




Abstract
The aim of this study is to evaluate the diagnostic value of genome sequencing in children with epilepsy, and to provide genome sequencing-based insights into the molecular genetic mechanisms of epilepsy to help establish accurate diagnoses, design appropriate treatments and assist in genetic counselling.
We performed genome sequencing on 320 Chinese children with epilepsy, and interpreted single-nucleotide variants and copy number variants of all samples. The complete pedigree and clinical data of the probands were established and followed up. The clinical phenotypes, treatments, prognoses and genotypes of the patients were analysed.
Age at seizure onset ranged from 1 day to 17 years, with a median of 4.3 years. Pathogenic/likely pathogenic variants were found in 117 of the 320 children (36.6%), of whom 93 (29.1%) had single-nucleotide variants, 22 (6.9%) had copy number variants and two had both single-nucleotide variants and copy number variants. Single-nucleotide variants were most frequently found in SCN1A (10/95, 10.5%), which is associated with Dravet syndrome, followed by PRRT2 (8/95, 8.4%), which is associated with benign familial infantile epilepsy, and TSC2 (7/95, 7.4%), which is associated with tuberous sclerosis. Among the copy number variants, there were three with a length <25 kilobases. The most common recurrent copy number variants were 17p13.3 deletions (5/24, 20.8%), 16p11.2 deletions (4/24, 16.7%), and 7q11.23 duplications (2/24, 8.3%), which are associated with epilepsy, developmental retardation and congenital abnormalities. Four particular 16p11.2 deletions and two 15q11.2 deletions were considered to be susceptibility factors contributing to neurodevelopmental disorders associated with epilepsy.
The diagnostic yield was 75.0% in patients with seizure onset during the first postnatal month, and gradually decreased in patients with seizure onset at a later age. Forty-two patients (13.1%) were found to be specifically treatable for the underlying genetic cause identified by genome sequencing. Three of them received corresponding targeted therapies and demonstrated favourable prognoses.
Genome sequencing provides complete genetic diagnosis, thus enabling individualized treatment and genetic counselling for the parents of the patients. Genome sequencing is expected to become the first choice of methods for genetic testing of patients with epilepsy.
Introduction
Epilepsy is one of the most common diseases of the CNS. It most prominently appears in childhood and is characterized by recurrent seizures. According to the World Health Organization, epilepsy is present in about 50 million people worldwide. There are a multitude of causes that can lead to epilepsies including infections, genetic abnormalities and structural, metabolic or immune abnormalities without clear underlying genetic components. In about half of cases, however, the causes remain entirely unknown.1
Among the genetic causes, a variety of sequence alterations can lead to epilepsies, such as single-nucleotide variants (SNVs), copy number variants (CNVs), small insertions or deletions (indels) and repeat expansions, each generating pathogenic alleles of one or multiple epilepsy genes. Many of these variants are associated not only with seizures but also other symptoms. Furthermore, different variants of the same gene can lead to distinct epileptic syndromes, and what appears as the same epileptic syndrome may be caused by variants of different genes.2 Since the identification of the first epilepsy gene in 1995,3 the number of genes associated with epilepsy4 and the types of genetic test identifying variants have significantly increased. Most patients with epilepsy have a good prognosis provided they are correctly treated, but the most appropriate treatment for any individual patient will depend on the underlying cause of the disease in that patient. Hence, genetic testing is very important to establish definitive diagnoses, accurate treatments and genetic counselling.
There are several techniques available for genetic testing, such as next generation sequencing, capture-based targeted sequencing, exome sequencing (exome sequencing) and genome sequencing (genome sequencing).5 Among these, genome sequencing has several advantages. First, it can sequence the entire genome of an individual and identify not only SNVs but also CNVs and small fragment insertions and deletions simultaneously, the last rendering it superior to chromosomal microarray analysis (CMA) that can only identify CNVs of larger sizes.6 Second, genome sequencing is better than capture-based targeted sequencing (which is limited to the identification of known genes) and exome sequencing, which misses potentially pathogenic non-coding and intronic SNVs (for instance a variation in an intronic part of SCN1A),7 non-coding repeat expansions (for instance in familial adult myoclonic epilepsy),8 SNVs in untranslated regions of mRNAs (unless specifically designed to identify them), non-coding RNAs and small and large structural variants. Third, library preparation for genome sequencing is simpler and faster than for capture-based targeted sequencing or exome sequencing. Fourth, genome sequencing allows for a single test to capture almost all genomic variations in an unbiased manner due to even coverage throughout the genome as DNA amplification can be kept to a minimum with no capture step needed for enrichment.9
Despite these numerous advantages, genome sequencing has not widely been applied to epilepsy patients (especially paediatric epilepsy patients) probably because of the higher costs associated with genome sequencing over other techniques. However, with costs gradually decreasing, genome sequencing should become an invaluable alternative to genetic testing by other methods even in clinical settings outside specific research projects.
Here, we performed genome sequencing on a cohort of 320 Chinese children with epilepsy and assess its diagnostic yield and clinical utility.
Materials and methods
Participant recruitment and eligibility criteria
A cohort of 320 paediatric patients with epilepsy was recruited from the Department of Neurology of Shenzhen Children’s Hospital (Shenzhen, China) during the period October 2016 to December 2017. All patients were evaluated clinically by paediatric neurologists and diagnosed based on the type of seizure, electroencephalographic findings and brain imaging results. The criteria for inclusion were age <18 years and diagnosis of epilepsy based on International League Against Epilepsy criteria (2017)10 and suspicion of underlying genetic causes. Excluded were patients with seizures probably due to acquired brain injuries, including (but not limited to): head trauma, brain tumours and CNS infections; patients with seizures probably due to cerebrovascular pathological changes; patients having undergone transplant surgery, stem cell therapy or transfusion of allogeneic blood products within the past 6 months; and patients with haemolytic disease.
The study was approved by the Institutional Review Board on Bioethics and Biosafety of BGI and the ethics committee of the Shenzhen Children’s Hospital. Written informed consent was obtained from all parents or legal guardians of the patients according to the Declaration of Helsinki principles.
Genome sequencing and analysis
Genomic DNA was extracted from 2–4 ml of peripheral blood collected from patients and their parents (if available) with the QIAamp DNA Blood Mini Kit (Qiagen). Patient samples were subjected to genome sequencing by BGI-Shenzhen performing paired-end 100 sequencing using the BGISEQ-500 platform as previously described.11,12 The patients were analysed as singletons by genome sequencing while parental samples of patients with SNVs deemed pathogenic or probably pathogenic were subjected to Sanger sequencing to evaluate potential parental origin of the variants. Pedigrees were obtained through interviews, and clinical data of probands were followed up for 2 years.
All samples were analysed for SNVs and CNVs. Deep sequencing data were aligned to the reference GRCh Build 37 (hg19) and variants were called using the Edico Dragen analysis pipeline.13 Edico Genome's Dragen Bio-IT Platform is based on the company's Dragen Bio-IT Processor, a bioinformatics application-specific integrated reference-based mapping, aligning, sorting, deduplication and variant calling.
Variants were annotated using bcfanno (v.1.4; https://github.com/shiquan/bcfanno; accessed 10 November 2021) in the Frequency data (The Exome Aggregation Consortium,14 Genome Aggregation Database,15 1000 Genomes16) gene-disease database (ClinVar),17 Clinical Genomic Database,18 Online Mendelian Inheritance in Man database (OMIM),19 and Human Gene Mutation Database (HGMD).20 The predictive programs SIFT (v.5.2.2),21 PolyPhen-2 (v.2.2.2),22 MutationTaster (NCBI 37/Ensembl 69)23 and PROVEAN (v.1.1.5)24 were used to access the pathogenic potential of the variants.
The procedure for SNVs filtering, classification and interpretation is shown in Supplementary Fig. 1A. First, SNVs with allele frequencies above 5% were excluded. Second, the filtered SNVs were divided into functional and non-functional region variants, and the non-functional region variants were filtered and interpreted by the ClinVar and HGMD databases. Third, variants from the functional regions and filtered variants from the non-functional regions were then assigned to one of four categories (pathogenic, likely pathogenic, uncertain significance, negative) based on clinical significance according to the American College of Medical Genetics and Genomics (ACMG) Guidelines Revisions.25,26 Fourth, combined with phenotypes, we searched the variants through OMIM, HGMD and PubMed databases, and the pathogenicity of a variant was then determined according to whether it belonged to a known epilepsy-associate gene or a known epilepsy phenotype in a syndrome caused by the gene. Finally, Sanger sequencing was done for all patients with pathogenic, likely pathogenic sequence alterations and uncertain significance favouring pathogenicity as well as for their parents for segregation analyses and for verification of the diagnostic value of the variants.
