




Abstract
The aetiology of late-onset neurodegenerative diseases is largely unknown. Here we investigated whether de novo somatic variants for semantic dementia can be detected, thereby arguing for a more general role of somatic variants in neurodegenerative disease. Semantic dementia is characterized by a non-familial occurrence, early onset (<65 years), focal temporal atrophy and TDP-43 pathology. To test whether somatic variants in neural progenitor cells during brain development might lead to semantic dementia, we compared deep exome sequencing data of DNA derived from brain and blood of 16 semantic dementia cases. Somatic variants observed in brain tissue and absent in blood were validated using amplicon sequencing and digital PCR. We identified two variants in exon one of the TARDBP gene (L41F and R42H) at low level (1–3%) in cortical regions and in dentate gyrus in two semantic dementia brains, respectively. The pathogenicity of both variants is supported by demonstrating impaired splicing regulation of TDP-43 and by altered subcellular localization of the mutant TDP-43 protein. These findings indicate that somatic variants may cause semantic dementia as a non-hereditary neurodegenerative disease, which might be exemplary for other late-onset neurodegenerative disorders.
Introduction
Multifactorial aetiology, including genetic and environmental factors, has been used to explain most late-onset neurodegenerative diseases. Only a small percentage of cases with autosomal dominant inheritance is caused by germline variants in specific genes, for example PSEN1 and APP variants in Alzheimer’s disease, MAPT and GRN in frontotemporal dementia (FTD) and C9orf72 and TARDBP in both amyotrophic lateral sclerosis and FTD (Ferrari et al., 2019; Greaves and Rohrer, 2019; Clarimon et al., 2020). There is an increasing interest in the potential pathogenic role of de novo variants in patients with neurodegenerative diseases with a negative family history (Leija-Salazar et al., 2018; Lodato and Walsh, 2019). A few cases with de novo germline variants have been identified in early-onset Alzheimer’s disease (Nicolas et al., 2018). For neurodevelopmental diseases, low-level (≤20% of cells) somatic variants in mTOR, AKT3 and CCND arising from the ventricular or subventricular zone have been identified by deep sequencing of candidate genes in affected brain tissue (Lee et al., 2012; Lin et al., 2012; Veltman and Brunner, 2012; Miller et al., 2013; Poduri et al., 2013; Hu et al., 2014; Jamuar et al., 2014; Kovacs et al., 2014; Mirzaa et al., 2014; Rogalski et al., 2014; Bushman et al., 2015; Lim et al., 2015; Lodato et al., 2015; Sala Frigerio et al., 2015; Wiseman et al., 2015; Hoekstra et al., 2016; Kim et al., 2016; Takata et al., 2016). The hypothesis is that post-zygotic variants (after fertilization) or late-somatic variants during brain development might explain the sporadic presentation of neurodegenerative diseases with a negative family history.
The most ideal approach to determine the role of late-somatic variants in neurodegenerative diseases would be the comparison between blood- and brain-derived DNA within the same patients. However, brain tissue for DNA isolation was often not available during life, and DNA derived from blood was often not collected during life in deceased patients. Recent brain-derived DNA studies without matched DNA samples from blood have tried to detect somatic variants in Alzheimer’s and Parkinson’s disease (Beck et al., 2004; Lin et al., 2012; Proukakis et al., 2014; Bushman et al., 2015; Lodato et al., 2015; Sala Frigerio et al., 2015; Wiseman et al., 2015; Coxhead et al., 2016; Hoekstra et al., 2016; Lee et al., 2018; Lodato et al., 2018; Mokretar et al., 2018; Nicolas et al., 2018; Park et al., 2019; Wei et al., 2019). A higher number of low-level mosaic variants in causative genes (APP, SNCA) have been detected in DNA of Alzheimer’s disease or Parkinson’s disease brains compared to controls (Lee et al., 2018; Mokretar et al., 2018). Only the study by Park et al. (2019) performing deep sequencing of hippocampal formation and matched blood tissues found an enrichment of somatic DNA variation in the Tau signalling pathway in Alzheimer’s disease patients compared to controls (Park et al., 2019). Specifically, a single carrier of a somatic variant in PIN1 was suggested as potential causal factor in the respective Alzheimer’s disease patient (Park et al., 2019).
In the present study, we uniquely investigated the presence of low-level somatic variants in the temporal cortex and dentate gyrus of brains of patients with semantic dementia, which were absent in their blood-derived DNA. Semantic dementia is a well-defined clinical and pathological subtype of FTD, mostly occurring before the age of 65 (Hodges et al., 1992; Irish et al., 2012; Mesulam et al., 2014). The disease is characterized by a circumscribed asymmetric atrophy of the anterior temporal cortex, suggesting a local disease process (Mummery et al., 2000; Kumfor et al., 2016). Severe neuronal loss with pathological TDP-43 protein accumulation in neurites and neurons in the temporal cortex and dentate gyrus of the hippocampus are the defining salient and consistent neuropathological features of semantic dementia, most commonly classified as FTD-TDP type C (Davies et al., 2005; Mackenzie et al., 2011; Leyton et al., 2016; Neumann and Mackenzie, 2019). Semantic dementia has a sporadic, non-familial occurrence, and a current lack of mechanistic insight in the disease process precludes a therapeutic strategy. We performed deep exome sequencing (310×–658×) of middle temporal gyrus and dentate gyrus tissue of semantic dementia patients with pathologically confirmed FTD-TDP type C, and compared data with blood DNA samples of the same patients. We identified somatic TARDBP variants in the brains of two semantic dementia patients that were absent in blood. These variants were validated using custom amplicon panel sequencing and digital droplet PCR. In addition, we confirmed the disruptive effects of these TARDBP variants by demonstrating altered cellular distribution of the mutant TDP-43 proteins. Our results indicate that somatic variants in TARDBP contribute to semantic dementia pathogenesis.
Materials and methods
Patient tissue DNA collection
For the present study, we used fresh-frozen brain samples from 16 semantic dementia patients with confirmed FTD-TDP type C pathology, obtained from the Netherlands Brain Bank (Table 1) (Mackenzie and Neumann, 2017). Informed consent was obtained from all patients for brain autopsy and the use of tissue and clinical information for research purposes. DNA was extracted from fresh frozen brain samples of middle temporal gyrus (n = 14) and from the dentate gyrus (n = 13). From all cases, DNA from blood was available, obtained during life in 12 patients from the Dutch FTD study and extracted from blood obtained at the time of autopsy in the remaining four cases (Seelaar et al., 2008, 2011). The average age at death was 69 (range 62–74), 50% of patients were female. Medical records and neuroimaging (either CT or MRI) were collected and reviewed, if available. For 14 patients the left hemisphere of the brain was fresh-frozen for research, versus the right hemisphere for two patients.
