




Abstract
KCNN2 encodes the small conductance calcium-activated potassium channel 2 (SK2). Rodent models with spontaneous Kcnn2 mutations show abnormal gait and locomotor activity, tremor and memory deficits, but human disorders related to KCNN2 variants are largely unknown. Using exome sequencing, we identified a de novo KCNN2 frameshift deletion in a patient with learning disabilities, cerebellar ataxia and white matter abnormalities on brain MRI. This discovery prompted us to collect data from nine additional patients with de novo KCNN2 variants (one nonsense, one splice site, six missense variants and one in-frame deletion) and one family with a missense variant inherited from the affected mother. We investigated the functional impact of six selected variants on SK2 channel function using the patch-clamp technique. All variants tested but one, which was reclassified to uncertain significance, led to a loss-of-function of SK2 channels. Patients with KCNN2 variants had motor and language developmental delay, intellectual disability often associated with early-onset movement disorders comprising cerebellar ataxia and/or extrapyramidal symptoms. Altogether, our findings provide evidence that heterozygous variants, likely causing a haploinsufficiency of the KCNN2 gene, lead to novel autosomal dominant neurodevelopmental movement disorders mirroring phenotypes previously described in rodents.
Introduction
Small conductance calcium-activated potassium channels (SK) are a subfamily of potassium channels comprising three members (SK1–3) encoded by different genes (KCNN1–3) in humans, all widely expressed in the brain (Bond et al., 2005; Faber, 2009). A fourth paralogue (KCNN4, IK/SK4 channel) is primarily expressed in non-neuronal tissues. SK channels share the same topology as voltage-gated potassium channels, with each subunit containing six hydrophobic transmembrane domains (S1–S6) and assembling to form homo- or hetero-tetrameric channels (Lee and MacKinnon, 2018). However, contrary to voltage-gated potassium channels, SK channel activation is voltage independent and depends on intracellular calcium levels. SK channels are activated via calmodulin, which is constitutively attached to a C-terminal region of each subunit (calmodulin binding domain, CaMBD), and triggers channel opening upon Ca2+ binding (Faber, 2009; Lee and MacKinnon, 2018). SK channels can be selectively blocked by the bee venom toxin apamin, which directly binds to the pore of the channel (Weatherall et al., 2010).
In neurons, SK channels play important roles in the regulation of membrane excitability by calcium and serve distinct physiological roles by coupling with different calcium sources. For instance, they contribute to regulate action potential firing by controlling the medium duration of the afterhyperpolarization phase when activated by the transient elevation of intracellular calcium during action potentials (Bond et al., 2005; Faber, 2009). At the postsynaptic level, SK channels can also be activated by calcium influx through N-methyl-d-aspartate (NMDA) receptors, or by a release of calcium from intracellular stores. At this juncture, SK channels play an important role in hampering excitatory synaptic transmission and synaptic plasticity, regulating dendritic excitability and modulating long-term potentiation (Faber, 2009). In vivo pharmacological blockade with apamin or overexpression of SK channels in animals have an impact on learning and memory, although there is a debate on whether their inhibition enhances or impairs learning processes (van der Staay et al., 1999; Hammond et al., 2006; Baker et al., 2011).
KCNN1 has not yet been associated with a human disorder whereas de novo gain-of-function variants in KCNN3 cause Zimmermann-Laband syndrome, a neurodevelopmental disorder characterized by intellectual disability and gingival enlargement (Bauer et al., 2019). One sporadic patient and one family with KCNN2 variants have independently been reported in the literature (Raghuram et al., 2017; Balint et al., 2020) but evidence is still lacking to definitely link KCNN2 to human genetic disorders. Yet, several mouse or rat models with spontaneous Kcnn2 mutations have been described. ‘Frissonnant’ mice exhibit abnormal gait and locomotor activity, early-onset tremors, and memory deficits (Callizot et al., 2001). This mouse line carries a homozygous deletion encompassing exons 1–2 of Kcnn2 (Szatanik et al., 2008). The recessive Jitter mutation (Kcnn2 Leu168Pro) is associated with stiff gait, hunched posture and tremors starting from 3 weeks of age (Mutagenetix database). More recently, a rat model (Tremor dominant Kyoto, Trdk) exhibiting tremor from weaning was shown to carry the dominant Kcnn2 Ile289Asn variant (Kuramoto et al., 2017). In this study, we assembled molecular and clinical data of 11 patients with KCNN2 variants and demonstrate that KCNN2 variants underlie a dominant channelopathy characterized by developmental delay and movement disorders.
