s disease (Figs 3 and 6). Neuroinflammation is an important contributor to Alzheimer’s disease pathogenesis (Valmikinathan and Verghese, 1966; McGeer and McGeer, 1999; Calsolaro and Edison, 2016). Although microglia are essential components of adult brain homeostasis, functioning in processes such as clearance of amyloid-β or cell debris (Chen and Trapp, 2016), chronic hyperactivation of microglia, termed ‘dark microglia’ (Bisht et al., 2016), can produce severe pathological features in the brain by releasing excessive pro-inflammatory factors including TNF-α, IL-1β, reactive oxygen species, and reactive nitrogen species. These may exacerbate neurodegeneration and recruit myeloid immune cells across the blood–brain barrier (Zhou et al., 2006; Qu et al., 2012). Accordingly, the modulation of microglial activation in Alzheimer’s disease may open new therapeutic avenues. Our data show that recombinant C8G could suppress neuroinflammation and thus alleviate disease severity in animal models of Alzheimer’s disease (Figs 3 and 6). Moreover, C8G treatment almost abrogated IL-1β expression in neuroinflammation and Alzheimer’s disease models. A recent study demonstrated that the NLRP3 inflammasome assembles inside microglia on activation, leading to increased cleavage and activity of caspase-1 and downstream IL-1β, which has been shown to be essential for the tau pathology (Ising et al., 2019). Thus, our results suggest that C8G may inhibit the inflammasome in microglia. In combination, the results of the present study suggest that C8G has the potential to be developed as a therapeutic treatment.
C8G inhibits the pro-inflammatory S1P signalling pathway in microglia
Based on a series of experiments, we deciphered the mechanism by which C8G inhibits LPS-induced microglial activation. We predicted that S1PR2 interacts with C8G, and the interaction was confirmed by a co-immunoprecipitation assay (Fig. 7A–C). We also experimentally confirmed increased S1P and SphK1 expression under neuroinflammatory conditions and Alzheimer’s disease (Fig. 7D–F). Finally, we revealed that C8G inhibits microglial RhoA/NF-κB activity dependent on S1P signalling (Supplementary Fig. 11). The LPS-induced Toll-like receptor (TLR) 4 signalling pathway reportedly activates SphK1, which phosphorylates sphingosine to generate S1P. Intracellular levels of S1P increase, which are then released into the extracellular space by the S1P transporter (Du et al., 2012). Furthermore, extracellular S1P acts as a ligand of S1PR2, which activates the RhoA/NF-κB pathway (Zhang et al., 2013). Taken together, our results suggest that C8G inhibits the LPS-induced microglial RhoA-ROCK-NF-κB activation pathway by blocking S1PR2.
Contradictory findings exist regarding the role of S1P and S1PR2 signalling in immune and inflammatory responses (Blankenbach et al., 2016). Although several studies have found that S1PR2 counteracts pro-inflammatory responses in chondrocytes (Stradner et al., 2013) and inhibits macrophage recruitment (Michaud et al., 2010), many other studies have reported that S1PR2 promotes pro-inflammatory signalling pathways and increases levels of COX-2, prostaglandin, and cytokines in various cell types (Siehler et al., 2001; Blom et al., 2010; O’Sullivan et al., 2014; Volzke et al., 2014). These contradictory observations suggest that the role of S1PR2 is dependent on the activation of different G-proteins, such as Gi, Gq, and G12/13, coupled with the receptor. For example, S1PR2 inhibits Akt signalling and endothelial cell migration via G12/13 activation (Sanchez et al., 2007; Green et al., 2011; Japtok et al., 2015), whereas it activates Akt to protect from myocardial ischaemia–reperfusion injury via Gi activation (Means et al., 2007). Moreover, Gq activation leads to phospholipase C stimulation and calcium mobilization in a Chinese hamster ovary cell line (Gonda et al., 1999). Therefore, various cellular responses are orchestrated by S1PR2 depending on the G-protein that it is coupled with (Nagahashi et al., 2014; Kwong et al., 2015). Questions regarding the role of S1PR2 in neuroinflammation remain. In the current study, a series of experiments indicated a pro-inflammatory S1PR2-G12/13 -RhoA-NF-κB axis in microglia, which was clearly inhibited by C8G (Supplementary Fig. 11). The anti-inflammatory role of C8G, as a S1PR2 inhibitor, seems to be specific to microglia. The combined results of the present study suggest that C8G is capable of modulating microglial activation by inhibiting pro-inflammatory S1PR2 signalling.
How does C8G affect the ligand binding activity of S1PR2? Our co-immunoprecipitation investigations, which included the deletion constructs of S1PR2, revealed that C8G binds to the N-terminus region of S1PR2 (Fig. 7B). A previous study reported that the hydrophobic tail of S1P interacts with a ligand-binding pocket within the membrane compartment of S1PRs, which is occluded by the N-terminus and ECL (Hanson et al., 2012). The N-terminus acts as a ‘lid', forming a helical cap that limits access to the amphipathic binding pocket, but it also contributes to binding interactions (Hanson et al., 2012). Based on this model, we speculate that C8G-binding to the N-terminus of S1PR2 may obstruct or weaken the interaction between S1P and its binding pocket in S1PR2.
Translational significance of the study and conclusions
Targeting neuroinflammation has been considered a potential therapeutic strategy for the treatment of Alzheimer’s disease and other neurodegenerative diseases (Valmikinathan and Verghese, 1966; Chitnis and Weiner, 2017; Clayton et al., 2017). However, several anti-inflammatory agents, including non-steroidal anti-inflammatory drugs (NSAIDs), have produced unsuccessful clinical results (Valmikinathan and Verghese, 1966). To overcome these limitations, anti-inflammatory treatment strategies require a better understanding of highly contextual glial responses and interglial crosstalk (Kettenmann et al., 2011; Jha et al., 2019). In the present study, we have suggested for the first time that C8G is an astrocyte-secreted anti-inflammatory molecule that modulates microglial activation by the inhibition of the S1PR2-RhoA-ROCK-NF-κB signalling pathway in neuroinflammation and Alzheimer’s disease. C8G seems to mediate an astrocyte–microglia crosstalk in the complex network of neuroinflammation. In previous reports, S1PR2 antagonists or genetic deletion of S1PR2 reportedly reduced microvascular permeability and vascular inflammation in vitro and in vivo (Lee et al., 2009a; Zhang et al., 2013) and protected the brain from stroke-induced ischaemic and inflammatory injury (Kim et al., 2015). In addition, the level of S1PR2 is reportedly enhanced in female experimental autoimmune encephalomyelitis (EAE) model mice, and its manipulation attenuated EAE disease severity (Cruz-Orengo et al., 2014). Therefore, anti-inflammatory C8G as an inhibitor of microglial S1PR2 signalling may be a useful intervention in not only neuroinflammatory (e.g. multiple sclerosis) and neurodegenerative diseases (e.g. Alzheimer’s disease and Parkinson’s disease) but also in cases of brain injury (e.g. stroke). Taken together, our findings suggest that C8G may be useful as an index of the severity of neuroinflammation in the pathological brain, and the immune-modulatory role of C8G could form the basis of therapeutic intervention in CNS injury and disorders.
Funding
This work was supported by a grant from the Basic Science Research Program through the National Research Foundation (NRF), which is funded by the Korean government (Ministry of Science, ICT and Future Planning, MSIP) (2016M3C7A1904148, NRF-2017R1A5A2015391).
Competing interests
The authors report no competing interests.
Supplementary material
Supplementary material is available at Brain online.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}