Patient characteristics
| Patient | Sex | Age at onset | Disease duration | Dominant side pathology | Brain tissue side | Dominant side | MTG | DG |
|---|---|---|---|---|---|---|---|---|
| SD01 | Female | 60 | 10 | Both | Left | No | Yes | No |
| SD02 | Male | 48 | 14 | Left | Left | Yes | No | Yes |
| SD03 | Female | 60 | 8 | Both | Left | NA | Yes | No |
| SD04 | Female | 45 | 20 | Both | Left | NA | Yes | Yes |
| SD05 | Male | 56 | 10 | Both | Left | No | Yes | Yes |
| SD06 | Male | 51 | 12 | Both | Left | No | Yes | Yes |
| SD07 | Female | 53 | 11 | Both | Left | No | No | Yes |
| SD08 | Male | 57 | 12 | Left | Left | Yes | Yes | Yes |
| SD09 | Female | 63 | 11 | Both | Left | NA | Yes | Yes |
| SD10 | Male | 55 | 13 | Left | Right | No | Yes | Yes |
| SD11 | Male | 51 | 15 | Both | Left | No | Yes | Yes |
| SD12 | Female | 60 | 12 | Both | Left | No | Yes | Yes |
| SD13 | Female | 63 | 9 | Left | Right | No | Yes | Yes |
| SD14 | Male | 57 | 15 | Both | Left | No | Yes | Yes |
| SD15 | Female | 66 | 8 | Left | Left | Yes | Yes | No |
| SD16 | Male | 61 | 13 | Both | Left | Yes | Yes | Yes |
| Patient | Sex | Age at onset | Disease duration | Dominant side pathology | Brain tissue side | Dominant side | MTG | DG |
|---|---|---|---|---|---|---|---|---|
| SD01 | Female | 60 | 10 | Both | Left | No | Yes | No |
| SD02 | Male | 48 | 14 | Left | Left | Yes | No | Yes |
| SD03 | Female | 60 | 8 | Both | Left | NA | Yes | No |
| SD04 | Female | 45 | 20 | Both | Left | NA | Yes | Yes |
| SD05 | Male | 56 | 10 | Both | Left | No | Yes | Yes |
| SD06 | Male | 51 | 12 | Both | Left | No | Yes | Yes |
| SD07 | Female | 53 | 11 | Both | Left | No | No | Yes |
| SD08 | Male | 57 | 12 | Left | Left | Yes | Yes | Yes |
| SD09 | Female | 63 | 11 | Both | Left | NA | Yes | Yes |
| SD10 | Male | 55 | 13 | Left | Right | No | Yes | Yes |
| SD11 | Male | 51 | 15 | Both | Left | No | Yes | Yes |
| SD12 | Female | 60 | 12 | Both | Left | No | Yes | Yes |
| SD13 | Female | 63 | 9 | Left | Right | No | Yes | Yes |
| SD14 | Male | 57 | 15 | Both | Left | No | Yes | Yes |
| SD15 | Female | 66 | 8 | Left | Left | Yes | Yes | No |
| SD16 | Male | 61 | 13 | Both | Left | Yes | Yes | Yes |
Contains clinical and pathological information on the patients examined in this study. Pathological diagnosis, as extracted from the reports from the Netherlands Brain Bank. The most affected side of the brain is reported according to the post-mortem pathological examination. Brain tissue side: the side of the brain fresh frozen and used in this study. Dominant side yes/no; whether the side studied was the one most affected according to neuroimaging (NA = not applicable, as both sides were equally affected). Middle temporal gyrus (MTG) and dentate gyrus (DG) indicate whether these areas were available and included in the study.
Patient characteristics
| Patient | Sex | Age at onset | Disease duration | Dominant side pathology | Brain tissue side | Dominant side | MTG | DG |
|---|---|---|---|---|---|---|---|---|
| SD01 | Female | 60 | 10 | Both | Left | No | Yes | No |
| SD02 | Male | 48 | 14 | Left | Left | Yes | No | Yes |
| SD03 | Female | 60 | 8 | Both | Left | NA | Yes | No |
| SD04 | Female | 45 | 20 | Both | Left | NA | Yes | Yes |
| SD05 | Male | 56 | 10 | Both | Left | No | Yes | Yes |
| SD06 | Male | 51 | 12 | Both | Left | No | Yes | Yes |
| SD07 | Female | 53 | 11 | Both | Left | No | No | Yes |
| SD08 | Male | 57 | 12 | Left | Left | Yes | Yes | Yes |
| SD09 | Female | 63 | 11 | Both | Left | NA | Yes | Yes |
| SD10 | Male | 55 | 13 | Left | Right | No | Yes | Yes |
| SD11 | Male | 51 | 15 | Both | Left | No | Yes | Yes |
| SD12 | Female | 60 | 12 | Both | Left | No | Yes | Yes |
| SD13 | Female | 63 | 9 | Left | Right | No | Yes | Yes |
| SD14 | Male | 57 | 15 | Both | Left | No | Yes | Yes |
| SD15 | Female | 66 | 8 | Left | Left | Yes | Yes | No |
| SD16 | Male | 61 | 13 | Both | Left | Yes | Yes | Yes |
| Patient | Sex | Age at onset | Disease duration | Dominant side pathology | Brain tissue side | Dominant side | MTG | DG |
|---|---|---|---|---|---|---|---|---|
| SD01 | Female | 60 | 10 | Both | Left | No | Yes | No |
| SD02 | Male | 48 | 14 | Left | Left | Yes | No | Yes |
| SD03 | Female | 60 | 8 | Both | Left | NA | Yes | No |
| SD04 | Female | 45 | 20 | Both | Left | NA | Yes | Yes |
| SD05 | Male | 56 | 10 | Both | Left | No | Yes | Yes |
| SD06 | Male | 51 | 12 | Both | Left | No | Yes | Yes |
| SD07 | Female | 53 | 11 | Both | Left | No | No | Yes |
| SD08 | Male | 57 | 12 | Left | Left | Yes | Yes | Yes |
| SD09 | Female | 63 | 11 | Both | Left | NA | Yes | Yes |
| SD10 | Male | 55 | 13 | Left | Right | No | Yes | Yes |
| SD11 | Male | 51 | 15 | Both | Left | No | Yes | Yes |
| SD12 | Female | 60 | 12 | Both | Left | No | Yes | Yes |
| SD13 | Female | 63 | 9 | Left | Right | No | Yes | Yes |
| SD14 | Male | 57 | 15 | Both | Left | No | Yes | Yes |
| SD15 | Female | 66 | 8 | Left | Left | Yes | Yes | No |
| SD16 | Male | 61 | 13 | Both | Left | Yes | Yes | Yes |
Contains clinical and pathological information on the patients examined in this study. Pathological diagnosis, as extracted from the reports from the Netherlands Brain Bank. The most affected side of the brain is reported according to the post-mortem pathological examination. Brain tissue side: the side of the brain fresh frozen and used in this study. Dominant side yes/no; whether the side studied was the one most affected according to neuroimaging (NA = not applicable, as both sides were equally affected). Middle temporal gyrus (MTG) and dentate gyrus (DG) indicate whether these areas were available and included in the study.
Whole exome sequencing
Blood-derived DNA of 16 patients and brain-derived DNA from middle temporal gyrus (n = 14) and/or dentate gyrus (n = 13) of semantic dementia brains (n = 16) was captured using Nimblegen’s SeqCap or MedExome library prep kits and sequenced to an average depth of 139×, 496× and 395×, respectively. Reads were mapped to the hg19 reference genome using BWA and processed using picard and GATK, following best practices. Candidate variants were called using thresholds to detect variants present in the brain (>5 reads), but absent in blood (≤1 read). The next three filtering steps for candidate variants were; (i) a custom signal to noise filter (S2N ≥5) as described in the Supplementary material; (ii) a minor allele frequency < 0.01% in the ExAC database; and (iii) a CADD score above 10.
Validation amplicon panel sequencing
We validated a selection of candidate variants (present in brain, absent in blood) to confirm true-positive variants, and two candidate genes (GRN and TARDBP) to exclude false negatives, by amplicon panel sequencing of the same DNA samples used in the discovery whole exome sequencing. All candidate variants in these targets were included in a custom amplicon panel (SWIFT, product code SW CP-ER6161) and sequenced to an average depth of 1601× on a MiSeq v3 with 600 cycles. A second round of amplicon panel sequencing was carried out for further classification of somatic variants of interest in DNA from additional cortex regions (middle frontal gyrus, superior parietal lobe) and cerebellum of two semantic dementia brains, and in DNA from middle temporal gyrus of 66 non-demented control brains from the Netherlands Brain Bank. Data analysis of the panel was done similar to the discovery. Candidate somatic variants were validated when: (i) read depth in the validation was at least 100; (ii) the variant allele count was at least 20 in DNA of the brain; (iii) the variant allele frequency was at least 1% in DNA of the brain; and (iv) variant allele frequency was <1% in blood of the same patient.