Material and methods
Genetic studies
Patient 1 was recruited from a series of individuals with leukoencephalopathies of unknown origin for whom exome sequencing was undertaken to identify a genetic cause (Fig. 1). Trio exome sequencing was performed using the SeqCap EZ MedExome Enrichment Kit (Roche). Libraries were sequenced as paired-end 150 bp reads on a NextSeq500 (Illumina). Bioinformatic analyses were conducted using BWA-0.7.12, picard-tools-1.121, GenomeAnalysisTK-2014.3-17-g0583013, SNPEff-4.2, and variant filtering was performed using the Polyweb interface (Paris-Descartes University, France). We collected clinical and molecular data from additional patients with KCNN2 variants through GeneMatcher (Sobreira et al., 2015) and the EuroEPINOMICS-RES consortium. Exome or genome sequencing was performed in trio or singleton for Patients 2 to 10 in diagnostic or research settings from blood samples (Table 1 and Supplementary Table 1). For singleton analyses, KCNN2 variants were validated and searched for in parents using Sanger sequencing. Variants were classified using the Intervar interface with manual adjustment (Li and Wang, 2017). KCNN2 variants are described on Refseq isoform NM_021614.3 (ENST00000264773.7), which encodes the well-characterized 579 amino acid SK2 channel subunit (Q9H2S1-1 in Uniprot). Of note, several other KCNN2 isoforms (four in UCSC and six in Ensembl) have been described. KCNN2 variants described in this study have been submitted to ClinVar (accessions SCV001371847–SCV001371856). Referring physicians filled out a table with detailed developmental, neurological and behavioural history, including brain MRI data when available. Informed written consent was obtained for each individual or his/her parents before blood sampling. Experiments were performed in accordance with European guidelines and legislation. This study received the ethics approval of Assistance Publique des Hôpitaux de Paris (RCB 2010-A01395-34). Ethics approval were locally obtained for genetic analyses and/or data sharing for Patients 2–10. Patient information was anonymized prior to data sharing and no documents that could establish patient identity were transferred.
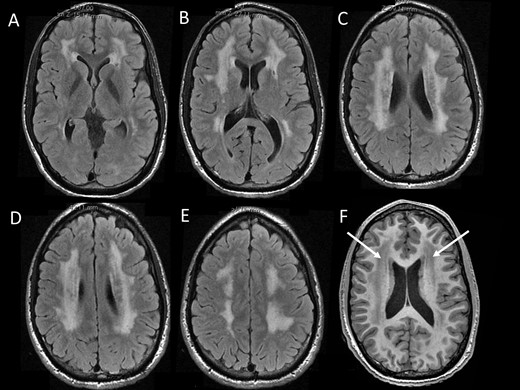
Brain MRI characteristics. MRI in Patient 1 at the age of 20 years showed diffuse periventricular white matter abnormalities with hyperintensenties on FLAIR sequences (A–E) and hypointensities (arrows) on T1-weighted sequences.
Molecular and clinical characteristics of patients with pathogenic or likely pathogenic KCNN2 variants
| Patient ID | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Male | Male | Female | Female | Male | Male | Male | Male | Female | Female |
| Age at study | 30 y | 9 y 2 m | 17 y | 2 y | 15 y | 5 y 9 m | 7 y 11 m | 60 y | 16 y | 7 y |
| cDNA change (NM_021614.3) | c.800_803del | c.480C>A | c.862_863delinsTC | c.962_964delTAT | c.1077 T > G | c.1082A>G | c.1084G>A | c.1162C>G | c.1254+2T>C | c.1295T>C |
| Protein change | p.(Tyr267*) | p.(Tyr160*) | p.(Ile288Ser) | p.(Leu321del) | p.(Ile359Met) | p.(Tyr361Cys) | p.(Gly362Ser) | p.(Leu388Val) | p.? | p.(Leu432Pro) |
| Inheritance | De novo | De novo | De novo | De novo | De novo | De novo | De novo | De novo | De novo | Inherited from affected mother |
| Identification method | WES (trio) | WES (trio) | WES (trio) | WES (trio) | WES (trio) | WES (trio) | WES (trio) | WGS (trio) | WES (singleton) | WES (singleton) |
| ACMG classification | Pathogenic | Pathogenic | Likely pathogenic | Pathogenic | Pathogenic | Likely pathogenic | Pathogenic | Pathogenic | Likely pathogenic | Pathogenic |
| Motor delay | No | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Language delay | No | Yes (regression) | Yes | Yes | Yes | Yes (regression) | Yes | Yes | Yes | No |
