




Mutations in SCN2A, a gene encoding the voltage-gated sodium channel Nav1.2, have been associated with a spectrum of epilepsies and neurodevelopmental disorders. Here, we report the phenotypes of 71 patients and review 130 previously reported patients. We found that (i) encephalopathies with infantile/childhood onset epilepsies (≥3 months of age) occur almost as often as those with an early infantile onset (<3 months), and are thus more frequent than previously reported; (ii) distinct phenotypes can be seen within the late onset group, including myoclonic-atonic epilepsy (two patients), Lennox-Gastaut not emerging from West syndrome (two patients), and focal epilepsies with an electrical status epilepticus during slow sleep-like EEG pattern (six patients); and (iii) West syndrome constitutes a common phenotype with a major recurring mutation (p.Arg853Gln: two new and four previously reported children). Other known phenotypes include Ohtahara syndrome, epilepsy of infancy with migrating focal seizures, and intellectual disability or autism without epilepsy. To assess the response to antiepileptic therapy, we retrospectively reviewed the treatment regimen and the course of the epilepsy in 66 patients for which well-documented medical information was available. We find that the use of sodium channel blockers was often associated with clinically relevant seizure reduction or seizure freedom in children with early infantile epilepsies (<3 months), whereas other antiepileptic drugs were less effective. In contrast, sodium channel blockers were rarely effective in epilepsies with later onset (≥3 months) and sometimes induced seizure worsening. Regarding the genetic findings, truncating mutations were exclusively seen in patients with late onset epilepsies and lack of response to sodium channel blockers. Functional characterization of four selected missense mutations using whole cell patch-clamping in tsA201 cells—together with data from the literature—suggest that mutations associated with early infantile epilepsy result in increased sodium channel activity with gain-of-function, characterized by slowing of fast inactivation, acceleration of its recovery or increased persistent sodium current. Further, a good response to sodium channel blockers clinically was found to be associated with a relatively small gain-of-function. In contrast, mutations in patients with late-onset forms and an insufficient response to sodium channel blockers were associated with loss-of-function effects, including a depolarizing shift of voltage-dependent activation or a hyperpolarizing shift of channel availability (steady-state inactivation). Our clinical and experimental data suggest a correlation between age at disease onset, response to sodium channel blockers and the functional properties of mutations in children with SCN2A-related epilepsy.
Introduction
The SCN2A gene encodes the voltage-gated sodium channel Nav1.2, one of the major neuronal sodium channels that play a role in the initiation and conduction of action potentials. Nav1.2 is expressed in axon initial segments and nodes of Ranvier of myelinated nerve fibres in early development, and in the adult brain in the axon initial segment and unmyelinated axons (Boiko et al., 2001, 2003; Kaplan et al., 2001; Liao et al., 2010b). Accordingly, SCN2A mutations have been mainly shown to affect the early developmental period (Catterall, 2014), but some mutations have also been found as causes of later onset neurological diseases (Kobayashi et al., 2012; Horvath et al., 2016), or a combination of both (Schwarz et al., 2016).
Since the first description of a patient with epilepsy caused by a SCN2A mutation and the findings of SCN2A mutations in benign (familial) neonatal/infantile seizures [B(F)NIS] (Sugawara et al., 2001; Heron et al., 2002), the phenotypic spectrum has expanded considerably. In particular, severe phenotypes with encephalopathy have been reported, including distinct epileptic syndromes such as Ohtahara syndrome (Nakamura et al., 2013; Allen et al., 2016), epilepsy of infancy with migrating focal seizures (EIMFS) (Howell et al., 2015), infantile spasms (Ogiwara et al., 2009; Wong et al., 2015) or West syndrome (Allen et al., 2013; Nakamura et al., 2013), as well as patients with unclassified severe epilepsy phenotypes. However, SCN2A mutations have also been found in patients with intellectual disability and/or autistic features without epilepsy, suggesting the possible involvement of the gene in the aetiology of autism spectrum disorders (Sanders et al., 2012; Li et al., 2016).
To date, the mechanisms for the phenotypic heterogeneity, ranging from benign to very severe clinical presentations, are poorly understood. Differences in functional effects of the mutations may account at least in part for the phenotypic diversity. In addition, the efficacy of antiepileptic drugs (AEDs), especially of sodium channel blockers (SCBs), could be influenced by the way in which specific SCN2A mutations affect Nav1.2 activity.
Therefore, we aimed to assess systematically the phenotypic spectrum and treatment effects in a large cohort of SCN2A-related disorders comprising 201 patients, 71 of whom were not reported previously. For some missense mutations that we selected based on specific clinical findings, and supported by previous reports from the literature, we were able to correlate phenotype and treatment responses to the specific biophysical effects of the mutations.