CNV calls were generated using the SpeedSeq SV pipeline (v.0.0.3a),27 which uses Lumpy (v.0.2.9)28 to process samples and SVtyper (v.0.0.2)27 to genotype variants. The pipeline outputs deletions, duplications and inversions, and identifies breakpoints that cannot be assigned to any of these three types of genomic alteration. Low coverage data were analysed based on a pipeline29 that calculates karyotype and ploidy based on the ratio of CNVs compared to reference genomes. Interpretation of CNVs was performed using in-house laboratory interpretation rules (Supplementary Fig. 1B), and re-evaluated using ACMG's latest CNV interpretation criteria.26 If a CNV involves a known epilepsy gene and the epilepsy phenotype was consistent with variants of that gene, we considered the CNV pathogenic. If a CNV did not involve a known epilepsy gene, we searched for what phenotypes associated with that CNV had ever been reported. If most of the reported phenotypes matched with the ones of our case and the CNV had been reported to be associated with epilepsy, then we considered this CNV to be pathogenic. Last, if a CNV did not involve a known epilepsy gene or was never reported to be associated with epilepsy, we considered this CNV to be of unknown clinical significance. We divided pathogenic/likely pathogenic CNVs into three categories30: recurrent CNVs with well-documented enrichment in epilepsy; CNVs that contained genes already implicated in epilepsy; and CNVs related to syndromes with epilepsy phenotypes reported in OMIM and literature. We also screened genome sequencing data for tandem repeat expansions using TRHist,31 which outputs information such as repeat units, length of tandem repeat expansion sequences and surrounding flanking sequences. Then we interpreted the results regarding whether any of the tandem repeat expansions were known pathogenic variants.
The results were combined with the patient's phenotype and disease progression (as evaluated by paediatric neurologists with extensive clinical experience) to yield a final genetic diagnosis. The chromosomal locations of diagnostic SNVs and CNVs were plotted using the R package ‘RIdeogram’.32
Statistical analysis
A chi-squared test of independence was performed using IBM SPSS v19 statistics software (SPSS 19.0; IBM Corp., Armonk, NY, USA) to evaluate the diagnostic yield across different patient groups. A P-value <0.05 was considered statistically significant.
Data availability
All sequencing data were produced by China National GeneBank, Shenzhen, China. The data that support the findings of this study have been deposited in the CNSA (https://db.cngb.org/cnsa/; accessed 10 November 2021) of CNGBdb with accession code CNP0000788.
Results
Demographics and clinical data
A total of 320 Chinese children were recruited and their demographic, clinical and neuroimaging characteristics are shown in Table 1 and Supplementary Table 1. The ratio of male to female was 1.48:1, most likely due to the fact that epilepsy is more frequent in males. Age at seizure onset ranged from 1 day to 17 years, with a median of 4.3 years. As shown in Supplementary Fig. 2A, focal seizures (219/320, 68.4%) were the most common seizure type among the 320 patients, followed by infantile spasms (100/320, 31.3%), generalized tonic seizures (31/320, 9.7%), myoclonic seizures (26/320, 8.1%), absence seizures (10/320, 3.1%), generalized tonic-clonic seizures (4/320, 1.3%) and generalized clonic seizures (1/320, 0.3%). In 30% of patients, there were two or more types of clinical seizure.
Comparison of general clinical information in patients with pathogenic or likely pathogenic variants and patients without causative variants
| Variables | Category total (n = 320) | Patients with pathogenic or likely pathogenic variants (n, %a) | Patients without causative variants (n, %b) | P-value |
|---|---|---|---|---|
| Total | 320 | 117 (36.6) | 203 (63.4) | |
| Age at seizure onset | ||||
| Neonatal (0–1 month) | 28 | 21 (17.9) | 7 (3.4) | 0.0002 |
| Infancy (1 month–1 year) | 179 | 65 (55.6) | 114 (56.2) | |
| Toddler (1–3 years) | 69 | 22 (18.8) | 47 (23.2) | |
| Early childhood (3–6 years) | 33 | 8 (6.8) | 25 (12.3) | |
| Middle childhood (6–12 years) | 10 | 1 (0.9) | 9 (4.4) | |
| Adolescent (12–18 years) | 1 | 0 (0.0) | 1 (0.5) | |
| Sex | ||||
| Male | 191 | 74 (63.2) | 117 (57.6) | 0.346 |
| Female | 129 | 43 (36.8) | 86 (42.4) | |
| DEE | 209 | 86 (73.5) | 123 (60.6) | 0.021 |
| Epilepsy classifications | ||||
| Generalized epilepsy | 101 | 28 (23.9) | 73 (36.0) | 0.033 |
| Focal epilepsy | 152 | 60 (51.3) | 92 (45.3) | 0.353 |
| Generalized and focal epilepsy | 67 | 29 (24.8) | 38 (18.7) | 0.203 |
| Epilepsy syndromes | ||||
| West syndrome | 68 | 25 (21.4) | 43 (21.2) | 0.969 |
| CSWS | 21 | 6 (5.1) | 15 (7.4) | 0.491 |
| Tuberous sclerosis | 16 | 12 (10.3) | 4 (2.0) | 0.002 |
| Ohtahara syndrome | 13 | 8 (6.8) | 5 (2.5) | 0.076 |
| Dravet syndrome | 11 | 11 (9.4) | 0 (0.0) | 1.14 × 10−5 |
| Lennox–Gastaut syndrome | 10 | 5 (4.3) | 5 (2.5) | 0.506 |
| Febrile seizures plus | 10 | 1 (0.9) | 9 (4.4) | 0.099 |
| Benign familial infantile epilepsy | 7 | 6 (5.1) | 1 (0.5) | 0.011 |
| Other phenotypes | ||||
| Developmental delay | 219 | 100 (85.5) | 119 (58.6) | 6.4 × 10−7 |
| Dystonia | 62 | 31 (26.5) | 31 (15.3) | 0.019 |
| Microcephaly | 33 | 14 (12.0) | 19 (9.4) | 0.577 |
| Premature | 30 | 13 (11.1) | 17 (8.4) | 0.419 |
| MGS | 20 | 10 (8.5) | 10 (4.9) | 0.197 |
| Congenital heart disease | 13 | 6 (5.1) | 7 (3.4) | 0.463 |
| Autism spectrum disorder | 13 | 7 (6.0) | 6 (3.0) | 0.240 |
| SGA | 11 | 4 (3.4) | 7 (3.4) | 0.989 |
| Lissencephaly | 17 | 11 (9.4) | 6(3.0) | 0.019 |
| Abnormal limbs | 15 | 10 (8.5) | 5 (2.5) | 0.024 |
| Brain MRI | ||||
| With findings | 170 | 77 (65.8) | 93 (45.8) | 0.007 |
| No findings | 122 | 36 (30.8) | 86 (42.4) | |
| Not determined | 28 | 4 (3.4) | 24 (11.8) | |
| Variables | Category total (n = 320) | Patients with pathogenic or likely pathogenic variants (n, %a) | Patients without causative variants (n, %b) | P-value |
|---|---|---|---|---|
| Total | 320 | 117 (36.6) | 203 (63.4) | |
| Age at seizure onset | ||||
| Neonatal (0–1 month) | 28 | 21 (17.9) | 7 (3.4) | 0.0002 |
| Infancy (1 month–1 year) | 179 | 65 (55.6) | 114 (56.2) | |
| Toddler (1–3 years) | 69 | 22 (18.8) | 47 (23.2) | |
| Early childhood (3–6 years) | 33 | 8 (6.8) | 25 (12.3) | |
| Middle childhood (6–12 years) | 10 | 1 (0.9) | 9 (4.4) | |
| Adolescent (12–18 years) | 1 | 0 (0.0) | 1 (0.5) | |
| Sex | ||||
| Male | 191 | 74 (63.2) | 117 (57.6) | 0.346 |
| Female | 129 | 43 (36.8) | 86 (42.4) | |
| DEE | 209 | 86 (73.5) | 123 (60.6) | 0.021 |
| Epilepsy classifications | ||||
| Generalized epilepsy | 101 | 28 (23.9) | 73 (36.0) | 0.033 |
| Focal epilepsy | 152 | 60 (51.3) | 92 (45.3) | 0.353 |
| Generalized and focal epilepsy | 67 | 29 (24.8) | 38 (18.7) | 0.203 |
| Epilepsy syndromes | ||||
| West syndrome | 68 | 25 (21.4) | 43 (21.2) | 0.969 |
| CSWS | 21 | 6 (5.1) | 15 (7.4) | 0.491 |
| Tuberous sclerosis | 16 | 12 (10.3) | 4 (2.0) | 0.002 |
| Ohtahara syndrome | 13 | 8 (6.8) | 5 (2.5) | 0.076 |
| Dravet syndrome | 11 | 11 (9.4) | 0 (0.0) | 1.14 × 10−5 |
| Lennox–Gastaut syndrome | 10 | 5 (4.3) | 5 (2.5) | 0.506 |
| Febrile seizures plus | 10 | 1 (0.9) | 9 (4.4) | 0.099 |
| Benign familial infantile epilepsy | 7 | 6 (5.1) | 1 (0.5) | 0.011 |
| Other phenotypes | ||||
| Developmental delay | 219 | 100 (85.5) | 119 (58.6) | 6.4 × 10−7 |
| Dystonia | 62 | 31 (26.5) | 31 (15.3) | 0.019 |
| Microcephaly | 33 | 14 (12.0) | 19 (9.4) | 0.577 |
| Premature | 30 | 13 (11.1) | 17 (8.4) | 0.419 |
| MGS | 20 | 10 (8.5) | 10 (4.9) | 0.197 |
| Congenital heart disease | 13 | 6 (5.1) | 7 (3.4) | 0.463 |
| Autism spectrum disorder | 13 | 7 (6.0) | 6 (3.0) | 0.240 |
| SGA | 11 | 4 (3.4) | 7 (3.4) | 0.989 |
| Lissencephaly | 17 | 11 (9.4) | 6(3.0) | 0.019 |
| Abnormal limbs | 15 | 10 (8.5) | 5 (2.5) | 0.024 |
| Brain MRI | ||||
| With findings | 170 | 77 (65.8) | 93 (45.8) | 0.007 |
| No findings | 122 | 36 (30.8) | 86 (42.4) | |
| Not determined | 28 | 4 (3.4) | 24 (11.8) | |
CSWS = epileptic encephalopathy with continuous spike-and-wave during sleep; DEE = developmental and/or epileptic encephalopathies; MGS = malformations of the genitourinary system; SGA = small for gestational age infant.