| Intellectual disability | Yes (mild) | Yes (severe) | Yes (mild) | Developmental delay | Yes (moderate) | Yes (severe) | Yes (mild) | Yes (moderate) | Yes (moderate) | Yes (mild) |
| Behavioural disturbances | Psychotic episodes in adulthood | ASD | ASD, anxiety, panic attacks | Normal at 2 y | ADHD, anxiety | Autistic features | Autistic features | Autistic features, ADHD | Autistic features, ADHD, anxiety | Normal at 7 y |
| Seizures (age at onset; type) | No | No | No | No | No but epileptiform activity on EEG | No | Yes (2 y 1 m; febrile, GTCS) | No | Yes (14 years, GTCS, nocturnal focal seizures) | No |
| Tremor | Yes (17 y; intention tremor) | No | Yes (10 y; intention and resting tremor) | No | Yes (10 y; intention and resting tremor) | No | No | Yes (mild tremor) | No | Yes (3 m; intention tremor) |
| Cerebellar ataxia (age at onset) | Yes (childhood) | No | No | No | No | Yes (since starting to walk) | No | Yes | No | Yes (12 m) |
| Extrapyramidal symptoms (age at onset) | No | No | Myoclonus dystonia | No | Bradykinesia, cogwheel rigidity (15 y) | No | No | Dyskinesia, parkinsonism, choreic movements | No | Dyskinesia (6 y) |
| Patient ID | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Male | Male | Female | Female | Male | Male | Male | Male | Female | Female |
| Age at study | 30 y | 9 y 2 m | 17 y | 2 y | 15 y | 5 y 9 m | 7 y 11 m | 60 y | 16 y | 7 y |
| cDNA change (NM_021614.3) | c.800_803del | c.480C>A | c.862_863delinsTC | c.962_964delTAT | c.1077 T > G | c.1082A>G | c.1084G>A | c.1162C>G | c.1254+2T>C | c.1295T>C |
| Protein change | p.(Tyr267*) | p.(Tyr160*) | p.(Ile288Ser) | p.(Leu321del) | p.(Ile359Met) | p.(Tyr361Cys) | p.(Gly362Ser) | p.(Leu388Val) | p.? | p.(Leu432Pro) |
| Inheritance | De novo | De novo | De novo | De novo | De novo | De novo | De novo | De novo | De novo | Inherited from affected mother |
| Identification method | WES (trio) | WES (trio) | WES (trio) | WES (trio) | WES (trio) | WES (trio) | WES (trio) | WGS (trio) | WES (singleton) | WES (singleton) |
| ACMG classification | Pathogenic | Pathogenic | Likely pathogenic | Pathogenic | Pathogenic | Likely pathogenic | Pathogenic | Pathogenic | Likely pathogenic | Pathogenic |
| Motor delay | No | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Language delay | No | Yes (regression) | Yes | Yes | Yes | Yes (regression) | Yes | Yes | Yes | No |
| Intellectual disability | Yes (mild) | Yes (severe) | Yes (mild) | Developmental delay | Yes (moderate) | Yes (severe) | Yes (mild) | Yes (moderate) | Yes (moderate) | Yes (mild) |
| Behavioural disturbances | Psychotic episodes in adulthood | ASD | ASD, anxiety, panic attacks | Normal at 2 y | ADHD, anxiety | Autistic features | Autistic features | Autistic features, ADHD | Autistic features, ADHD, anxiety | Normal at 7 y |
| Seizures (age at onset; type) | No | No | No | No | No but epileptiform activity on EEG | No | Yes (2 y 1 m; febrile, GTCS) | No | Yes (14 years, GTCS, nocturnal focal seizures) | No |
| Tremor | Yes (17 y; intention tremor) | No | Yes (10 y; intention and resting tremor) | No | Yes (10 y; intention and resting tremor) | No | No | Yes (mild tremor) | No | Yes (3 m; intention tremor) |
| Cerebellar ataxia (age at onset) | Yes (childhood) | No | No | No | No | Yes (since starting to walk) | No | Yes | No | Yes (12 m) |
| Extrapyramidal symptoms (age at onset) | No | No | Myoclonus dystonia | No | Bradykinesia, cogwheel rigidity (15 y) | No | No | Dyskinesia, parkinsonism, choreic movements | No | Dyskinesia (6 y) |
ACMG = American College of Medical Genetics and Genomics; ADHD = attention-deficit hyperactivity disorder; GTCS = generalized tonic-clonic seizures; m = months; WES = whole exome sequencing; WGS = whole genome sequencing; y = years. Detailed molecular and clinical data of all 11 patients, including treatments, are available in Supplementary Table 1.