Validation of TARDBP variants
We performed additional validation using digital droplet PCR of two TARDBP somatic variant carriers. In short, custom LNA FAM+HEX probes for each variant were designed and optimized by TATAA Biocenter. Synthetic DNA fragments (gBlocksTM) with these variants were generated to serve as positive controls and as a dilution ladder for technical evaluation of the assay. Negative controls were water and DNA of middle temporal gyrus from two unrelated non-demented controls. Each assay was tested on five brain regions of the carrier (medial temporal gyrus, medial frontal gyrus, superior parietal gyrus, dentate gyrus and cerebellum), blood and the two negative controls. Droplets were generated using Bio-Rad’s Droplet Generation Oil for Probes (cat#1863005) in combination with the qPCR Droplet PCR supermix (no dUTP, Bio-Rad cat#1863024) on a Bio-Rad QX200 Droplet Generator. The PCR plate was measured using the QX200 Droplet Reader (Bio-Rad) and analysed with the Quantasoft Analysis Pro software (Bio-Rad). Reactions with fewer than 10 000 accepted droplets were not used in the analysis. Sensitivity rates of the assays were established using 0.1%, 1.0% and 2.5% spiked positive control gBlocksTM mutation fragments and subsequently used to estimated variant allele frequencies by the ratio of FAM-positive droplets over HEX-positive droplets.
Germline variants in blood and brain
To exclude (de novo) germline variants in 12 FTD (CHMP2B, DPP6, FUS, GRN, MAPT, OPTN, SQSTM1, TARDBP, TBK1, TREM2, UNC13A and VCP) candidate genes we performed regular germline variant calling using GATK’s Haplotypecaller using best practices (van Rooij et al., 2017; Ferrari et al., 2019; Greaves and Rohrer, 2019; Clarimon et al., 2020). Variants were annotated using annovar and were manually evaluated based on exonic function, CADD score, frequency in GnomAD, variant allele frequency and presence in the other tissues of the same patient.
Functional analysis of somatic TARDBP variants
The functional impact of both somatic TARDBP variants on the TDP-43 protein was assessed by a previously published add-back splicing assay and by immunofluorescent microscopy of TDP-43 in HeLa cells (D'Ambrogio et al., 2009). In short, the splicing assay contains a minigene construct containing CFTR exon 9 carrying a mutation (C155T) in an exonic splicing enhancer sequence in order to have a ∼50% chance of in- or out-splicing of exon 9. Using wild-type TDP-43 as positive control, and complete loss-of-function F4L mutated TDP-43 as negative control, the relative impact of L41F and R42H on TDP-43 function could be ascertained. To obtain P-values, an unpaired t-test was carried out using GraphPad software (GraphPad Software, La Jolla California, USA). For the immunofluorescence assays, HeLa cells were transfected with wild-type TDP-43 or with TDP-43 carrying variants L41F or R42H. Nuclei were located by chromatic staining of DAPI, and co-localization of TDP-43 is identified by FLAG-TDP-43 protein, as published previously (Mompean et al., 2017). FLAG TDP-43 staining was quantified using regions of interest for nuclear and cytoplasmic signal using Fiji ImageJ software. The percentage of nuclear and cytoplasmic fluorescent signal was measured for nine cells each for the wild-type, L41F and R42H TDP-43 expressing cells. Statistical tests were performed using two-way ANOVA in GraphPad for nuclear-cytoplasmic TDP-43 localization within each cell-line, as well as between the wild-type and the L41F or R42H TDP-43 transfected cells.
Cell-type specificity of the somatic R42H TARDBP variant
For the R42H TARDBP variant carrier, we performed fluorescence-activated nuclear sorting (FANS) on the frontal lobe and parietal lobe, then isolated DNA from the nuclei with QIAamp DNA Micro Kit (QIAGEN). Using NeuN and Olig2 as cell surface markers, we separated neurons (NeuN-positive) and oligodendrocytes (Olig2-positive) from microglia, astrocytes and any other nuclei (double negative). Parietal cortex tissue from a dementia patient unrelated to this study was similarly sorted and used as negative control. Each resulting DNA sample was amplicon sequenced and analysed using the described procedures.
Data availability
All main results are available through Tables 1, 2 and Supplementary Tables 1–3. Additional data from the raw results are available on request from the authors.
Summary of results of eight validated somatic variants
| Variant | Gene | Function | Sample | Tissue | S2N | MAF | CADD | WES-Blood | WES-Brain | P-value | Panel-Blood | Panel-Brain | P-value | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| REF | ALT | VAF | REF | ALT | VAF | REF | ALT | VAF | REF | ALT | VAF | ||||||||||
| chr1:11073909: G/A | TARDBP | Nonsyn | SD14 | MTG | 9.8 | 0 | 20 | 87 | 0 | 0.000 | 765 | 10 | 0.013 | 0.6 | 5126 | 0 | 0.000 | 18719 | 271 | 0.014 | 9 × 10−29 |
| chr1:11073909: C/T | TARDBP | Nonsyn | SD10 | DG | NA | 0 | 28 | 138 | 0 | 0.000 | 1037 | 0 | 0.000 | 1 | 9349 | 4 | 0.000 | 7533 | 152 | 0.020 | 3 × 10−47 |
| chr9:130928555: T/G | CIZ1 | Nonsyn | SD11 | DG | 20.8 | 0 | 16 | 39 | 0 | 0.000 | 323 | 54 | 0.143 | 0.005 | 134 | 0 | 0.000 | 346 | 35 | 0.092 | 3 × 10−5 |
| chr17:4883215: A/G | CAMTA2 | Nonsyn | SD09 | DG | 11.1 | 0 | 18 | 58 | 1 | 0.017 | 420 | 42 | 0.091 | 0.07 | 146 | 1 | 0.007 | 1058 | 24 | 0.022 | 0.4 |
| chr17:34192311: T/G | HEATR9 | Nonsyn | SD11 | DG | 7.6 | 0 | 13 | 72 | 0 | 0.000 | 278 | 21 | 0.070 | 0.02 | 3232 | 24 | 0.007 | 18031 | 329 | 0.018 | 3 × 10−6 |
| chr17:70119726: C/A | SOX9 | Nonsyn | SD14 | DG | 9.0 | 0 | 20 | 56 | 1 | 0.018 | 405 | 15 | 0.036 | 0.7 | 1676 | 5 | 0.003 | 1133 | 76 | 0.063 | 9 × 10−24 |
| chr19:51133405: A/G | SYT3 | Nonsyn | SD11 | DG | 8.5 | 0 | 14 | 14 | 0 | 0.000 | 227 | 25 | 0.099 | 0.4 | 103 | 0 | 0.000 | 1593 | 29 | 0.018 | 0.4 |
| chr19:51133405: A/G | SYT3 | Nonsyn | SD09 | DG | 12.4 | 0 | 14 | 21 | 0 | 0.000 | 236 | 40 | 0.145 | 0.09 | 289 | 1 | 0.003 | 1829 | 29 | 0.016 | 0.2 |
| Variant | Gene | Function | Sample | Tissue | S2N | MAF | CADD | WES-Blood | WES-Brain | P-value | Panel-Blood | Panel-Brain | P-value | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| REF | ALT | VAF | REF | ALT | VAF | REF | ALT | VAF | REF | ALT | VAF | ||||||||||
| chr1:11073909: G/A | TARDBP | Nonsyn | SD14 | MTG | 9.8 | 0 | 20 | 87 | 0 | 0.000 | 765 | 10 | 0.013 | 0.6 | 5126 | 0 | 0.000 | 18719 | 271 | 0.014 | 9 × 10−29 |
| chr1:11073909: C/T | TARDBP | Nonsyn | SD10 | DG | NA | 0 | 28 | 138 | 0 | 0.000 | 1037 | 0 | 0.000 | 1 | 9349 | 4 | 0.000 | 7533 | 152 | 0.020 | 3 × 10−47 |
| chr9:130928555: T/G | CIZ1 | Nonsyn | SD11 | DG | 20.8 | 0 | 16 | 39 | 0 | 0.000 | 323 | 54 | 0.143 | 0.005 | 134 | 0 | 0.000 | 346 | 35 | 0.092 | 3 × 10−5 |
| chr17:4883215: A/G | CAMTA2 | Nonsyn | SD09 | DG | 11.1 | 0 | 18 | 58 | 1 | 0.017 | 420 | 42 | 0.091 | 0.07 | 146 | 1 | 0.007 | 1058 | 24 | 0.022 | 0.4 |
| chr17:34192311: T/G | HEATR9 | Nonsyn | SD11 | DG | 7.6 | 0 | 13 | 72 | 0 | 0.000 | 278 | 21 | 0.070 | 0.02 | 3232 | 24 | 0.007 | 18031 | 329 | 0.018 | 3 × 10−6 |
| chr17:70119726: C/A | SOX9 | Nonsyn | SD14 | DG | 9.0 | 0 | 20 | 56 | 1 | 0.018 | 405 | 15 | 0.036 | 0.7 | 1676 | 5 | 0.003 | 1133 | 76 | 0.063 | 9 × 10−24 |
| chr19:51133405: A/G | SYT3 | Nonsyn | SD11 | DG | 8.5 | 0 | 14 | 14 | 0 | 0.000 | 227 | 25 | 0.099 | 0.4 | 103 | 0 | 0.000 | 1593 | 29 | 0.018 | 0.4 |
| chr19:51133405: A/G | SYT3 | Nonsyn | SD09 | DG | 12.4 | 0 | 14 | 21 | 0 | 0.000 | 236 | 40 | 0.145 | 0.09 | 289 | 1 | 0.003 | 1829 | 29 | 0.016 | 0.2 |
All eight variants passed the validation criteria. WES-Blood/WES-Brain shows the read counts for variant and wild-type from exome sequencing. Panel-Blood/Panel-Brain shows the counts for the amplicon panel. ALT = number of reads carrying the alternative allele; MAF = minor allele frequency; Nonsyn = non-synonymous; REF = number of reads carrying the reference allele; S2N = signal to noise; VAF = variant allele frequency. P-values were obtained by Fisher’s exact tests of the counts between blood and brain.