Per cent among the 117 cases.
Per cent among the 203 cases.
Comparison of general clinical information in patients with pathogenic or likely pathogenic variants and patients without causative variants
| Variables | Category total (n = 320) | Patients with pathogenic or likely pathogenic variants (n, %a) | Patients without causative variants (n, %b) | P-value |
|---|---|---|---|---|
| Total | 320 | 117 (36.6) | 203 (63.4) | |
| Age at seizure onset | ||||
| Neonatal (0–1 month) | 28 | 21 (17.9) | 7 (3.4) | 0.0002 |
| Infancy (1 month–1 year) | 179 | 65 (55.6) | 114 (56.2) | |
| Toddler (1–3 years) | 69 | 22 (18.8) | 47 (23.2) | |
| Early childhood (3–6 years) | 33 | 8 (6.8) | 25 (12.3) | |
| Middle childhood (6–12 years) | 10 | 1 (0.9) | 9 (4.4) | |
| Adolescent (12–18 years) | 1 | 0 (0.0) | 1 (0.5) | |
| Sex | ||||
| Male | 191 | 74 (63.2) | 117 (57.6) | 0.346 |
| Female | 129 | 43 (36.8) | 86 (42.4) | |
| DEE | 209 | 86 (73.5) | 123 (60.6) | 0.021 |
| Epilepsy classifications | ||||
| Generalized epilepsy | 101 | 28 (23.9) | 73 (36.0) | 0.033 |
| Focal epilepsy | 152 | 60 (51.3) | 92 (45.3) | 0.353 |
| Generalized and focal epilepsy | 67 | 29 (24.8) | 38 (18.7) | 0.203 |
| Epilepsy syndromes | ||||
| West syndrome | 68 | 25 (21.4) | 43 (21.2) | 0.969 |
| CSWS | 21 | 6 (5.1) | 15 (7.4) | 0.491 |
| Tuberous sclerosis | 16 | 12 (10.3) | 4 (2.0) | 0.002 |
| Ohtahara syndrome | 13 | 8 (6.8) | 5 (2.5) | 0.076 |
| Dravet syndrome | 11 | 11 (9.4) | 0 (0.0) | 1.14 × 10−5 |
| Lennox–Gastaut syndrome | 10 | 5 (4.3) | 5 (2.5) | 0.506 |
| Febrile seizures plus | 10 | 1 (0.9) | 9 (4.4) | 0.099 |
| Benign familial infantile epilepsy | 7 | 6 (5.1) | 1 (0.5) | 0.011 |
| Other phenotypes | ||||
| Developmental delay | 219 | 100 (85.5) | 119 (58.6) | 6.4 × 10−7 |
| Dystonia | 62 | 31 (26.5) | 31 (15.3) | 0.019 |
| Microcephaly | 33 | 14 (12.0) | 19 (9.4) | 0.577 |
| Premature | 30 | 13 (11.1) | 17 (8.4) | 0.419 |
| MGS | 20 | 10 (8.5) | 10 (4.9) | 0.197 |
| Congenital heart disease | 13 | 6 (5.1) | 7 (3.4) | 0.463 |
| Autism spectrum disorder | 13 | 7 (6.0) | 6 (3.0) | 0.240 |
| SGA | 11 | 4 (3.4) | 7 (3.4) | 0.989 |
| Lissencephaly | 17 | 11 (9.4) | 6(3.0) | 0.019 |
| Abnormal limbs | 15 | 10 (8.5) | 5 (2.5) | 0.024 |
| Brain MRI | ||||
| With findings | 170 | 77 (65.8) | 93 (45.8) | 0.007 |
| No findings | 122 | 36 (30.8) | 86 (42.4) | |
| Not determined | 28 | 4 (3.4) | 24 (11.8) | |
| Variables | Category total (n = 320) | Patients with pathogenic or likely pathogenic variants (n, %a) | Patients without causative variants (n, %b) | P-value |
|---|---|---|---|---|
| Total | 320 | 117 (36.6) | 203 (63.4) | |
| Age at seizure onset | ||||
| Neonatal (0–1 month) | 28 | 21 (17.9) | 7 (3.4) | 0.0002 |
| Infancy (1 month–1 year) | 179 | 65 (55.6) | 114 (56.2) | |
| Toddler (1–3 years) | 69 | 22 (18.8) | 47 (23.2) | |
| Early childhood (3–6 years) | 33 | 8 (6.8) | 25 (12.3) | |
| Middle childhood (6–12 years) | 10 | 1 (0.9) | 9 (4.4) | |
| Adolescent (12–18 years) | 1 | 0 (0.0) | 1 (0.5) | |
| Sex | ||||
| Male | 191 | 74 (63.2) | 117 (57.6) | 0.346 |
| Female | 129 | 43 (36.8) | 86 (42.4) | |
| DEE | 209 | 86 (73.5) | 123 (60.6) | 0.021 |
| Epilepsy classifications | ||||
| Generalized epilepsy | 101 | 28 (23.9) | 73 (36.0) | 0.033 |
| Focal epilepsy | 152 | 60 (51.3) | 92 (45.3) | 0.353 |
| Generalized and focal epilepsy | 67 | 29 (24.8) | 38 (18.7) | 0.203 |
| Epilepsy syndromes | ||||
| West syndrome | 68 | 25 (21.4) | 43 (21.2) | 0.969 |
| CSWS | 21 | 6 (5.1) | 15 (7.4) | 0.491 |
| Tuberous sclerosis | 16 | 12 (10.3) | 4 (2.0) | 0.002 |
| Ohtahara syndrome | 13 | 8 (6.8) | 5 (2.5) | 0.076 |
| Dravet syndrome | 11 | 11 (9.4) | 0 (0.0) | 1.14 × 10−5 |
| Lennox–Gastaut syndrome | 10 | 5 (4.3) | 5 (2.5) | 0.506 |
| Febrile seizures plus | 10 | 1 (0.9) | 9 (4.4) | 0.099 |
| Benign familial infantile epilepsy | 7 | 6 (5.1) | 1 (0.5) | 0.011 |
| Other phenotypes | ||||
| Developmental delay | 219 | 100 (85.5) | 119 (58.6) | 6.4 × 10−7 |
| Dystonia | 62 | 31 (26.5) | 31 (15.3) | 0.019 |
| Microcephaly | 33 | 14 (12.0) | 19 (9.4) | 0.577 |
| Premature | 30 | 13 (11.1) | 17 (8.4) | 0.419 |
| MGS | 20 | 10 (8.5) | 10 (4.9) | 0.197 |
| Congenital heart disease | 13 | 6 (5.1) | 7 (3.4) | 0.463 |
| Autism spectrum disorder | 13 | 7 (6.0) | 6 (3.0) | 0.240 |
| SGA | 11 | 4 (3.4) | 7 (3.4) | 0.989 |
| Lissencephaly | 17 | 11 (9.4) | 6(3.0) | 0.019 |
| Abnormal limbs | 15 | 10 (8.5) | 5 (2.5) | 0.024 |
| Brain MRI | ||||
| With findings | 170 | 77 (65.8) | 93 (45.8) | 0.007 |
| No findings | 122 | 36 (30.8) | 86 (42.4) | |
| Not determined | 28 | 4 (3.4) | 24 (11.8) | |
CSWS = epileptic encephalopathy with continuous spike-and-wave during sleep; DEE = developmental and/or epileptic encephalopathies; MGS = malformations of the genitourinary system; SGA = small for gestational age infant.