Molecular and clinical characteristics of patients with pathogenic or likely pathogenic KCNN2 variants
| Patient ID | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Male | Male | Female | Female | Male | Male | Male | Male | Female | Female |
| Age at study | 30 y | 9 y 2 m | 17 y | 2 y | 15 y | 5 y 9 m | 7 y 11 m | 60 y | 16 y | 7 y |
| cDNA change (NM_021614.3) | c.800_803del | c.480C>A | c.862_863delinsTC | c.962_964delTAT | c.1077 T > G | c.1082A>G | c.1084G>A | c.1162C>G | c.1254+2T>C | c.1295T>C |
| Protein change | p.(Tyr267*) | p.(Tyr160*) | p.(Ile288Ser) | p.(Leu321del) | p.(Ile359Met) | p.(Tyr361Cys) | p.(Gly362Ser) | p.(Leu388Val) | p.? | p.(Leu432Pro) |
| Inheritance | De novo | De novo | De novo | De novo | De novo | De novo | De novo | De novo | De novo | Inherited from affected mother |
| Identification method | WES (trio) | WES (trio) | WES (trio) | WES (trio) | WES (trio) | WES (trio) | WES (trio) | WGS (trio) | WES (singleton) | WES (singleton) |
| ACMG classification | Pathogenic | Pathogenic | Likely pathogenic | Pathogenic | Pathogenic | Likely pathogenic | Pathogenic | Pathogenic | Likely pathogenic | Pathogenic |
| Motor delay | No | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Language delay | No | Yes (regression) | Yes | Yes | Yes | Yes (regression) | Yes | Yes | Yes | No |
| Intellectual disability | Yes (mild) | Yes (severe) | Yes (mild) | Developmental delay | Yes (moderate) | Yes (severe) | Yes (mild) | Yes (moderate) | Yes (moderate) | Yes (mild) |
| Behavioural disturbances | Psychotic episodes in adulthood | ASD | ASD, anxiety, panic attacks | Normal at 2 y | ADHD, anxiety | Autistic features | Autistic features | Autistic features, ADHD | Autistic features, ADHD, anxiety | Normal at 7 y |
| Seizures (age at onset; type) | No | No | No | No | No but epileptiform activity on EEG | No | Yes (2 y 1 m; febrile, GTCS) | No | Yes (14 years, GTCS, nocturnal focal seizures) | No |
| Tremor | Yes (17 y; intention tremor) | No | Yes (10 y; intention and resting tremor) | No | Yes (10 y; intention and resting tremor) | No | No | Yes (mild tremor) | No | Yes (3 m; intention tremor) |
| Cerebellar ataxia (age at onset) | Yes (childhood) | No | No | No | No | Yes (since starting to walk) | No | Yes | No | Yes (12 m) |
| Extrapyramidal symptoms (age at onset) | No | No | Myoclonus dystonia | No | Bradykinesia, cogwheel rigidity (15 y) | No | No | Dyskinesia, parkinsonism, choreic movements | No | Dyskinesia (6 y) |
| Patient ID | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Male | Male | Female | Female | Male | Male | Male | Male | Female | Female |
| Age at study | 30 y | 9 y 2 m | 17 y | 2 y | 15 y | 5 y 9 m | 7 y 11 m | 60 y | 16 y | 7 y |
| cDNA change (NM_021614.3) | c.800_803del | c.480C>A | c.862_863delinsTC | c.962_964delTAT | c.1077 T > G | c.1082A>G | c.1084G>A | c.1162C>G | c.1254+2T>C | c.1295T>C |
| Protein change | p.(Tyr267*) | p.(Tyr160*) | p.(Ile288Ser) | p.(Leu321del) | p.(Ile359Met) | p.(Tyr361Cys) | p.(Gly362Ser) | p.(Leu388Val) | p.? | p.(Leu432Pro) |
| Inheritance | De novo | De novo | De novo | De novo | De novo | De novo | De novo | De novo | De novo | Inherited from affected mother |
| Identification method | WES (trio) | WES (trio) | WES (trio) | WES (trio) | WES (trio) | WES (trio) | WES (trio) | WGS (trio) | WES (singleton) | WES (singleton) |
| ACMG classification | Pathogenic | Pathogenic | Likely pathogenic | Pathogenic | Pathogenic | Likely pathogenic | Pathogenic | Pathogenic | Likely pathogenic | Pathogenic |
| Motor delay | No | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Language delay | No | Yes (regression) | Yes | Yes | Yes | Yes (regression) | Yes | Yes | Yes | No |
| Intellectual disability | Yes (mild) | Yes (severe) | Yes (mild) | Developmental delay | Yes (moderate) | Yes (severe) | Yes (mild) | Yes (moderate) | Yes (moderate) | Yes (mild) |
| Behavioural disturbances | Psychotic episodes in adulthood | ASD | ASD, anxiety, panic attacks | Normal at 2 y | ADHD, anxiety | Autistic features | Autistic features | Autistic features, ADHD | Autistic features, ADHD, anxiety | Normal at 7 y |
| Seizures (age at onset; type) | No | No | No | No | No but epileptiform activity on EEG | No | Yes (2 y 1 m; febrile, GTCS) | No | Yes (14 years, GTCS, nocturnal focal seizures) | No |
| Tremor | Yes (17 y; intention tremor) | No | Yes (10 y; intention and resting tremor) | No | Yes (10 y; intention and resting tremor) | No | No | Yes (mild tremor) | No | Yes (3 m; intention tremor) |
| Cerebellar ataxia (age at onset) | Yes (childhood) | No | No | No | No | Yes (since starting to walk) | No | Yes | No | Yes (12 m) |
| Extrapyramidal symptoms (age at onset) | No | No | Myoclonus dystonia | No | Bradykinesia, cogwheel rigidity (15 y) | No | No | Dyskinesia, parkinsonism, choreic movements | No | Dyskinesia (6 y) |
ACMG = American College of Medical Genetics and Genomics; ADHD = attention-deficit hyperactivity disorder; GTCS = generalized tonic-clonic seizures; m = months; WES = whole exome sequencing; WGS = whole genome sequencing; y = years. Detailed molecular and clinical data of all 11 patients, including treatments, are available in Supplementary Table 1.