Summary of results of eight validated somatic variants
| Variant | Gene | Function | Sample | Tissue | S2N | MAF | CADD | WES-Blood | WES-Brain | P-value | Panel-Blood | Panel-Brain | P-value | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| REF | ALT | VAF | REF | ALT | VAF | REF | ALT | VAF | REF | ALT | VAF | ||||||||||
| chr1:11073909: G/A | TARDBP | Nonsyn | SD14 | MTG | 9.8 | 0 | 20 | 87 | 0 | 0.000 | 765 | 10 | 0.013 | 0.6 | 5126 | 0 | 0.000 | 18719 | 271 | 0.014 | 9 × 10−29 |
| chr1:11073909: C/T | TARDBP | Nonsyn | SD10 | DG | NA | 0 | 28 | 138 | 0 | 0.000 | 1037 | 0 | 0.000 | 1 | 9349 | 4 | 0.000 | 7533 | 152 | 0.020 | 3 × 10−47 |
| chr9:130928555: T/G | CIZ1 | Nonsyn | SD11 | DG | 20.8 | 0 | 16 | 39 | 0 | 0.000 | 323 | 54 | 0.143 | 0.005 | 134 | 0 | 0.000 | 346 | 35 | 0.092 | 3 × 10−5 |
| chr17:4883215: A/G | CAMTA2 | Nonsyn | SD09 | DG | 11.1 | 0 | 18 | 58 | 1 | 0.017 | 420 | 42 | 0.091 | 0.07 | 146 | 1 | 0.007 | 1058 | 24 | 0.022 | 0.4 |
| chr17:34192311: T/G | HEATR9 | Nonsyn | SD11 | DG | 7.6 | 0 | 13 | 72 | 0 | 0.000 | 278 | 21 | 0.070 | 0.02 | 3232 | 24 | 0.007 | 18031 | 329 | 0.018 | 3 × 10−6 |
| chr17:70119726: C/A | SOX9 | Nonsyn | SD14 | DG | 9.0 | 0 | 20 | 56 | 1 | 0.018 | 405 | 15 | 0.036 | 0.7 | 1676 | 5 | 0.003 | 1133 | 76 | 0.063 | 9 × 10−24 |
| chr19:51133405: A/G | SYT3 | Nonsyn | SD11 | DG | 8.5 | 0 | 14 | 14 | 0 | 0.000 | 227 | 25 | 0.099 | 0.4 | 103 | 0 | 0.000 | 1593 | 29 | 0.018 | 0.4 |
| chr19:51133405: A/G | SYT3 | Nonsyn | SD09 | DG | 12.4 | 0 | 14 | 21 | 0 | 0.000 | 236 | 40 | 0.145 | 0.09 | 289 | 1 | 0.003 | 1829 | 29 | 0.016 | 0.2 |
| Variant | Gene | Function | Sample | Tissue | S2N | MAF | CADD | WES-Blood | WES-Brain | P-value | Panel-Blood | Panel-Brain | P-value | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| REF | ALT | VAF | REF | ALT | VAF | REF | ALT | VAF | REF | ALT | VAF | ||||||||||
| chr1:11073909: G/A | TARDBP | Nonsyn | SD14 | MTG | 9.8 | 0 | 20 | 87 | 0 | 0.000 | 765 | 10 | 0.013 | 0.6 | 5126 | 0 | 0.000 | 18719 | 271 | 0.014 | 9 × 10−29 |
| chr1:11073909: C/T | TARDBP | Nonsyn | SD10 | DG | NA | 0 | 28 | 138 | 0 | 0.000 | 1037 | 0 | 0.000 | 1 | 9349 | 4 | 0.000 | 7533 | 152 | 0.020 | 3 × 10−47 |
| chr9:130928555: T/G | CIZ1 | Nonsyn | SD11 | DG | 20.8 | 0 | 16 | 39 | 0 | 0.000 | 323 | 54 | 0.143 | 0.005 | 134 | 0 | 0.000 | 346 | 35 | 0.092 | 3 × 10−5 |
| chr17:4883215: A/G | CAMTA2 | Nonsyn | SD09 | DG | 11.1 | 0 | 18 | 58 | 1 | 0.017 | 420 | 42 | 0.091 | 0.07 | 146 | 1 | 0.007 | 1058 | 24 | 0.022 | 0.4 |
| chr17:34192311: T/G | HEATR9 | Nonsyn | SD11 | DG | 7.6 | 0 | 13 | 72 | 0 | 0.000 | 278 | 21 | 0.070 | 0.02 | 3232 | 24 | 0.007 | 18031 | 329 | 0.018 | 3 × 10−6 |
| chr17:70119726: C/A | SOX9 | Nonsyn | SD14 | DG | 9.0 | 0 | 20 | 56 | 1 | 0.018 | 405 | 15 | 0.036 | 0.7 | 1676 | 5 | 0.003 | 1133 | 76 | 0.063 | 9 × 10−24 |
| chr19:51133405: A/G | SYT3 | Nonsyn | SD11 | DG | 8.5 | 0 | 14 | 14 | 0 | 0.000 | 227 | 25 | 0.099 | 0.4 | 103 | 0 | 0.000 | 1593 | 29 | 0.018 | 0.4 |
| chr19:51133405: A/G | SYT3 | Nonsyn | SD09 | DG | 12.4 | 0 | 14 | 21 | 0 | 0.000 | 236 | 40 | 0.145 | 0.09 | 289 | 1 | 0.003 | 1829 | 29 | 0.016 | 0.2 |
All eight variants passed the validation criteria. WES-Blood/WES-Brain shows the read counts for variant and wild-type from exome sequencing. Panel-Blood/Panel-Brain shows the counts for the amplicon panel. ALT = number of reads carrying the alternative allele; MAF = minor allele frequency; Nonsyn = non-synonymous; REF = number of reads carrying the reference allele; S2N = signal to noise; VAF = variant allele frequency. P-values were obtained by Fisher’s exact tests of the counts between blood and brain.
Results
Deep whole exome sequencing
All DNA samples from middle temporal gyrus (n = 14), dentate gyrus (n = 13) and blood (n = 16) were sequenced to an average depth of 496 (range 429–658), 395 (range 310–520) and 139 (range 72–229), respectively.