Per cent among the 117 cases.
Per cent among the 203 cases.
As shown in Supplementary Fig. 2B, many patients carried a diagnosis of an epileptic syndrome, such as West syndrome diagnosed in 68 of the 320 patients (21.3%); Ohtahara syndrome found in 13 patients (4.1%); Dravet syndrome found in 11 patients (3.4%); Lennox–Gastaut syndrome found in 10 patients (3.1%); and benign familial infantile epilepsy found in seven patients (2.2%). Furthermore, 245 of the 320 patients (76.6%) had comorbidities, such as global developmental delay in 219 (68.4%), and brain MRI detected potentially epileptogenic abnormalities in 170 (53.1%) of the patients, including brain malformations, brain dysplasia and brain atrophy (Table 1).
We also screened for non-coding repeat expansions but found no known pathogenic repeat expansion variants in genes like CSTB,33,34 SAMD12,35 STARD7,36 TNRC6A,37 RAPGEF2,38 MARCH6,39 YEATS240 and C9orf72,41 or in any of the epilepsy genes identified in this research.
Detection rates of single-nucleotide and copy number variants
By simultaneously analysing SNVs and CNVs in the 320 patients, pathogenic or likely pathogenic variants were found in 117 patients (36.6%). In 95 patients (29.7%), we detected a total of 107 pathogenic or likely pathogenic SNVs (Supplementary Table 1), and in 24 patients (7.5%), there were a total of 25 pathogenic or likely pathogenic CNVs (Supplementary Table 2). Note that this table lists 30 patients because it also includes six cases in which particular 15q11.2 and 16p11.2 deletion syndromes were considered susceptibility factors; see below). Concomitant pathogenic SNVs and CNVs were identified in two of the 117 patients (0.6%), including (in one of them) a compound heterozygosity with an SNV on one chromosome and a CNV on the corresponding part of the other.
A map of the chromosomal distribution of the SNVs and CNVs in the patient cohort is shown in Fig. 1. Interestingly, 31 of 132 (23.5%) pathogenic or likely pathogenic variants, comprising 29 SNVs and 2 CNVs, were located on chromosome 16. Moreover, 17 of the pathogenic or likely pathogenic variants (12.9%) were located on chromosome 2, including 15 SNVs and 2 CNVs.
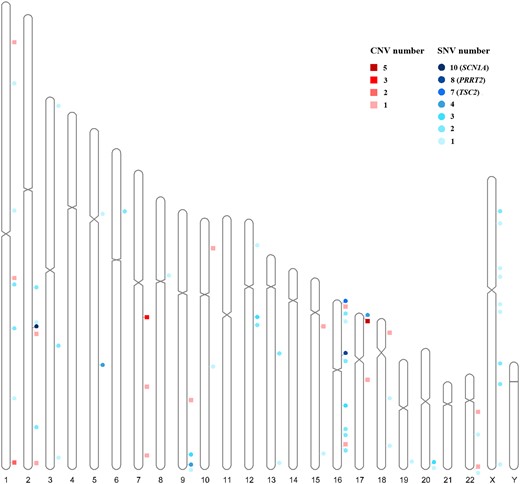
Distribution across chromosomes of associated diagnostic SNVs and diagnostic CNVs. Circles represent SNVs; squares represent CNVs; CNV number represents diagnostic CNV number in the region; SNV number represents diagnostic SNV number in a gene.
Of the 320 patients, 233 also underwent urine screening and tandem mass spectrometry of whole blood. Although no significant metabolic abnormalities were found in these samples, the genetic analysis revealed alterations in genes associated with metabolic diseases in nine of them (4% of the 233 patients).
Comparison of clinical and genetic diagnoses
Table 1 shows a comparison of the clinical findings in patients in which pathogenic/likely pathogenic variants were identified versus those in which no such variants were found. Two aspects are noteworthy. One concerned the age at onset of epileptic seizures. Interestingly, a genetic cause was overall more readily discoverable in patients whose seizure onset occurred at a younger age (average onset at 12.84 months versus 21.70 months in patients in which no genetic cause was found; P = 0.0002). Furthermore, a division of the patients into different age cohorts showed that the detection rates of pathogenic/likely pathogenic variants steadily decreased as the age at seizure onset increased (Fig. 2).
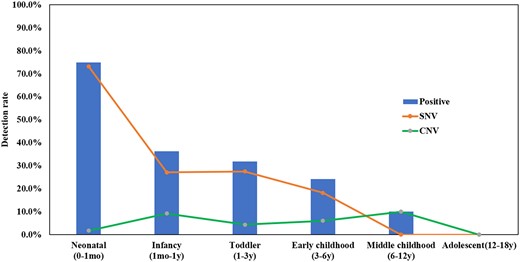
Detection rates at different ages of seizure onset. Detection rate: The ratio of the total number of positive samples in the age range of seizure onset. mo = month(s); y = years.
The other aspect concerned the association of a genetic cause with the diagnosis of an epileptic syndrome. In fact, many of our patients were diagnosed with epileptic syndromes, and patients diagnosed with tuberous sclerosis syndrome, Dravet syndrome and benign familial infantile epilepsy were more likely to show an underlying genetic cause than no such cause (P < 0.05) (Tables 1 and 2). Patients with developmental and/or epileptic encephalopathies were also more likely to show an underlying genetic cause (P = 0.021). The likelihood of association of a given SNV or CNV with the different phenotypic subgroups is shown in the heat maps of Supplementary Fig. 3A and B.