Electrophysiology
The first six non-truncating variants identified were introduced into a plasmid containing the human KCNN2 NM_021614.3 cDNA (Origene) using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). Plasmids were verified by Sanger sequencing. Chinese hamster ovary (CHO)-K1 cells were transiently cotransfected with 4 µg of plasmid expressing the wild-type (WT) or mutant human KCNN2 (hKCNN2) and a plasmid expressing the green fluorescent protein (pEGFP) at a ratio of 75:1, using the NeonTM transfection system (Invitrogen). Currents were recorded from green fluorescent cells using the whole-cell configuration of the patch-clamp technique 16 to 24 h after transfection at room temperature. Voltage-clamp recordings were performed using an Axopatch 200B amplifier and a Digidata 1440 Analogue/Digital interface (Axon Instruments). Data were low-pass filtered at 2 kHz, digitized at 10 kHz and analysed offline using Clampfit software. Currents were recorded during a 400 ms voltage ramp from −120 mV to +60 mV applied from a holding potential of −80 mV every 5 s after establishment of the whole-cell configuration. The resistance of a typical patch pipette was 3–4 MΩ when filled with the intracellular solution. Series resistance was not compensated, and no leak subtraction was performed. Data were not corrected offline for voltage error and liquid junction potential. The pipette solution contained (in mM): K-gluconate 144, MgCl2 1.15, CaCl2 0.85, MgATP 2, EGTA 1, HEPES, 10, pH adjusted to 7.2 with KOH. This composition gives 1.97 µM calcium-free at 25°C based on the Maxchelator program. The extracellular solution contained (in mM): NaCl 140, KCl 2.5, MgCl2 1, CaCl2 2, HEPES 10, glucose 10, pH adjusted to 7.3 with NaOH.
Data availability
The raw data that support the findings of this study are available from the corresponding authors, upon request.
Results
Trio exome sequencing in a 30-year-old male with learning disabilities, cerebellar ataxia and white matter abnormalities (Fig. 1) led to the identification of a de novo 4-nucleotide deletion introducing a premature termination codon in KCNN2 (c.800_803del, p.Tyr267*). Through collaborative efforts, we identified nine additional patients with de novo KCNN2 variants and an affected mother-daughter pair with an inherited variant (p.Leu432Pro) in the CaMBD domain (Fig. 2 and Table 1). All 11 variants were absent from gnomAD. De novo variants include one nonsense (p.Tyr160*), one splice site (c.1254+2T>C), six missense variants (p.Glu30Gln, p.Ile288Ser, p.Ile359Met, p.Tyr361Cys, p.Gly362Ser, p.Leu388Val), and one deletion of a single amino acid (p.Leu321del). Except for p.Glu30Gln located in the N-terminal domain, de novo amino acid substitutions and deletions cluster in or near functional SK2 domains, i.e. between S4 and S5 (p.Ile288Ser), in S5 (p.Leu321del) or S6 (p.Leu388Val) transmembrane domains, or in the pore region (p.Ile359Met, p.Tyr361Cys, p.Gly362Ser). Missense variants and p.Leu321del alter amino acids highly conserved in mammals, and except for p.Glu30Gln, they also affect amino acids conserved in SK1 (KCNN1) and SK3 (KCNN3) channels (Supplementary Fig. 1).
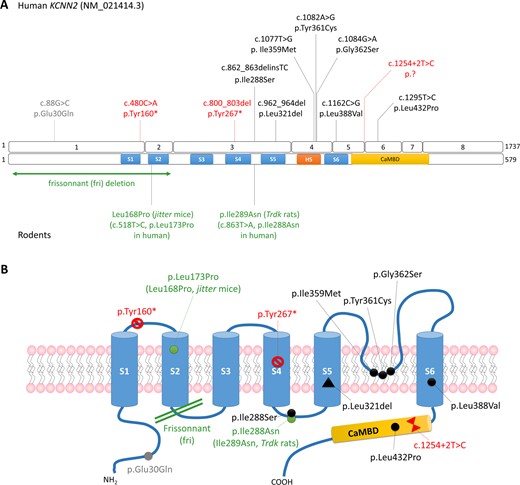
Overview of KCNN2 pathogenic variants. (A) Schematic representation of the KCNN2 variants identified in patients in this study (top) and mutations described in rodents (bottom) on the gene (isoform NM_021414.3) and on the protein. (B) Schematic representation of the SK2 channel showing the localization of the pathogenic variants identified in patients (red: truncating variants; black: pathogenic or likely pathogenic missense variants; grey: de novo variant of unknown significance) and rodents (in green).