Exclusion of causal germline variants
Germline variant analysis in the whole exome sequencing data of all semantic dementia patients did not result in known pathogenic variants in any of the 12 known FTD genes. One patient was identified as germline carrier of the V90A variant in TARDBP, which was also reported in controls and thus considered of uncertain significance (Supplementary Table 1) (Borroni et al., 2010; Lattante et al., 2013; Caroppo et al., 2016).
Discovery and validation of somatic variants in semantic dementia brains
After signal to noise, minor allele frequency and CADD score filtering we retained on average 172 variants for dentate gyrus and 57 for middle temporal gyrus per patient (Fig. 1 and Supplementary Fig. 1). We detected variants in 1450 genes from the dentate gyrus and/or middle temporal gyrus of at least one semantic dementia patient and absent in blood. To confirm true-positive variants, we selected a set of 305 variants for validation in a panel of amplicon sequencing based on one of the two following criteria: (i) somatic variants present in at least five brains (resulting in 252 variants in a total of 128 genes); or (ii) variants in candidate genes involved in neurodevelopmental or neurodegenerative diseases (resulting in 53 variants in 51 genes present in one to four brains). Amongst the 51 candidate genes fulfilling the second criterion were single variant carriers in TARDBP (R42H) and in GRN.
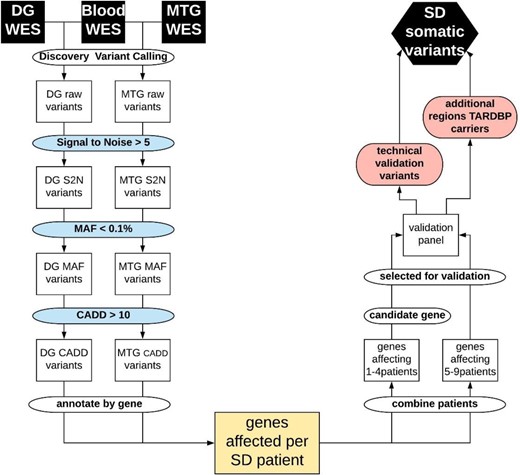
Flowchart of data filtering and analysis. From top left: Raw somatic variant calling using blood and dentate gyrus (DG) or medial temporal gyrus (MTG) deep exome sequencing data (WES), signal to noise filter (S2N), minor allele frequency filter (MAF), CADD score filter, annotating and grouping per gene, resulting in the genes affected in each patient with semantic dementia (SD). Right: Grouping genes affecting multiple patients (>5) or affecting candidate genes in fewer patients (one to four) to be included in the validation amplicon panel. To excluded false negative findings in the WES data in FTD-TDP known germline causal genes GRN and TARDBP, all exons in these genes were included in the validation panel. The first validation round was performed on the same tissues as the discovery WES to confirm true positive variants from the WES, or identify false negative findings in GRN or TARDBP. The second round of validation further classified true positive variants in additional brain tissues and non-demented controls.
We identified eight true-positive variants in the panel of amplicon sequencing (≥100× depth in both brain and blood, variant observed ≥20 times in the brain, variant allele frequency of ≥1% in brain and ≤1% in blood). Seven of those were previously detected with exome sequencing, whereas the eighth variant was not detected in exome sequencing but identified though rescreening of the TARDBP gene in the amplicon sequencing data (Table 2). The non-synonymous variant (R42H) in TARDBP; chr1:11073909-G/A with a CADD score of 20 was the most significantly replicated variant (271 of 18 990 sequenced fragments in middle temporal gyrus, and none of 5126 fragments in blood) and completely absent from gnomAD (variant allele frequency of 1.4% in the middle temporal gyrus of a single semantic dementia brain).
A second non-synonymous variant in the same exon; chr1:11073905-C/T (L41F) in the TARDBP gene was detected in the dentate gyrus of another patient with variant allele frequency of 2.0% in the amplicon panel sequencing data [152 of 7533 fragments, P = 2.8 × 10−47, odds ratio (OR) = 47, 95% confidence interval (CI) = 18–175, compared to blood]. This variant with a CADD score of 28 was also absent from the population databases, and was not observed in blood-derived DNA or any of other brain regions of the same patient (Fig. 2). Both variants observed in a single patient each were taken forward for further validation by digital PCR and functional testing, as germline variants in TARDBP are known to cause FTD and/or amyotrophic lateral sclerosis with TDP-43 pathology (Borroni et al., 2010; Lattante et al., 2013; Caroppo et al., 2016).
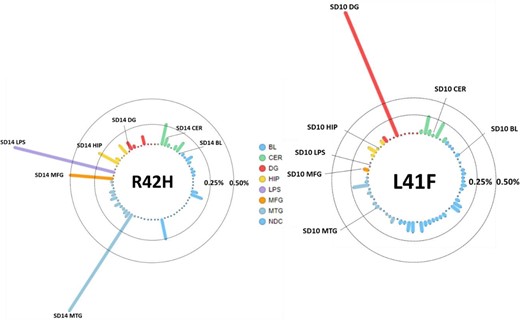
Allele frequencies for L41F and R42H in all tested amplicon panel samples. Each column is a sample, the tissues represented by colour; blood (BL, blue), cerebellum (CER, green), dentate gyrus (DG, red), hippocampus (HIP, orange), middle temporal gyrus (MTG, purple), middle frontal gyrus (MFG, salmon) and superior parietal lobe (LPS, pink). The vertical axis shows the variant allele frequency in that respective tissue, with lines representing the 0.25% and 0.50% thresholds. The tissues with highest variant allele frequency are labelled with the patient identifier and respective tissue.
Validation of TARDBP variant R42H by amplicon sequencing, digital droplet PCR
After confirming presence of this variant in middle temporal gyrus (271 fragments of 18 990, P = 8.9 × 10−29) and absence in 5123 sequenced fragments from blood, we validated this variant in other cortical regions of the same brain. We observed this variant with similar frequency in the parietal lobe (1.2%, 12 of 973 fragments, P = 2.3 × 10−10), in the frontal lobe (0.5%, 11 of 2,122 fragments, P = 1.3 × 10−6), and at lower frequency in the hippocampus (0.3%, 8 of 3021 fragments, P = 3.6 × 10−4) and cerebellum (0.1%, 3 of 2881 fragments, P = 4.7 × 10−2), although the variant allele frequency observed in hippocampus and cerebellum were within the range observed in the other samples, as shown in Fig. 2A. The variant was not observed among temporal cortex samples of 66 non-demented controls (0.03%, total 28 fragments of 106 635, likely representing random sequencing errors). The R42H variant was then sequenced in only neuronal nuclei (NeuN-positive), oligodendrocyte nuclei (Olig2-positive) or the nuclear fraction containing, amongst others, astrocytes and microglia (and other NeuN/Olig2 double negative CNS cell nuclei) in both frontal and parietal lobe of the R42H carrier to an average depth of 3579×. The R42H variant was detected in 2.4% of the neurons in the parietal lobe and 1.1% in the frontal lobe (74 and 42 fragments of 3093 and 3806 in total, respectively). These frequencies were doubled compared the bulk parietal and frontal tissue (1.2% and 0.5%, respectively). The variant was not observed in the control sample (<0.1%) and at three to four times lower frequencies in the oligodendrocytes or double negative nuclear fraction (<0.5% in the parietal lobe and <0.4% in the frontal lobe, respectively).
Validation using digital droplet PCR confirmed the amplicon sequencing results, as shown by the allelic discrimination plots (Fig. 3). The variant was observed in 242 droplets of 13 048 non-empty droplets (variant allele frequency = 1.9%) in the temporal lobe which was significantly higher than the negative controls; blood of the same patient (variant allele frequency = 0.1%, 1 of 809 droplets, P = 1.1 × 10−5) and temporal lobe of two non-demented controls (variant allele frequency = 0.04%, 3 of 7698 droplets, P = 1.5 × 10−44). Similarly, the variant was observed at significantly higher levels compared to the controls in the frontal lobe (variant allele frequency = 1.3%, 126 droplets of 9549, P = 6.7 × 10−4 and P = 1.1 × 10−28) and parietal lobe (variant allele frequency = 0.6%, 21 of 3697 droplets, P = 0.16 and P = 3.5 × 10−8) and cerebellum (variant allele frequency = 0.1%, 13 of 9231 droplets P = 1.0 and P = 0.04).