Mutated genes associated with clinically diagnosed syndromes
| Clinically diagnosed syndrome | Diagnosis yield | Associated genes mutated |
|---|---|---|
| West syndrome | 36.8% (25/68) | PAFAH1B1 (4), UBA5 (2), SUOX (2), DCX (1), DEPDC5 (1), GNAO1 (1), HCN1 (1), KCNH1 (1), KCNQ2 (1), PRKCG (1), TSC2 (1), WDR45 (1), WWOX (1), 17p13.3 del (5), 9q31.1 dup (1), 7q11.22q11.23 del (1), 7q11.22q11.23 dup (1), 16p13.3 del (1), 16q23.1 dup (1)a |
| Tuberous sclerosis | 75.0% (12/16) | TSC2 (7), TSC1 (4), 16p13.3 del including TSC2 (1) |
| Dravet syndrome | 100.0% (11/11) | SCN1A (10), 2q24.3 del including SCN1A (1) |
| Ohtahara syndrome | 61.5% (8/13) | ABAT (2), CDKL5 (2), GNAO1 (2), KCNT1 (1), STXBP1 (1), WWOX (1) |
| Epileptic encephalopathy with continuous spike-and-wave during sleep | 28.6% (6/21) | TSC1 (2), FGF12 (1), GRIN2A (1); 7q11.21q21.3 dup (1), 15q11.2q13.1 del (1) |
| Benign familial infantile epilepsy | 85.7% (6/7) | PRRT2 (6) |
| Lennox–Gastaut syndrome | 50.0% (5/10) | ALDH7A1 (2), SCN8A (1), STXBP1 (1) 7q11.22q11.23 dup (1), 22q13.33 del (1) |
| Myotonic epliepsy in infancy | 50.0% (2/4) | GRIN2B (1), KCNA2 (1) |
| Febrile seizures plus | 10.0% (1/10) | SETBP1 (1) |
| Panayiotopoulos syndrome | 25.0% (1/4) | 1q43q44 del (1) |
| Childhood epilepsy with centrotemporal spikes | 25.0% (1/4) | KIF4A (1) |
| Epilepsy of infancy with migrating focal seizures | 33.3% (1/3) | KCNQ2 (1) |
| Epliespy with myoclonic-atonc seizures | 100.0% (1/1) | ITPR1 (1) |
| Early myoclonic encephalopathy | 100.0% (1/1) | EEF1A2 (1) |
| Childhood absence epilepsy | 0.0% (0/3) | ― |
| Clinically diagnosed syndrome | Diagnosis yield | Associated genes mutated |
|---|---|---|
| West syndrome | 36.8% (25/68) | PAFAH1B1 (4), UBA5 (2), SUOX (2), DCX (1), DEPDC5 (1), GNAO1 (1), HCN1 (1), KCNH1 (1), KCNQ2 (1), PRKCG (1), TSC2 (1), WDR45 (1), WWOX (1), 17p13.3 del (5), 9q31.1 dup (1), 7q11.22q11.23 del (1), 7q11.22q11.23 dup (1), 16p13.3 del (1), 16q23.1 dup (1)a |
| Tuberous sclerosis | 75.0% (12/16) | TSC2 (7), TSC1 (4), 16p13.3 del including TSC2 (1) |
| Dravet syndrome | 100.0% (11/11) | SCN1A (10), 2q24.3 del including SCN1A (1) |
| Ohtahara syndrome | 61.5% (8/13) | ABAT (2), CDKL5 (2), GNAO1 (2), KCNT1 (1), STXBP1 (1), WWOX (1) |
| Epileptic encephalopathy with continuous spike-and-wave during sleep | 28.6% (6/21) | TSC1 (2), FGF12 (1), GRIN2A (1); 7q11.21q21.3 dup (1), 15q11.2q13.1 del (1) |
| Benign familial infantile epilepsy | 85.7% (6/7) | PRRT2 (6) |
| Lennox–Gastaut syndrome | 50.0% (5/10) | ALDH7A1 (2), SCN8A (1), STXBP1 (1) 7q11.22q11.23 dup (1), 22q13.33 del (1) |
| Myotonic epliepsy in infancy | 50.0% (2/4) | GRIN2B (1), KCNA2 (1) |
| Febrile seizures plus | 10.0% (1/10) | SETBP1 (1) |
| Panayiotopoulos syndrome | 25.0% (1/4) | 1q43q44 del (1) |
| Childhood epilepsy with centrotemporal spikes | 25.0% (1/4) | KIF4A (1) |
| Epilepsy of infancy with migrating focal seizures | 33.3% (1/3) | KCNQ2 (1) |
| Epliespy with myoclonic-atonc seizures | 100.0% (1/1) | ITPR1 (1) |
| Early myoclonic encephalopathy | 100.0% (1/1) | EEF1A2 (1) |
| Childhood absence epilepsy | 0.0% (0/3) | ― |
del = deletion; dup = duplication.
16q23.1 dup was an intragenic variant in WWOX.
Mutated genes associated with clinically diagnosed syndromes
| Clinically diagnosed syndrome | Diagnosis yield | Associated genes mutated |
|---|---|---|
| West syndrome | 36.8% (25/68) | PAFAH1B1 (4), UBA5 (2), SUOX (2), DCX (1), DEPDC5 (1), GNAO1 (1), HCN1 (1), KCNH1 (1), KCNQ2 (1), PRKCG (1), TSC2 (1), WDR45 (1), WWOX (1), 17p13.3 del (5), 9q31.1 dup (1), 7q11.22q11.23 del (1), 7q11.22q11.23 dup (1), 16p13.3 del (1), 16q23.1 dup (1)a |
| Tuberous sclerosis | 75.0% (12/16) | TSC2 (7), TSC1 (4), 16p13.3 del including TSC2 (1) |
| Dravet syndrome | 100.0% (11/11) | SCN1A (10), 2q24.3 del including SCN1A (1) |
| Ohtahara syndrome | 61.5% (8/13) | ABAT (2), CDKL5 (2), GNAO1 (2), KCNT1 (1), STXBP1 (1), WWOX (1) |
| Epileptic encephalopathy with continuous spike-and-wave during sleep | 28.6% (6/21) | TSC1 (2), FGF12 (1), GRIN2A (1); 7q11.21q21.3 dup (1), 15q11.2q13.1 del (1) |
| Benign familial infantile epilepsy | 85.7% (6/7) | PRRT2 (6) |
| Lennox–Gastaut syndrome | 50.0% (5/10) | ALDH7A1 (2), SCN8A (1), STXBP1 (1) 7q11.22q11.23 dup (1), 22q13.33 del (1) |
| Myotonic epliepsy in infancy | 50.0% (2/4) | GRIN2B (1), KCNA2 (1) |
| Febrile seizures plus | 10.0% (1/10) | SETBP1 (1) |
| Panayiotopoulos syndrome | 25.0% (1/4) | 1q43q44 del (1) |
| Childhood epilepsy with centrotemporal spikes | 25.0% (1/4) | KIF4A (1) |
| Epilepsy of infancy with migrating focal seizures | 33.3% (1/3) | KCNQ2 (1) |
| Epliespy with myoclonic-atonc seizures | 100.0% (1/1) | ITPR1 (1) |
| Early myoclonic encephalopathy | 100.0% (1/1) | EEF1A2 (1) |
| Childhood absence epilepsy | 0.0% (0/3) | ― |
| Clinically diagnosed syndrome | Diagnosis yield | Associated genes mutated |
|---|---|---|
| West syndrome | 36.8% (25/68) | PAFAH1B1 (4), UBA5 (2), SUOX (2), DCX (1), DEPDC5 (1), GNAO1 (1), HCN1 (1), KCNH1 (1), KCNQ2 (1), PRKCG (1), TSC2 (1), WDR45 (1), WWOX (1), 17p13.3 del (5), 9q31.1 dup (1), 7q11.22q11.23 del (1), 7q11.22q11.23 dup (1), 16p13.3 del (1), 16q23.1 dup (1)a |
| Tuberous sclerosis | 75.0% (12/16) | TSC2 (7), TSC1 (4), 16p13.3 del including TSC2 (1) |
| Dravet syndrome | 100.0% (11/11) | SCN1A (10), 2q24.3 del including SCN1A (1) |
| Ohtahara syndrome | 61.5% (8/13) | ABAT (2), CDKL5 (2), GNAO1 (2), KCNT1 (1), STXBP1 (1), WWOX (1) |
| Epileptic encephalopathy with continuous spike-and-wave during sleep | 28.6% (6/21) | TSC1 (2), FGF12 (1), GRIN2A (1); 7q11.21q21.3 dup (1), 15q11.2q13.1 del (1) |
| Benign familial infantile epilepsy | 85.7% (6/7) | PRRT2 (6) |
| Lennox–Gastaut syndrome | 50.0% (5/10) | ALDH7A1 (2), SCN8A (1), STXBP1 (1) 7q11.22q11.23 dup (1), 22q13.33 del (1) |
| Myotonic epliepsy in infancy | 50.0% (2/4) | GRIN2B (1), KCNA2 (1) |
| Febrile seizures plus | 10.0% (1/10) | SETBP1 (1) |
| Panayiotopoulos syndrome | 25.0% (1/4) | 1q43q44 del (1) |
| Childhood epilepsy with centrotemporal spikes | 25.0% (1/4) | KIF4A (1) |
| Epilepsy of infancy with migrating focal seizures | 33.3% (1/3) | KCNQ2 (1) |
| Epliespy with myoclonic-atonc seizures | 100.0% (1/1) | ITPR1 (1) |
| Early myoclonic encephalopathy | 100.0% (1/1) | EEF1A2 (1) |
| Childhood absence epilepsy | 0.0% (0/3) | ― |
del = deletion; dup = duplication.
16q23.1 dup was an intragenic variant in WWOX.
A breakdown of the syndrome-associated genes in which we identified pathogenic/likely pathogenic variants is shown in Table 2. Of the 320 patients, 68 were clinically diagnosed as West syndrome, with 28 distinct variants detected among them (detection rate: 36.8%). Four SNVs were found in PAFAH1B1; two each in UBA5 and SUOX, and one each in DCX, DEPDC5, GNAO1, HCN1, KCNH1, KCNQ2, PRKCG, TSC2, WDR45 and WWOX, respectively. Furthermore, the following CNVs were found in cases of West syndrome: five 17p13.3 deletions; one 9q31.1 duplication; one 7q11.22q11.23 deletion; one 7q11.22q11.23 duplication; one 16p13.3 deletion and one 16q23.1 duplication that includes WWOX. Sixteen cases were clinically diagnosed as tuberous sclerosis complex, for which a total of 12 variants were detected (detection rate: 75%) (seven variants in TSC2, four variants in TSC1 and one 16p13.3 deletion that included TSC2). Eleven cases were clinically diagnosed as Dravet syndrome, 10 of which carrying a variant of SCN1A and one a 2q24.3 deletion that included SCN1A (detection rate: 100%). Thirteen cases were clinically diagnosed as Ohtahara syndrome, with eight variants in six genes (detection rate: 61.5%) (two variants each in ABAT, CDKL5 and GNAO1 and one each in KCNT1, STXBP1 and WWOX). Other epilepsy syndromes and the genes mutated for the corresponding syndromes are presented in Table 2.