To investigate the functional consequences of KCNN2 variants, whole cell currents were recorded from CHO-K1 cells transfected with plasmids expressing the wild-type or six selected mutants using the patch-clamp technique. Large currents recorded upon voltage ramp from cell transfected with hKCNN2 wild-type were generated within a few seconds of whole cell formation and increased to a steady state by 2 min (Fig. 3A and B). This current was sensitive to the specific blocker of SK2 channel apamin (Supplementary Fig. 2). Currents recorded from cells transfected with hKCNN2 p.Glu30Gln also increased with calcium dialysis after the establishment of whole cell configuration, whereas only endogenous currents were recorded from cells transfected with all other mutants (p.Leu321del, p.Ile359Met, p.Gly362Ser, p.Leu388Val and p.Leu432Pro) (Fig. 3A and B). Current densities from cells transfected with hKCNN2 p.Glu30Gln were not statistically different from those recorded from cells expressing hKCNN2 wild-type (Fig. 3C, P = 0.121, Mann-Whitney Rank Sum test). Altogether, these findings indicate that all variants studied, except p.Glu30Gln, lead to a loss-of-function of SK2 homomeric channels.
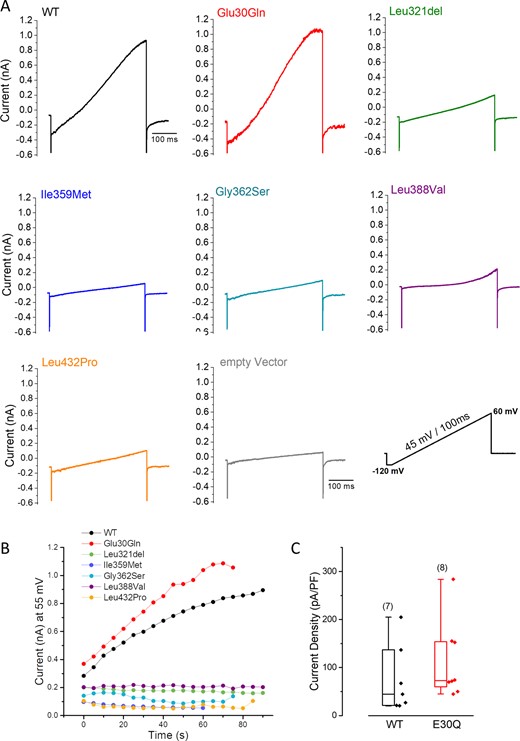
Impact of six selected KCNN2 variants on SK2 channel functional expression. (A) Representative traces of whole cell current recorded from CHO-K1 cells transiently transfected with human wild-type (WT) or mutant hKCNN2 plasmid during a voltage ramp protocol using the patch-clamp technique. Each current trace is the steady-state current obtained after calcium dialysis following the establishment of the whole cell configuration. hKCNN2 p.Leu321del (n = 5), p.Ile359Met (n = 5), p.Gly362Ser (n = 5), p.Leu388Val (n = 8) and p.Leu432Pro (n = 9) mutants do not conduct functional SK2 currents whereas hKCNN2 p.Glu30Gln (n = 8) conducts a SK2 current similar to that expressed by hKCNN2 wild-type (n = 22). (B) Time course of current amplitudes at 55 mV recorded from the cells shown in A, voltage ramp protocols being applied every 5 s. (C) Box plots of the current density for wild-type and p.Glu30Gln (E30Q) showing the median (horizontal line), the 25–75% percentile range (box) and range within 1.5 IQR (whiskers). Numbers of experiment from different cells are shown in parentheses. Statistical analysis between groups was performed with the Kruskal-Wallis ANOVA on ranks (Mann-Whitney rank-sum test, P = 0.121).
Excluding Patient 11, whose variant p.Glu30Gln was reclassified to unknown significance (Supplementary Table 1), this patient series comprises 10 subjects, including six males and four females, with pathogenic or likely pathogenic KCNN2 variants, aged from 2 to 60 years (mean: 17.0 years, median: 12.0 years) (Table 1 and Supplementary Table 1). Nine patients presented with developmental delay, including delays in acquiring motor milestones and a speech delay or a regression of language abilities. All patients had intellectual disability although the degree of cognitive impairment was variable. Four patients exhibited mild intellectual disability, three patients had moderate intellectual disability and two had severe intellectual disability. Autism spectrum disorder (ASD) or autistic features were present in two and four patients, respectively. Movement disorders were observed in six patients, including tremor in five. Four patients presented with cerebellar ataxia, with an early onset from childhood to adolescence, whereas four patients exhibited extrapyramidal symptoms with dyskinesia (n = 2), bradykinesia/parkinsonism (n = 2), myoclonus-dystonia (n = 1) and choreic movements (n = 1). Other neurological features included motor tics (head jerking, eye blinking) (n = 3), nystagmus (n = 2), peripheral neuropathy (n = 2), pyramidal signs (n = 1) and microcephaly (n = 1). The four patients (Patients 2, 4, 7 and 9) without movement disorders were under the age of 16 years at the time of the study. Movement disorders might appear later on and progress over time, as observed in Patient 5, who developed rigidity after initially presenting with tremor. Two patients had epilepsy: Patient 7 (p.Gly362Ser) had one febrile seizure at 20 months of age and afebrile generalized seizures from the age of 25 months, and Patient 9 (c.1254+2T>C) had a generalized clonic-tonic seizure (GTCS) at 14 years followed by nocturnal focal seizures. Brain MRI data were available for six patients, with three displaying white matter abnormalities. Only sparse information was available for the mother of Patient 9 who also carried the p.Leu432Pro variant. She was reported to have mild intellectual disability, a dyskinetic movement disorder with dystonia, and an unspecified psychiatric disorder. She had a brother with similar symptoms and ASD, who was unavailable for genetic testing. Noticeably, Patients 1 and 8, who were aged 30 and 60 years, also had psychotic episodes, suggesting that psychiatric features can be part of the clinical picture of this new disorder in older subjects.