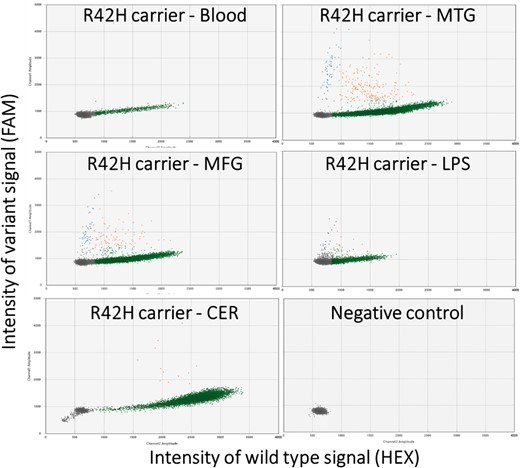
Allelic discrimination plots of the digital droplet PCR for the R42H TARDBP somatic variant. Each marker represents a single droplet and its respective wild-type (horizontal axis) and variant (vertical axis) signal intensity. Five different tissues of the carrier were tested: blood, middle temporal gyrus (MTG), middle frontal gyrus (MFG), lateral parietal lobe (LPS), cerebellum (CER) and a negative control of water is shown. The grey droplets are considered empty, green droplets are wild-type only, orange is both wild-type and variant alleles, and blue droplets were harbouring only the variant allele.
Validation of TARDBP variant L41F by amplicon sequencing, digital droplet PCR
The second TARDBP somatic variant in the same exon was detected in the dentate gyrus with a variant allele frequency of 2.0% in the amplicon panel sequencing data (152 of 7533 fragments, P = 2.8 × 10−47) compared to blood (Fig. 2B). The variant was not observed among temporal cortex samples of 66 non-demented controls (0.04%, total 39 fragments of 106 632, likely representing random sequencing errors). Validation with digital droplet PCR confirmed absence of the variant in blood, cerebellum, frontal lobe, temporal lobe and parietal lobe. Because of the low quantity of DNA from laser-capture microdissection-derived dentate gyrus, this tissue could not be tested using digital PCR. This may have also influenced the whole exome sequencing (WES) result, in which many PCR duplicates were observed for the dentate gyrus data. We did not find any other somatic variants in the TARDBP gene in any of the other semantic dementia brains (average coverage across the gene of 1116) and also not in middle temporal gyrus of non-demented control samples (average coverage of 103× across the gene).
Clinicopathological description of the two cases with somatic TARDBP variants
Both patients carrying the TARDBP L41F or R42H somatic variant developed progressive problems with word finding and language comprehension, and visual agnosia at the age of 55 and 57, respectively. Compulsive-obsessive behaviour, loss of initiative and emotional lability were salient features in both patients, similar to the other 14 patients. Profound left-sided temporal atrophy was observed by neuroimaging (CT, MRI) 2 and 3 years after onset in both TARDBP carriers, in contrast to asymmetric but bilateral atrophy in the other semantic dementia patients (Fig. 4). Neuropathological examination after death (68 and 72 years, respectively) showed severe anterior temporal atrophy, left more pronounced than right in the L41F carrier and more symmetrical in R42H. Microscopically, neuropathological changes were consistent with TDP-pathology type C, with severe neuron loss, gliosis in the temporal cortex with long thick threads and round cytoplasmic inclusions in granular cells of the hippocampus. For the L41F carrier, DNA of the middle temporal gyrus from the right hemisphere was available in the Netherlands Brain Bank and used for all DNA analyses, for the carrier of the R42H variant this was the middle temporal gyrus of the left hemisphere.
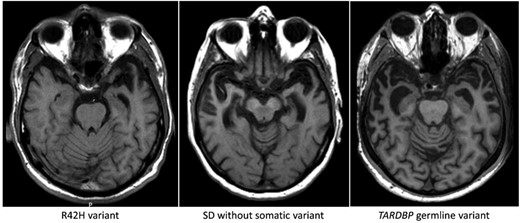
Axial T1-weighted MRI of the semantic dementia patient carrying somatic variant R42H, showing profound leftsided temporal atrophy 3 years after disease onset. Pathological examination 15 years after disease onset showed atrophy of both temporal poles. The middle image is from a patient without a somatic variant (4 years after onset) showing atrophy of both temporal lobes. Right: A patient with the germline (p.I383V) TARDBP variant, showing a similar atrophy pattern bilaterally (4 years after onset).
Functional analysis of TARDBP variants
TDP-43 (encoded by TARDBP) is a protein involved in RNA splicing (Buratti and Baralle, 2001; D'Ambrogio et al., 2009). Therefore, the impact of both TARDBP variants on the activity and localization TDP-43 was established in two assays; splicing regulation and cellular localization. The splicing assay contains a minigene construct containing CFTR exon 9 carrying a mutation (C155T) in an exonic splicing enhancer sequence in order to have ∼50% chance of in- or out-splicing of exon 9. The splicing is mediated by TDP-43 binding to the UG-repeat sequences near the 3′ start site. Thus, when the function of TDP-43 is lost upon targeted small interfering RNA (siRNA) treatment, a decrease to ∼20% of exon 9 skipping is observed. Exon 9 skipping is then rescued by adding back wild-type TDP-43 whose transcript has been made resistant to siRNA treatment. As negative control, we used a construct containing a TDP-43 that carries variant F4L, which is also resistant to the siRNA treatment but cannot bind RNA (Buratti and Baralle, 2001). In the presence of these positive and negative controls, the impact of uncharacterized TDP-43 variants can then be evaluated by comparing the amount of exon 9 skipping of each expressed variant. Both variants significantly decreased exon 9 skipping compared to wild-type TDP-43, as shown in Fig. 5. Splicing impairment was stronger for the L41F variant than for the R42H variant, in accordance with the predicted impact with CADD scores of 28 and 20, respectively. The impact on TDP-43 function was smaller for both variants compared to the siRNA-resistant TDP-43 variant F4L, which blocks RNA binding completely. Immunofluorescent staining demonstrated significantly altered localization of the R42H mutant TDP-43 protein compared to wild-type TDP-43 (Fig. 6). In the wild-type cells, 78% of the fluorescent signal was nuclear (n = 9), versus 71% for the L41F cells (P = 0.54) and 52% for the R42H cells (P = 0.0004). Only in the R42H TDP-43 expressing cells was TDP-43 no longer significantly localized in nuclei compared to cytoplasm. Region of interest measurements and statistical results are provided in Supplementary Table 2.