Materials and methods
Previously unpublished patients
Seventy-one previously unreported patients with a SCN2A mutation were included in this study. Patients were referred through a network of collaborating clinicians and geneticists. Mutations in SCN2A were identified in research or diagnostic laboratories and assumed to be pathogenic, if they were non-synonymous, splice-site altering, nonsense or frameshift changes, predicted damaging by one or more prediction software (PolyPhen-2, SIFT and MutationTaster), seen less than twice in >60 000 controls in the exome aggregation consortium browser (exac.broadinstitute.org), and either occurred de novo, or were inherited from an affected parent, an unaffected mosaic parent or previously reported as pathogenic in other patients. Sanger sequencing was used to confirm all mutations and perform segregation analysis. The study was approved by the local ethics committees.
Referring physicians were provided with a standardized phenotyping sheet to assess clinical characteristics, EEG, and neuroimaging findings. Seizures were diagnosed according to the International League Against Epilepsy commission on classification (Berg et al., 2010), and were assigned, whenever possible, to defined epileptic syndromes. Data on cognitive development and neurological features were recorded at age at onset and at last evaluation. Based on the presence and severity of epilepsy, cognitive status and age at onset of epilepsy patients were classified into the following groups: (i) B(F)NIS, defined as neonatal/infantile onset seizures with a seizure offset during infancy/early childhood, and/or autosomal-dominant inheritance, and normal cognitive development; (ii) encephalopathy with early infantile epilepsy, defined as seizure onset before the age of 3 months, and impaired cognitive development; (iii) encephalopathy with infantile/childhood epilepsy, defined as seizure onset at the age of at least 3 months, and impaired cognitive development; and (iv) intellectual disability and/or autism without epilepsy.
Antiepileptic treatment data were retrospectively assessed by standardized questionnaires. The effect on seizures was classified according to the judgement of the treating physicians into seizure freedom, seizure reduction, no effect or seizure worsening. Particular attention was given to the effects of SCBs, defined as AEDs that reduce the activity of sodium channels by stabilizing an inactivated state. SCBs included phenytoin, carbamazepine, oxcarbazepine, lacosamide, lamotrigine and zonisamide. To provide a general overview in our retrospective analysis, we specifically assessed whether patients were on an SCB by the time that seizure reduction, seizure freedom, or aggravation of seizures occurred.
Frequency of SCN2A-related disorders
To estimate the frequency of SCN2A mutations causing the reported phenotypes in the general population, we used the electronic population databases of National Statistics at the Statens Serum Institute (Denmark) to calculate the birth cohort from 2007 to 2014. The Danish Epilepsy Centre is the only tertiary hospital in Denmark specialized in the treatment of epilepsy, and the majority of patients with presumed genetic epilepsy are referred to this centre for genetic testing.
Literature review
We searched PubMed using the term ‘SCN2A’ and included all relevant patient-related information in our SCN2A dataset. Last search date was 1 June 2016. Papers not available in English, Italian or Danish were excluded. Cases with deletions and duplications spanning SCN2A as well as neighbouring genes were excluded. For patients with little or no clinical information, we listed the phenotype mentioned in the respective publication.
Mutagenesis
To engineer the mutations into the adult splice variant of the human Nav1.2 channel, site-directed mutagenesis was performed using Quickchange® II XL (Agilent Technologies; primers are available upon request) as described previously (Schwarz et al., 2016). Transfection of the α-subunit together with pCLH-hβ1-EGFP and pCLH-hβ2-CD8 in tsA201 cells using Mirus TransIT®-LT1 reagent was performed in a standard way as described previously (Liao et al., 2010a; Lauxmann et al., 2013; Schwarz et al., 2016).
Electrophysiology
Standard whole-cell patch clamp recordings were performed using an Axopatch 200B amplifier, a Digidata 1320A digitizer and pCLAMP 8 data acquisition software (Axon Instruments), as described before (Schwarz et al., 2016). Borosilicate glass pipettes had a final tip resistance of 1–2 MΩ when filled with internal recording solution containing (in mM): 130 CsF, 5 NaCl, 2 MgCl2, 5 EGTA, HEPES (pH 7.4, 290 mOsm). The bath solution contained (in mM): 140 NaCl, 4 KCl, 1 MgCl2, 2 CaCl2, 5 HEPES, 4 dextrose (pH 7.4, 300 mOsm). We carefully checked that the maximal voltage error due to residual series resistance after up to 95% compensation was always <5 mV. Voltage clamp protocols to study channel kinetics were performed as described previously (Schwarz et al., 2016) and are provided in detail in the Supplementary material.
Data and statistical analysis
Traces were displayed off-line with Clampfit software of pClamp 10.0 (Axon Instruments). Graphics were generated using a combination of Microsoft Excel, and Origin (version 9.1; OriginLab Inc., USA) software, statistics were performed using SigmaStat 3.1 (Systat Software GmbH, Germany). All data were tested for normal distribution. For statistical evaluation, ANOVA on ranks (Kruskal-Wallis-test) with Dunn’s post hoc test for not normally distributed data or one-way ANOVA (Bonferroni post hoc test) was used when datasets were normally distributed. All data are shown as means ± standard error of the mean (SEM), n gives the number of cells. We applied the χ2 test to estimate the significance of the differences in AED treatment effects in the two groups of epilepsy with encephalopathy with early and late onset.