Discussion
Genetic channelopathies are a group of disorders resulting from pathogenic variants in any of the 400 human genes encoding ion channels. More than 30 neurological disorders related to ion channels primarily expressed in the brain have been described so far. These ‘cerebral channelopathies’ commonly include intellectual disability, epilepsy and cerebellar ataxia as primary features (Imbrici et al., 2016; Spillane et al., 2016). In this study, we describe a new channelopathy resulting from pathogenic variants in KCNN2. This dominant disorder is characterized by developmental delay and intellectual disability, often associated with an early-onset movement disorder manifesting mainly as cerebellar ataxia and/or extrapyramidal symptoms.
A de novo frameshift variant (c.581dupA, p.Leu195Valfs*10) was previously reported in a female patient with developmental delay, epilepsy, spasticity, cerebellar ataxia and dystonia (Raghuram et al., 2017). However, this patient also had a homozygous in-frame deletion in ZNF135 and the cause of her phenotype remains uncertain. This phenotype being remarkably similar to those described in our series, the KCNN2 frameshift likely accounts for at least part of her disorder. More recently, a missense variant (c.1112G>A, p.Gly371Glu) located between the S5 and S6 segments in the pore region was shown to segregate with myoclonus-dystonia and anxiety in a three-generation family (Balint et al., 2020). No functional study was performed and the impact of this variant on SK2 channels therefore remains unknown. Nevertheless, myoclonus-dystonia was also diagnosed in Patient 3 and it clinically overlaps with movement disorders observed in other patients, in particular tremor, dyskinesia, parkinsonism and motor tics, further supporting both the pathogenicity of the p.Gly371Glu variant, and myoclonus-dystonia as part of the clinical spectrum of KCNN2 disorders.
Our results suggest that heterozygous KCNN2 variants predominantly lead to a loss-of-function of the SK2 channel (i.e. haploinsufficiency). Interestingly, p.Ile288Ser (Patient 3) alters the same amino acid than the mutation present in Trdk rats (Kcnn2 I289N, corresponding to KCNN2 p.Ile288Asn), which also significantly reduced SK2 currents (Kuramoto et al., 2017). The observation of three truncating variants that either trigger nonsense-mediated decay or truncate the channel before functional domains further supports haploinsufficiency as the main mechanism. The electrophysiological study performed by Raghuram et al. (2017) confirmed that, if expressed in patient’s cells, truncated channels would not be functional. This variant spectrum, comprising both truncating and missense variants altering key functional domains of the channel, is observed in other channelopathies associated with haploinsufficiency, such as Dravet syndrome (Depienne et al., 2009). The intolerance of KCNN2 to haploinsufficiency is strongly supported by a significant depletion of truncating variants in this gene in gnomAD [pLI = 0.99; putative loss-of-function observed/expected = 0.09 (0.04–0.27)]. Only 10 individuals with consistent loss-of-function variants were observed out of the 141 456 individuals included in this database, and half, located at the C-terminal end, spare functional SK domains, contrasting with the early location of truncating variants found in patients. We cannot exclude, however, that some missense variants reported herein may also have gain-of-function or dominant-negative effects on wild-type SK2 or other SK subunits, as observed for many missense variants in ion channels, including SK3/KCNN3 (Bauer et al., 2019).
Contrary to variants altering key functional domains, we could not confirm the pathogenicity of p.Glu30Gln, located in the less conserved N-terminal region of the channel. This patient has a more severe epileptic encephalopathy, possibly related to a yet unidentified cause. However, this patient also shares common features with patients of this series, including tremor, dystonia, dyskinesia and seizures. Further investigations are needed to determine the possible contribution of KCNN2 p.Glu30Gln to this epileptic encephalopathy phenotype.