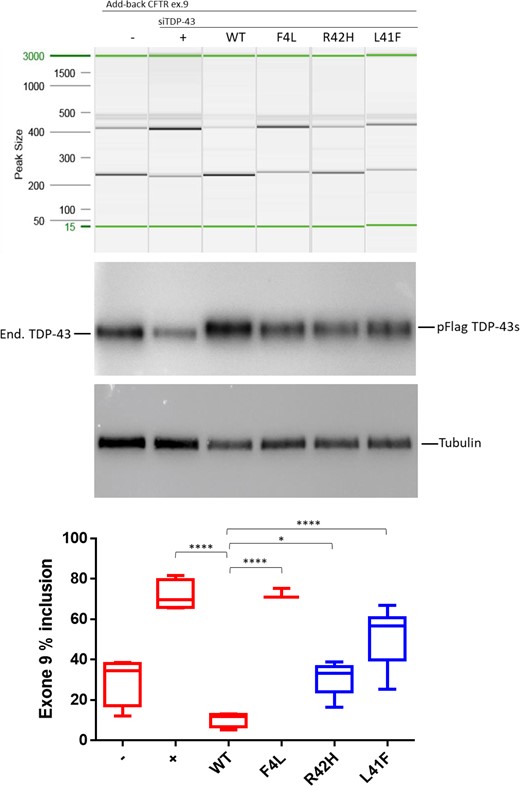
Impact of both TARDBP variants on the splicing regulation functionality of TDP-43, demonstrated by splice-in/out add-back assay of CFTR exon 9. From left to right: The first two lanes show the baseline measurement with both splicing in and out of exon 9 in the absence (-) or presence of TDP-43 siRNA (+). Lane 3 shows that addition of si-resistant wild-type TDP-43 can rescue the splicing functionality (WT) but this cannot be achieved by a TDP-43 carrying the F4L mutation that does not allow the protein to bind RNA (lane 4). Lanes 5 and 6 show the results obtained after the addition of mutated TDP-43 carrying the predicted damaging variants (R42H and L41F). Middle: Western blots showing equal expression of the flagged-TDP-43 wild-type and mutants (pFlag-TDP-43s) following knockdown of the endogenous protein (end. TDP-43). Tubulin was used as an internal control. Bottom: Quantification of the ratio of CFTR exon 9 inclusion. The standard deviation and P-values are reported for three independent experiments. Unpaired t-test was performed for statistical analysis (*P < 0.05).
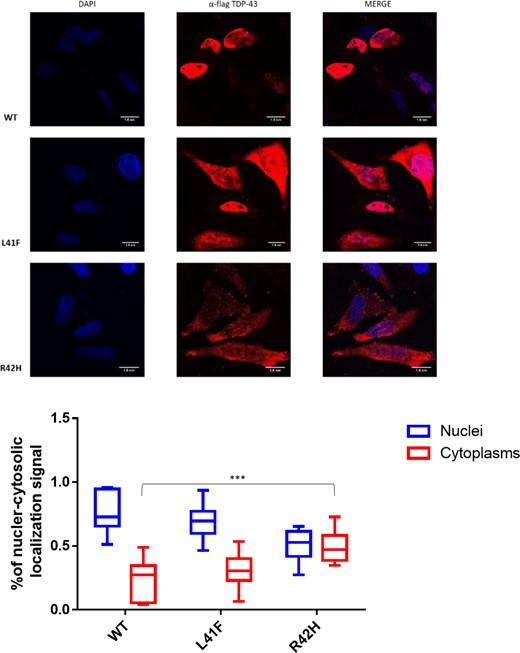
Impact of TARDBP variants on the localization of flagged-TDP-43 wild-type and mutant proteins overexpressed in HeLa cells. The overexpressed proteins were visualized using anti-Flag polyclonal antibody in a 100 nm/pixel field. Scale bar = 10 nm. Top row: Wild-type Flag TDP-43, followed by Flagged TDP-43s carrying both variants; L41F and R42H. Left column: DAPI staining to indicate the chromatin in the nucleus in blue. Middle column: TDP-43 stained in red with a Flag-specific antibody. Right column: Merged images demonstrating TDP-43 localization in the nucleus for wild-type TDP-43, whilst localizing also in the cytoplasm for both TDP43 with variant R42H and L41F. Bottom: Box plots showing fluorescent TDP-43 signal is quantified in the nucleus and cytoplasm for nine cells of each line. The average ratio of nuclear and cytosolic signal is plotted and compared between groups. ****P < 0.0001 and ***P < 0.001 as calculated by two-way ANOVAs between the groups illustrated.
Discussion
The present study identified the occurrence of two low-level pathogenic somatic variants in the TARDBP gene in brains of patients with semantic dementia. These two variants in the first exon of the gene are absent from public databases and significantly affect TDP-43 function and localization. Moreover, the temporal lobe atrophy observed by MRI neuroimaging 3 years after onset in one of the two somatic TARDBP variant carriers resembled classical FTD due to germline TARDBP variants.
The observed low level (1–3%) of TARDBP somatic variants in brain-derived DNA was in accordance with the hypothesis that somatic variants occurred in one or more clones of neurons acquired in a single neural progenitor cells during brain development. Subsequently, the pathophysiological process arising from neurons carrying the somatic variants would then result in focal neurodegeneration later in life. The low percentage may further be attributed to by selective loss of neurons that carried the somatic variants in the affected brain region. The presence of somatic variants shared by (a) clone(s) of neurons in the temporal cortex or dentate gyrus was in contrast to recent studies, which investigated post-mitotic somatic mosaicism (pathogenic single-nucleotide variants and somatic copy-number variations) of known germline disease genes in individual cells (Lee et al., 2018; Lodato et al., 2018; Mokretar et al., 2018). Such post-mitotic somatic variants increased with age in the latter studies and were found in significantly higher number in Alzheimer’s disease or Parkinson’s disease brains compared to controls (Lee et al., 2018; Lodato et al., 2018; Mokretar et al., 2018). Although these somatic DNA variations for age-associated brain diseases were potential interesting, their causal role could not be determined for sure (Lodato et al., 2018).
Post-mitotic variants are a less likely cause for semantic dementia in patients as the disease occurs at a relatively young onset age (<65 years) and its prevalence does not increase with age (Hodges et al., 2010; Landin-Romero et al., 2016). Therefore, our sequencing of DNA from bulk tissue, aiming to identify variants shared by neurons, and estimating their variant-allele-frequencies resembled the study of Park et al. (2019) in which somatic variants were found per brain region (hippocampal formation) in both Alzheimer’s disease patients and controls.
The presence of single somatic variants (R42H and L41F) in the TARDBP gene in several neocortical regions (temporal, frontal and parietal) or dentate gyrus strongly points to the initial occurrence of somatic mosaicism in a single neural progenitor cell (Zilles et al., 2013; Palomero-Gallagher and Zilles, 2019). Somatic variants in neurons arising from the ventricular or subventricular zone have also been shown in childhood or adult neurological diseases (Lee et al., 2012; Lim et al., 2015). By using blood-derived DNA from the same patients as control tissue, we could exclude somatic variants occurring from non-ectodermal lineages (Leija-Salazar et al., 2018). Somatic TARDBP variants could be excluded from 66 non-demented controls by using temporal cortex-derived DNA. As the specific somatic variant (R42H) was absent in both hippocampus and cerebellum of the same patient, the variant must have occurred in neural progenitor cells of the lateral segment of the pallium, which develops into the neocortex (Zilles et al., 2013; Palomero-Gallagher and Zilles, 2019). The variant was enriched (twice as frequent compared to bulk cells) in the neuronal subpopulation of the parietal and frontal lobes, further suggesting the neural origin. A low signal (<20% of signal in the neuronal fraction) of the variant in the other nuclear fractions is a likely due to some residual neuronal nuclei present in the NeuN-negative fraction. Based on these results, we estimate that the R42H variant is present in 5.6%, 4.8% and 2.2% of the neurons in the temporal, parietal and frontal lobes, respectively. The second variant (L41F) was only detected in the hippocampus, suggesting that it occurred in neural progenitor cells of the medial segment of the pallium. The asymmetric onset of the disease pathology in these cases did not necessarily require the occurrence of the somatic variants after developmental separation of both hemispheres, as germline variants have also been associated with other asymmetric neurodegenerative disease processes (Stiles and Jernigan, 2010; Caroppo et al., 2016; Gonzalez-Sanchez et al., 2018). Although of interest, due to the collection procedure in the Netherlands Brain Bank, freezing only one hemisphere, the occurrence of absence of the variants in the other hemisphere could not be tested. The similarity in clinical and pathological phenotype (i.e. severe temporal atrophy, TDP-43-positive inclusions) between the somatic TARDBP variant carriers and germline TARDBP variant carriers supports the potential pathogenicity of these variants (Caroppo et al., 2016; Gonzalez-Sanchez et al., 2018).