Single-nucleotide variants
The 107 pathogenic or likely pathogenic SNVs that we identified in 95 patients were assigned to a total of 52 different genes, among which the most frequently mutated one was SCN1A (10/95, 10.5%), followed by PRRT2 (8/95, 8.4%) and TSC2 (7/95, 7.4%). As shown in Table 3, four patients (4.2%) were found with mutations in PAFAH1B1 and TSC1, respectively; three patients (3.2%) with mutations in GNAO1, STXBP1, SCN8A and KCNQ2; and two patients (2.1%) with mutations in ZEB2, PCDH19, CDKL5, WWOX, ALDH7A1, DCX and SYNGAP1.
Pathogenic/likely pathogenic genes identified in 320 patients with seizures
| Number of patients per gene | Gene/syndromes |
|---|---|
| 1 | AARS, ABAT, ALG11, ARX, BCKDK, COG4, CHD2, COL4A1, CYP27A1, DARS2, DEPDC5, EEF1A2, FGF12, GRIN2A, GRIN2B, HCN1, ITPR1, KAT6A, KCNA2, KCNH1, KCNMA1, KCNT1, KIF4A, MECP2, PRKCG, RTTN, SCN3A, SETBP1, SHANK3, SLC2A1, SMC1A, SUOX, TAF1, TARS2, UBA5, WDR45 |
| 2 | ZEB2, WWOX, SYNGAP1, PCDH19, DCX, CDKL5, ALDH7A1 |
| 3 | STXBP1, SCN8A, KCNQ2, GNAO1 |
| 4 | TSC1, PAFAH1B1 |
| 7 | TSC2 |
| 8 | PRRT2 |
| 10 | SCN1A |
| Number of patients per gene | Gene/syndromes |
|---|---|
| 1 | AARS, ABAT, ALG11, ARX, BCKDK, COG4, CHD2, COL4A1, CYP27A1, DARS2, DEPDC5, EEF1A2, FGF12, GRIN2A, GRIN2B, HCN1, ITPR1, KAT6A, KCNA2, KCNH1, KCNMA1, KCNT1, KIF4A, MECP2, PRKCG, RTTN, SCN3A, SETBP1, SHANK3, SLC2A1, SMC1A, SUOX, TAF1, TARS2, UBA5, WDR45 |
| 2 | ZEB2, WWOX, SYNGAP1, PCDH19, DCX, CDKL5, ALDH7A1 |
| 3 | STXBP1, SCN8A, KCNQ2, GNAO1 |
| 4 | TSC1, PAFAH1B1 |
| 7 | TSC2 |
| 8 | PRRT2 |
| 10 | SCN1A |
Pathogenic/likely pathogenic genes identified in 320 patients with seizures
| Number of patients per gene | Gene/syndromes |
|---|---|
| 1 | AARS, ABAT, ALG11, ARX, BCKDK, COG4, CHD2, COL4A1, CYP27A1, DARS2, DEPDC5, EEF1A2, FGF12, GRIN2A, GRIN2B, HCN1, ITPR1, KAT6A, KCNA2, KCNH1, KCNMA1, KCNT1, KIF4A, MECP2, PRKCG, RTTN, SCN3A, SETBP1, SHANK3, SLC2A1, SMC1A, SUOX, TAF1, TARS2, UBA5, WDR45 |
| 2 | ZEB2, WWOX, SYNGAP1, PCDH19, DCX, CDKL5, ALDH7A1 |
| 3 | STXBP1, SCN8A, KCNQ2, GNAO1 |
| 4 | TSC1, PAFAH1B1 |
| 7 | TSC2 |
| 8 | PRRT2 |
| 10 | SCN1A |
| Number of patients per gene | Gene/syndromes |
|---|---|
| 1 | AARS, ABAT, ALG11, ARX, BCKDK, COG4, CHD2, COL4A1, CYP27A1, DARS2, DEPDC5, EEF1A2, FGF12, GRIN2A, GRIN2B, HCN1, ITPR1, KAT6A, KCNA2, KCNH1, KCNMA1, KCNT1, KIF4A, MECP2, PRKCG, RTTN, SCN3A, SETBP1, SHANK3, SLC2A1, SMC1A, SUOX, TAF1, TARS2, UBA5, WDR45 |
| 2 | ZEB2, WWOX, SYNGAP1, PCDH19, DCX, CDKL5, ALDH7A1 |
| 3 | STXBP1, SCN8A, KCNQ2, GNAO1 |
| 4 | TSC1, PAFAH1B1 |
| 7 | TSC2 |
| 8 | PRRT2 |
| 10 | SCN1A |
As shown in Fig. 3, of the total of 107 SNVs, 52 (48.6%) were missense variations, 29 (27.1%) frameshift variations, 15 (14.0%) nonsense variations, 8 (7.5%) splice variations and one each (0.9%) in frame variation, synonymous variation affecting splicing and intron variation. The parental origin of the variants was assessed by Sanger sequencing of the parents and is indicated (as far as it could be determined) in Supplementary Table 1. Based on OMIM and the literature, it is suggested that 68 variants (63.6%, 68/107) could be inherited in an autosomal dominant fashion, 27 (25.2%, 27/107) in an autosomal recessive fashion and 12 (11.2%, 12/107) in an X-linked fashion (Fig. 3, Supplementary Table 3). Many of the SNVs were indeed of parental origin; however, 60 of the 107 SNVs (55.1%) were variants not present in either parent and hence were considered de novo.
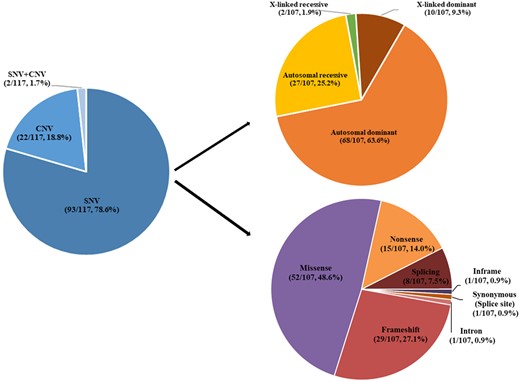
Variation type in 117 patients with epilepsy.
Further analysis revealed the following additional details. Of the 10 SCN1A variants that we detected, one was inherited from the father, but the other nine appeared de novo in the corresponding patients. Likewise, all four TSC1 and seven TSC2 variants appeared de novo. However, after verification by droplet digintal PCR (ddPCR), the father of the heterozygous TSC2 Patient SZCH0052 was mosaic, suggesting that this patient inherited the variant from the father. Eight PRRT2 variants were of parental origin. Of these, five exhibited the same frameshift variation in a previously identified variation hotspot with incomplete penetrance [NM_001256442.1:c.649dup (p.Arg217Profs8)],42 and one a different variant of codon 217.
One additional interesting case concerned Patient SZCH0237 who, as mentioned above, is a compound heterozygote for variants of the BCKDK gene. Seizure onset in this patient was at 6 years of age and was combined with neurodevelopmental disorders and autism. The BCKDK gene is associated with a rare and treatable recessive disease called branched-chain ketoacid dehydrogenase kinase deficiency (OMIM #614923), described so far in only three consanguineous families with autism, epilepsy and intellectual disability.43
Copy number variants
Among 24 patients of our cohort, we identified 25 CNVs (1 patient, Patient SZCH0370, harboured two distinct CNVs) (Supplementary Table 2). Their sizes ranged from 121 base pairs (bp) to 30.47 megabase pairs (Mbp) and encompassed 18 deletions (72.0%) and seven duplications (28.0%). According to our interpretation rules (Supplementary Fig. 1B), 19 CNVs (76.0%) were considered pathogenic and six (24.0%) likely pathogenic. An additional four 16p11.2 deletions and two 15q11.2 BP1-BP2 deletions were considered susceptibility factors for the phenotypes due to reduced penetrance and variable expressivity of clinical features.