Remarkably, patients with KCNN2 variants have phenotypes highly similar to those described in Kcnn2 mutant mouse and rat models (Callizot et al., 2001; Szatanik et al., 2008; Kuramoto et al., 2017). All showed rapid constant tremor and abnormal (ataxic and/or stiff) gait, mirroring movement disorders observed in patients, as well as memory deficits and behavioural disturbances (e.g. lack-lustre coat in Jitter mice), reminiscent of intellectual disability, autistic and psychiatric features. A major difference, however, is that Kcnn2 pathogenic variants are recessive in mice whereas they are dominant in rats and humans. The reason why KCNN2 is more sensitive to gene dosage in these species remains unexplained. Possible explanations include distinct baseline Kcnn2 expression levels, gene regulation, or compensatory mechanisms in response to Kcnn2/KCNN2 haploinsufficiency. ‘Frissonnant’ mice and Trdk rats have been proposed as models for Parkinson’s disease (Callizot et al., 2001) and essential tremor (Kuramoto et al., 2017), and interestingly parkinsonism and tremor are part of the clinical features seen in patients with KCNN2 variants. Another interesting observation is that tremors observed in ‘frissonnant’ mice respond to dopamine (Callizot et al., 2001), as also reported for one patient (Raghuram et al., 2017), suggesting that dopamine or dopamine agonists may be used to treat movement disorders in patients with KCNN2 variants. Yet, levodopa was not beneficial to treat myoclonus-dystonia in family members with p.Gly371Glu (Balint et al., 2020), and was not tested in any of our patients.
The exact pathophysiological mechanisms involved in this novel KCNN2 disorder have to be studied in more details, but given the neurological manifestations observed in patients, brain regions affected by KCNN2 haploinsufficiency are likely multiple and include cerebellum, where KCNN2 is highly expressed (Ballesteros-Merino et al., 2012; GTEx Consortium, 2015), basal ganglia and cerebral cortex. The mechanisms linking KCNN2 alteration and leukoencephalopathy remains unclear. According to the Protein Atlas Database, KCNN2 is mainly expressed in neurons of the cortex and cerebellum, but the SK2 protein is also detected in endothelial cells, where alterations may also cause leukodystrophies (Belleri and Presta, 2016).
The intellectual disability observed in KCNN2 patients indicates that germline alteration of SK2 channel does impair memory and learning in humans. The results contrast with previous findings that inhibition of SK channels with apamin enhances learning while overexpression of SK2 channel impairs learning in mice (Hammond et al., 2006). Increased Kcnn2 expression was observed in the cortex of a mouse model for foetal alcohol spectrum disorder and pharmacological blockade of SK2 channels was suggested as a possible treatment strategy (Mohammad et al., 2020). However, our results suggest that this strategy could be potentially hazardous in humans.
Recently, an association between a common intronic variant (rs13188074) of KCNN2 and ASD was reported (Alonso-Gonzalez et al., 2019). Autistic features were present in half of the patients in our series, confirming the association of KCNN2 variants with ASD with incomplete penetrance. KCNN2 variants may also predispose to additional psychiatric features that become more obvious with age since all older patients experienced psychotic episodes.
In conclusion, our study provides compelling evidence that heterozygous KCNN2 variants lead to a novel syndrome associating impairment of cognitive development and movement disorders in humans. This new channelopathy is clinically variable and its characteristics lie at the interface of genetics, paediatrics, neurology and psychiatry.
Web resources
Ensembl, https://www.ensembl.org/
GeneMatcher, https://www.genematcher.org/gnomAD, http://gnomad.broadinstitute.org/
InterVar, http://wintervar.wglab.org/
Maxchelator, https://somapp.ucdmc.ucdavis.edu/pharmacology/bers/maxchelator/
Mutagenetix, https://mutagenetix.utsouthwestern.edu/phenotypic/phenotypic_rec.cfm?pk=88
Protein Atlas, https://www.proteinatlas.org/
UCSC, https://genome.ucsc.edu/
Uniprot, https://www.uniprot.org/
Acknowledgements
The authors thank the patients and their family for their participation in this study, the DNA and cell bank of ICM for DNA extraction, and Chiara Fallerini for GeneMatcher management and Sanger sequencing in Patient 2. Electrophysiological experiments have been carried at the electrophysiology core facility of ICM.
Funding
This study was financially supported by AP-HP (PHRC SPECMET II), the “Investissements d’avenir” program ANR-10-IAIHU-06 (IHU-A-ICM), the ‘Biopsy labex’, INSERM, and University Hospital Essen. F.K received support from the DFG Research Unit FOR2488 ‘ProtectMove’. S.W. received support from FWO-FKM (1861419N). This study made use of the ‘Cell lines and DNA bank of Rett Syndrome, X-linked mental retardation and other genetic diseases’, a member of the Telethon Network of Genetic Biobanks (project no. GTB18001), funded by Telethon Italy and of the EuroBioBank network.
Competing interests
S.Y. is an employee of GeneDx, Inc., a wholly owned subsidiary of OPKO Health, Inc. D.N.S. is a full-time employee of Ambry Genetics. Exome sequencing is one of Ambry’s commercially available tests. R.J.L. is a clinical laboratory director at the Greenwood Genetic Center, which receives fee income from clinical laboratory testing. Other authors declare no competing interests.
Supplementary material
Supplementary material is available at Brain online.


{kind=link}
{kind=link}
{kind=link}