Both TARDBP variants identified (L41F and R42H) are located in the first exon of TARDBP and are non-synonymous changes predicted to impact the N-terminal domain of the protein with CADD scores of 28 and 20, respectively (Chang et al., 2012; Zhang et al., 2013; Sasaguri et al., 2016). Both variants are absent in human germline population databases ExAC and gnomAD; in fact, only eight germline variants in the first exon of TARDBP (amino acids 1–79) are described in the gnomAD database (120 000 participants), all extremely rare (<0.003%, 20 carriers across all eight variants combined). Our findings, identifying somatic variants in the N-terminal domain (amino acids 41 and 42) of TARDBP, are in contrast with all germline TARDBP gene variants for familial amyotrophic lateral sclerosis, and occasionally for familial FTD, reported in the glycine-rich region (GRR domain) between amino acids 262 and 414 of the TDP-43 protein (Barmada and Finkbeiner, 2010; Borroni et al., 2010; Lattante et al., 2013; Caroppo et al., 2016; Wang et al., 2016).
Our functional assays convincingly demonstrate a disruptive effect of both variants on normal TARDBP protein function. The impact on TDP-43 activity via CFTR minigene splicing was stronger for L41F than for R42H, with ∼75% and 40% decrease of TDP-43 activity, respectively, compared to wild-type (D'Ambrogio et al., 2009; Mompean et al., 2017). Also, the redistribution of mutant TDP-43 in HeLa cells, from mostly nuclear in unaffected control to both cytoplasmic and nuclear for the R42H variants, supports the cellular pathogenicity. Together, both assays suggest that a correctly folded N-terminal domain of TDP-43 is required for nuclear localization and function, and that neurons carrying these somatic variants have dysfunctional TDP-43 and redistribution of TDP-43 protein to the cytoplasm as observed in FTD and amyotrophic lateral sclerosis brains (Chang et al., 2012; Ihara et al., 2013; Zhang et al., 2013; Qin et al., 2014; Romano et al., 2015; Sasaguri et al., 2016; Mompean et al., 2017; Weskamp and Barmada, 2018). The resulting impact on TDP-43 function in shuttling RNA from the nucleus to the cytoplasm might lead to the protein aggregates observed in semantic dementia brains and subsequent pathogenicity for the cells and tissue in which the variants are present (Barmada and Finkbeiner, 2010; Igaz et al., 2011).
It is unclear how dysfunction of a small percentage of affected neurons (2–6%, double the variant allele frequency) would lead or contribute to extensive degeneration of the temporal lobe and widespread pathology (10–15% of neurons) in the dentate gyrus. Potentially, neuronal dysfunction within one brain region can accumulate until neuron-neuron signalling is sufficiently impaired to functionally disrupt the entire region. Another consideration is that the current study consider mosaicism in bulk DNA of all neurons in the temporal lobe and/or dentate gyrus, whereas many subtypes of neurons exist in these regions, leaving the possibility that the small number of affected neurons in these patients are enriched for a specific neuronal subtype. In Alzheimer’s disease, for instance, a selective loss of parvalbumin-positive GABAergic interneurons (∼3% of the total neuronal population) has been observed (Brady and Mufson, 1997), and the selective dysfunction of these neurons has been causally linked to global brain network changes and progressive amyloid pathology (Verret et al., 2012; Iaccarino et al., 2016; Hijazi et al., 2019), indicating that small populations of affected neurons can indeed contribute to more widespread neurodegenerative processes. The challenges in interpreting selective neuronal dysfunction in the context of widespread neurodegeneration are exemplary of the overall discussion on how neurodegeneration starts and progresses (often differently between patients) throughout the brain, regardless of initial cause of the disease. Further work is needed to fully understand these processes and place the contribution of developmental and post-mitotic somatic DNA variation in the context of disrupted brain function. Cell-specific studies of semantic dementia brains carrying these somatic TARDBP variants may determine in which neuronal subtypes the somatic variants were present.
An important question that remains is why somatic variants were not found in all 14 brains with semantic dementia. There are several potential explanations, some of which include limitations of this study: (i) the bioinformatic filtering steps (absent in blood, CADD score > 10) may have been too stringent and removed potentially causing somatic variants; (ii) pathogenic non-coding variants may have been not detected by the present exome sequencing, and low-level copy number variants missed by the present approach; (iii) causal somatic variants may have become undetectable (disappeared) due to neuron loss in medial temporal gyrus during the neurodegenerative process; (iv) the disease may have originated from causal somatic variants that were only present in the temporal cortex or dentate gyrus opposite to the side of the examined fresh-frozen brain samples, even though we expected that somatic variants occurred prior to the hemisphere separation in brain development; and (v) multifactorial genetic or non-genetic factors may be responsible for most of the semantic dementia cases. Finally, we may have overlooked a relevant variant in the WES data by first focusing on shared variants or damaging variants in candidate genes, which may be less likely true variants. Additionally, the low-level (<0.5%) error rate of the sequencing requires stringent filtering, which may exclude variants that could be detected through panel sequencing, and further investigation of the data may uncover additional relevant variants, as was observed for the L41F variant. Pathogenicity of the remaining six variants confirmed by panel sequencing validation must be validated by future studies.
An interesting issue is whether somatic variants present in TDP-43 related genes may trigger dysregulation in the TDP-43 pathway. In analogy to this, Park et al. (2019) reported a significant enrichment of somatic variants in the PI3K-AKT, MAPK and AMPK pathways in Alzheimer’s disease brains versus control brains. Using a KEGG pathway over-representation analysis, they hypothesized that multiple disease-causing somatic variants converge onto pathways that potentially affect tau phosphorylation. In our view, the next step would be to perform amplicon panel sequencing of a set of FTD-TDP related genes on both semantic dementia brains and controls in order to detect potential additional causal somatic variants in the TDP-43 pathway. Moreover, investigating other series of semantic dementia brains may support our findings, and may give a better estimation of their frequency in semantic dementia. Finally, the present findings raise the question whether somatic variants may be causative in other types of FTD, for example somatic variants in MAPT causing sporadic Pick’s disease. Overall, it seems warranted to carry out such targeted deep sequencing in all well-defined dementia subtypes.
Finally, although our unbiased deep sequencing approach yielded a substantial number of false-positive variants, despite extensive efforts to identify the most likely true variants, it also resulted in the detection of true-positive variants in a well-known candidate gene causative for FTD with TDP-43 pathology. In our view, future studies may choose between two alternative approaches: (i) targeted deep sequencing of bulk tissue of a large number of candidate genes in one way or another related to the pathophysiology; or (ii) single-cell whole genome sequencing generating more reliable data on true-positive variants.
In conclusion, low-level somatic pathogenic variants in the TARDBP gene are an underlying genetic cause of non-familial semantic dementia. This phenomenon needs investigation in other cases of semantic dementia, as well as in other early-onset neurodegenerative diseases, for example non-familial FTD with tau pathology. Moreover, in other neurodegenerative diseases, such as Alzheimer’s disease or Parkinson’s disease, somatic variants may also play a causal or contributing role, and deserve further investigation. Further investigation of somatic variants in known disease genes is warranted, specifically in patients without positive family history and with clearly defined focal neurodegeneration. Our findings have implications for understanding of neurodegenerative disease and the specific role of germline versus somatic variants therein. Also, negative germline variant testing might be insufficient for some diseases, and may require DNA from the appropriate tissue instead to detect somatic variants in order to determine disease causes. Finally, studying the properties of somatic disease-causing genetic variants may reveal novel underlying disease processes and point towards new therapeutic strategies.
Acknowledgements
We would like to thank the Netherlands Brain Bank and all donors that have provided the material to perform this research. Several authors of this publication are members of the European Reference Network for Rare Neurological Diseases – Project ID No 739510.
Funding
We would like to thank the funding agencies for this project; Netherlands Organization for Scientific Research (NWO) through the ZonMw Memorabel grants (project #733050811, #733050816), the Alzheimer Nederland organization, the Gieskes-Strijbis Foundation, AriSLA (project PathensTDP) and the Beneficientia Stiftung from Lichtenstein.
Competing interests
The authors declare no competing interests.
Supplementary material
Supplementary material is available at Brain online


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}