As shown in Fig. 1, the CNVs were distributed over 10 chromosomes. Many were associated with known syndromes, the most common being the chromosome 17p13.3 deletion syndrome, also known as Miller–Dieker lissencephaly syndrome (MIM#247200), which encompasses the deletion of PAFAH1B1, a known lissencephaly gene (OMIM #607432). In fact, we have identified 17 patients with lissencephaly, five of which harbouring CNVs affecting PAFAH1B1 (Supplementary Tables 2 and 4), in addition to the four mentioned above with SNVs in PAFAH1B1 (Supplementary Tables 1 and 4). Patients with PAFAH1B1 gene variants in our cohort all presented with infantile spasms and had more severe alterations in the posterior brain regions (posterior-to-anterior gradient), which was consistent with previous studies.44-46
The most common recurrent CNVs were the 17p13.3 deletion (5/24, 20.8%), 16p11.2 deletions (4/24, 16.7%) and 7q11.23 duplications (2/24, 8.3%), all of which associated with epilepsy, developmental retardation and congenital abnormalities.
These results indicate that most of the chromosomal abnormalities identified in our patient cohort could be assigned to known syndromes in which seizures are at least one symptom. Apart from the 17p13.3 deletion syndrome, other CNVs may also cover epileptogenic genes, such as the 1.5-Mb deletion at 2q24.3 in Patient SZCH0255, which includes several genes. Two of them (SCN1A and SCN9A) are known epilepsy genes, and deletion of either is known to cause epilepsy.7,47
Interestingly, among the 25 CNVs, the respective sizes of three CNVs were below 25 kilobase pairs (kbp). The detection of small fragments of CNVs is a major advantage of genome sequencing because chromosomal microarrays can currently not detect CNVs below 25 kbp.48 As shown in Supplementary Fig. 4, we verified one of these small deletions (the 1651-bp deletion in chromosome 16p13.3 of Patient SZCH0053) by PCR analysis that yielded, in addition to an expected wild-type band, a band of 237 bp predicted to occur only in presence of the CNV. The deletion seemed to have appeared de novo as the parents showed only the wild-type band. The analyses of additional CNVs revealed that the CNV fragment length correlated with a patient’s age at seizure onset (Supplementary Fig. 5).
Effect on potential targeted therapies
As mentioned before, seizures in many patients may be just one of the manifestations of an underlying neurogenetic disease. Genome sequencing can help identify the pathogenic causes for the seizures and thereby help to design treatment modalities that specifically target these underlying causes. In our cohort, there were 42 patients (13.1%)—summarized in Table 4—diagnosed with a disorder whose conditions improved to varying degrees under targeted therapy or avoidance of specific drugs. Here we highlight two patients for whom specific treatments led to improvements in the seizure symptomatology.
Gene variants associated with potential targeted therapies
| Gene | Sample number | Recommended drugs | Aggravating drugs |
|---|---|---|---|
| SCN1A | 10 | Valproate,70 benzodiazepines,70 cannabinoids,71 stiripentol,72 ketogenic diet,73 fenfluramine74 | Sodium channel blockers, e.g. carbamazepine, oxcarbazepine, lamotrigine, and phenytoin70,75 |
| SCN8A | 3 | Sodium channel blockers, e.g. carbamazepine, oxcarbazepine, lacosamide, lamotrigine, rufinamide, oxcarbazepine, and phenytoin76,77 | NA |
| KCNT1 | 1 | Quinidine78 | NA |
| TSC1 | 4 | Everolimus,79,80 vigabatrin81 | NA |
| TSC2 | 7 | Everolimus,79 vigabatrin81 | NA |
| PCDH19 | 2 | Bromide, clobazam82 | Carbamazepine, oxcarbazepine, rufinamide82 |
| ALDH7A1 | 2 | Pyridoxine (vitamin B6),83 folinic acid84 | NA |
| HCN1 | 1 | NA | Lacosamide and phenytoin85 |
| KCNMA1 | 1 | Vaproate and/or lamotrigine,86 valproate and/or levetiracetam87,88 | Ethosuximide87 |
| PRRT2 | 8 | Carbamazepine89 | NA |
| SLC2A1 | 1 | Ketogenic diet90,91 | NA |
| CDKL5 | 2 | Ketogenic diet92 | NA |
| Gene | Sample number | Recommended drugs | Aggravating drugs |
|---|---|---|---|
| SCN1A | 10 | Valproate,70 benzodiazepines,70 cannabinoids,71 stiripentol,72 ketogenic diet,73 fenfluramine74 | Sodium channel blockers, e.g. carbamazepine, oxcarbazepine, lamotrigine, and phenytoin70,75 |
| SCN8A | 3 | Sodium channel blockers, e.g. carbamazepine, oxcarbazepine, lacosamide, lamotrigine, rufinamide, oxcarbazepine, and phenytoin76,77 | NA |
| KCNT1 | 1 | Quinidine78 | NA |
| TSC1 | 4 | Everolimus,79,80 vigabatrin81 | NA |
| TSC2 | 7 | Everolimus,79 vigabatrin81 | NA |
| PCDH19 | 2 | Bromide, clobazam82 | Carbamazepine, oxcarbazepine, rufinamide82 |
| ALDH7A1 | 2 | Pyridoxine (vitamin B6),83 folinic acid84 | NA |
| HCN1 | 1 | NA | Lacosamide and phenytoin85 |
| KCNMA1 | 1 | Vaproate and/or lamotrigine,86 valproate and/or levetiracetam87,88 | Ethosuximide87 |
| PRRT2 | 8 | Carbamazepine89 | NA |
| SLC2A1 | 1 | Ketogenic diet90,91 | NA |
| CDKL5 | 2 | Ketogenic diet92 | NA |
Gene variants associated with potential targeted therapies
| Gene | Sample number | Recommended drugs | Aggravating drugs |
|---|---|---|---|
| SCN1A | 10 | Valproate,70 benzodiazepines,70 cannabinoids,71 stiripentol,72 ketogenic diet,73 fenfluramine74 | Sodium channel blockers, e.g. carbamazepine, oxcarbazepine, lamotrigine, and phenytoin70,75 |
| SCN8A | 3 | Sodium channel blockers, e.g. carbamazepine, oxcarbazepine, lacosamide, lamotrigine, rufinamide, oxcarbazepine, and phenytoin76,77 | NA |
| KCNT1 | 1 | Quinidine78 | NA |
| TSC1 | 4 | Everolimus,79,80 vigabatrin81 | NA |
| TSC2 | 7 | Everolimus,79 vigabatrin81 | NA |
| PCDH19 | 2 | Bromide, clobazam82 | Carbamazepine, oxcarbazepine, rufinamide82 |
| ALDH7A1 | 2 | Pyridoxine (vitamin B6),83 folinic acid84 | NA |
| HCN1 | 1 | NA | Lacosamide and phenytoin85 |
| KCNMA1 | 1 | Vaproate and/or lamotrigine,86 valproate and/or levetiracetam87,88 | Ethosuximide87 |
| PRRT2 | 8 | Carbamazepine89 | NA |
| SLC2A1 | 1 | Ketogenic diet90,91 | NA |
| CDKL5 | 2 | Ketogenic diet92 | NA |
| Gene | Sample number | Recommended drugs | Aggravating drugs |
|---|---|---|---|
| SCN1A | 10 | Valproate,70 benzodiazepines,70 cannabinoids,71 stiripentol,72 ketogenic diet,73 fenfluramine74 | Sodium channel blockers, e.g. carbamazepine, oxcarbazepine, lamotrigine, and phenytoin70,75 |
| SCN8A | 3 | Sodium channel blockers, e.g. carbamazepine, oxcarbazepine, lacosamide, lamotrigine, rufinamide, oxcarbazepine, and phenytoin76,77 | NA |
| KCNT1 | 1 | Quinidine78 | NA |
| TSC1 | 4 | Everolimus,79,80 vigabatrin81 | NA |
| TSC2 | 7 | Everolimus,79 vigabatrin81 | NA |
| PCDH19 | 2 | Bromide, clobazam82 | Carbamazepine, oxcarbazepine, rufinamide82 |
| ALDH7A1 | 2 | Pyridoxine (vitamin B6),83 folinic acid84 | NA |
| HCN1 | 1 | NA | Lacosamide and phenytoin85 |
| KCNMA1 | 1 | Vaproate and/or lamotrigine,86 valproate and/or levetiracetam87,88 | Ethosuximide87 |
| PRRT2 | 8 | Carbamazepine89 | NA |
| SLC2A1 | 1 | Ketogenic diet90,91 | NA |
| CDKL5 | 2 | Ketogenic diet92 | NA |
An 8-month-old female (Patient SZCH0292) developed seizures on the 13th day after birth that were refractory to anti-epileptic drugs. This patient was a compound heterozygote carrying two missense variants of the ALDH7A1 gene (OMIM #107323), c.1547A>G (p.Tyr516Cys) (NM_001182.4) and c.965C >T (p.Ala322Val) (NM_001182.4). ALDH7A1 deficiency leads to pyridoxal phosphate deficiency associated with pyridoxin-responsive epilepsy that can be alleviated by high doses of pyridoxine (vitamin B6).49 With this treatment, the patient became seizure-free and had a good prognosis.
A 3-year-old male (Patient SZCH0283) had seizures at 3 months of age, combined with ataxia and mental retardation. We detected a heterozygous frameshift variant on SLC2A1 associated with deficiencies in the glucose transporter-1 (GLUT1) protein (OMIM *138140, GLUT1 deficiency syndrome, infantile onset) [NM_006516.2:c.912del (p.Gln304Hisfs36)]. After treatment with a ketogenic diet, the male was seizure-free and showed normal intellectual and motor development.
Recurrence risk
Among the 95 patients with pathogenic/likely pathogenic SNVs, 29 (30.5%) patients inherited their variants from one or both parents. This result suggests that in these families, there is a risk of recurrence among other family members.
Nevertheless, among the 60 patients that harboured variants that appeared to have occurred de novo, three families (5.0%) had two children with the same birth defects. The most likely explanation for recurrence of de novo mutations is parental mosaicism in germ cells that is missed or absent in DNA usually derived from peripheral blood leukocytes and that might be more prevalent than previously predicted.50 Indeed, in one case (Patient SZCH0052), we found that the patient’s father was mosaic for the TSC2 variant detected in the child and so had probably transmitted it through the germline. Thus, genome sequencing can occasionally help to assess the risk for recurrence, even in cases where an original mutation seems to have arisen de novo.
Discussion
Despite the advantages of genome sequencing over other genetic methods, most large-scale genetic studies of paediatric epilepsy cases have been conducted using exome sequencing or capture-based targeted sequencing,51–54 probably because of cost considerations. Hence, the present genome sequencing study of 320 children with epilepsy remains one of few studies conducted on this scale thus underscoring its enhanced diagnostic yield and its feasibility in applying findings to clinical settings.
In our cohort, we found that 117 (36.6%) patients carried one (or in some cases more than one) pathogenic or likely pathogenic genetic variant. These variants included many of the possible genetic alterations such as SNVs in coding and non-coding sequences and CNVs as well as small indels, although no repeat expansions. Many of these alterations would be missed by exome sequencing or targeted capture methods. Nevertheless, a direct quantitative comparison of the diagnostic yields (provided by the different methods across different studies) is hampered by differences in patient cohorts, sequence coverage and calling methods for what constitutes a pathogenic or likely pathogenic variant. Nevertheless, an extended comparison of genome sequencing with possible results of exome sequencing or capture-based targeted methods, as shown in Supplementary Fig. 6, would indicate that the increased diagnostic yield obtained by genome sequencing was due to the detection of CNVs and intronic SNVs. In fact, our study showed that the diagnostic yield of SNVs was 29.1% (6.9% for CNVs) thus comparing favourably with similar studies based on exome sequencing or targeted capture methods.55–59
However, in the present study, four 16p11.2 deletions and two 15q11.2 BP1-BP2 deletions were considered to represent susceptibility loci for a variety of neurodevelopmental diseases, including seizures. Maya and coworkers60 showed that 15q11.2 BP1-BP2 deletions are common among affected and unaffected populations, with a calculated penetrance of 2.18% over the background risk. Rosenfeld and coworkers61 estimated that the risk of an abnormal phenotype ranged from 10.4% for 15q11.2 deletions to 46.8% for proximal 16p11.2 deletions. Because of the variable phenotypes and low penetrance, the pathogenicity of the deletions is low or not determined. Therefore, these CNVs may not be considered sufficient to be causal in epilepsy of the patients.
In the present study, we also found two special cases combining two types of variant. The first case was a 1-year-old female (Patient SZCH0677) presenting with seizures at 10 months of age and mental retardation and carrying a SYNGAP1 [NM_001130066.1:c.1543C>T (p.Arg515Cys)] variant and a 1542 kb deletion in 17q12 as part of the chromosome 17q12 deletion syndrome. Further analysis revealed that the SYNGAP1 variant can result in autosomal dominant mental retardation-5 (OMIM #612621), which is characterized by moderate to severe intellectual disability with delayed psychomotor development. Most of these patients develop variable types of seizure.62 Furthermore, 17q12 deletion syndrome is also associated with mental retardation, seizures and renal diseases. Hence, both the SYNGAP1 variant and the 17q12 deletion may contribute to the patient’s phenotypes.
The second case was a 2-year-old male (Patient SZCH0268) who carried an SNV and an intragenic partial CNV. He presented with seizures in the first month of life, hypertonia, mental retardation and corpus callosum dysplasia. Genome sequencing revealed a splice variant (NM_001291997.1:c.453-1G>C) of the WWOX gene as well as a partial duplication of its exon 6–exon 8. Mutations in WWOX (MIM*605131) are associated with epileptic encephalopathy, early infantile epilepsy (OMIM #616211), and the alleles described thus far show autosomal recessive inheritance. A combined SNV and CNV compound heterozygosity in WWOX was also reported in an unrelated patient.63 Combined with clinical features, it is likely, therefore, that the SNV/CNV combination in our patient was the cause of the disease. In conclusion, a traditional assay designed to discover either SNVs or CNVs, but not both together, would easily miss detection of such particular genetic constellations.
Apart from the advantages that genome sequencing generally offers in detecting CNVs, it also is superior over CMA techniques, specifically in the discovery of small indels.64 In fact, of all structural rearrangements that we discovered, three were indels below 25 kbp, the usual detection limit of CMA. Furthermore, genome sequencing can identify potentially pathogenic alterations in the UTRs of genes, as well as in intronic regions, such as in the highly conserved intronic region termed 20N of the epilepsy gene SCN1A.7 Last, it can detect alterations in the vast number of non-coding RNAs.
In this study, we found that the diagnostic yield of genome sequencing depended on a patient’s age at the first occurrence of a seizure, gradually decreasing with increasing age of seizure onset. This observation suggests that the earlier the onset, the more likely the cause was genetic.52 In fact, in patients with seizure onset within the first month of life, our detection rate (75%) was similar to that found in a previous study indicating that 83% of newborns with early-onset epilepsy had genetic aetiologies,65 but exceeded that seen in other studies on this age group.66,67
A particularly important contribution of genetic testing is to provide information about specific therapies targeted to an individual epilepsy patient. Thus, vitamin B6 therapy was successfully used to treat two patients with pyridoxine-dependent epilepsy, and one patient with GLUT1 deficiency syndrome was treated with a ketogenic diet. Before these two cases were identified molecularly, several therapeutic regimens had been used without real benefit to the patients; only the molecular findings made targeted and effective treatments possible as these treatments not only improved the seizure phenotype but also psychomotor development. Importantly, therefore, early detection and treatment initiation is critical for patients with early infantile epilepsy and is predictive for long-term cognitive and developmental outcomes.68,69 At present, treatment of epilepsy remains mainly empirical. Our findings of new genetic variants and potential therapeutic strategies (Table 4) will contribute to the establishment of personalized precision medicine and treatment stratification for individual patients. Genetic results can improve therapy by anti-epileptic drugs selection and precise medication approaches. However, the change of clinical managements still relies on patient data or clinical studies. Further clinical studies need to be conducted to verify the potential therapies targeted on different cellular mechanisms or channel functions.
In summary, early genome sequencing testing can provide an accurate molecular diagnosis in a timely manner, predict possible phenotypes, initiate appropriate therapies and follow-up, predict prognosis and assess the risk of recurrence, which are all extremely helpful for the clinical management of these patients.
Acknowledgements
The authors are deeply grateful to China National GeneBank (CNGB) as well as the patients and clinicians who participated in this work.
Funding
This research was supported by grants from the Science, Technology and Innovation Commission of Shenzhen Municipality (Grant No. KJYY20151116165726645), the Shenzhen Municipal Government of China (Grant No. JCY20170817145047361), and Sanming Project of Medicine in Shenzhen (Grant No. SZSM201812005).
Competing interests
The authors report no competing interests.
Supplementary material
Supplementary material is available at Brain online.
References
Epi25 Collaborative. Electronic address: [email protected]; Epi25 Collaborative.
Liang JS, Wang JS, Lin LJ, et al. Genetic diagnosis in children with epilepsy and developmental delay/mental retardation using targeted gene panel analysis.
Author notes
Dongfang Zou, Lin Wang, Jianxiang Liao and Hongdou Xiao contributed equally to this work.


{kind=link}
{kind=link}
{kind=link}