Results
Clinical characteristics and treatment response of the previously unpublished patients: B(F)NIS
| Patient | Mutation/ inheritance | Age at seizure onset | Epilepsy synd- rome | Initial seizure type | Other seizure types | EEG | MRI | Cognition onset/ follow-up | Neurol- ogical features | Additi- onal features | Age at last follow-up | Seizure outcome (offset: age) | Treatment effects | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Seizure- free | Seizure reduction | No effect | ||||||||||||||
| 1 | A202V/maternal | 1 d | BFNS | F, GTC | NAV | N | NA/N | N | 4 y 6 m | Sz free (2 m) | PB, PHT, TPM, CBZ | VPA, LEV | ||||
| 2 | G828V/de novo | 2 d | BNS | F, C, AU (cluster) | TCS | MF spikes | N | NA/N | N | 14 m | Sz free (2 m) | PHT→OXC | PB, PP, TPM, LEV | |||
| 3 | Q1531K/de novo | 3 d | BNS | S, C, GTC | GTC | NAV | NA | NA/N | N | 14 m | Sz free (5 d) | PB | ||||
| 4 | S863F/de novo | 5 d | BNS→ other | GTC | GTCS, SE, FD | NA→ MF spikes, ESES-like | N | NA/N | N | ADS | 11 y | Sz free (3 m → 10 y) | OXC→ST | LTG, LEV | ||
| 5 | D343G/de novo | 6 d | BNS | GTC (cluster) | F | N | N | NA/N | N | 18 m | Sz free (6 m) | OXC | LEV, PB, PP, CLB | |||
| 6 | F207S/de novo | 9 d | BNS | C | TCS | Spikes par l | N | NA/N | N | 18 m | Sz free (6 m) | PHT | TPM | PB, CLZ, OXC | ||
| 7 | V261M/de novo | 9 d | BNS | GTC | N | N | NA/N | N | 20 m | Sz free (3 m) | CLB | PHT, LEV | B6, VPA | |||
| 8 | R36G/maternal | 16 m | BFIS | F (cluster) | None | NA | N/N | N | 3 y | Sz free (2 y) | ||||||
| 9 | R36G/maternal | 23 m | BFIS | F | C | Slowing | NA | N/N | N | 4 y 5 m | Sz free (NAV) | ZNS | ||||
| Patient | Mutation/ inheritance | Age at seizure onset | Epilepsy synd- rome | Initial seizure type | Other seizure types | EEG | MRI | Cognition onset/ follow-up | Neurol- ogical features | Additi- onal features | Age at last follow-up | Seizure outcome (offset: age) | Treatment effects | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Seizure- free | Seizure reduction | No effect | ||||||||||||||
| 1 | A202V/maternal | 1 d | BFNS | F, GTC | NAV | N | NA/N | N | 4 y 6 m | Sz free (2 m) | PB, PHT, TPM, CBZ | VPA, LEV | ||||
| 2 | G828V/de novo | 2 d | BNS | F, C, AU (cluster) | TCS | MF spikes | N | NA/N | N | 14 m | Sz free (2 m) | PHT→OXC | PB, PP, TPM, LEV | |||
| 3 | Q1531K/de novo | 3 d | BNS | S, C, GTC | GTC | NAV | NA | NA/N | N | 14 m | Sz free (5 d) | PB | ||||
| 4 | S863F/de novo | 5 d | BNS→ other | GTC | GTCS, SE, FD | NA→ MF spikes, ESES-like | N | NA/N | N | ADS | 11 y | Sz free (3 m → 10 y) | OXC→ST | LTG, LEV | ||
| 5 | D343G/de novo | 6 d | BNS | GTC (cluster) | F | N | N | NA/N | N | 18 m | Sz free (6 m) | OXC | LEV, PB, PP, CLB | |||
| 6 | F207S/de novo | 9 d | BNS | C | TCS | Spikes par l | N | NA/N | N | 18 m | Sz free (6 m) | PHT | TPM | PB, CLZ, OXC | ||
| 7 | V261M/de novo | 9 d | BNS | GTC | N | N | NA/N | N | 20 m | Sz free (3 m) | CLB | PHT, LEV | B6, VPA | |||
| 8 | R36G/maternal | 16 m | BFIS | F (cluster) | None | NA | N/N | N | 3 y | Sz free (2 y) | ||||||
| 9 | R36G/maternal | 23 m | BFIS | F | C | Slowing | NA | N/N | N | 4 y 5 m | Sz free (NAV) | ZNS | ||||
ADS = attention deficit disorder; AU = autonomic seizures; BFIS = benign familial infantile seizures; BNS = benign neonatal seizures; BFNS = benign familial neonatal seizures; C = clonic; F = focal; FD = focal dyscognitive; GTC = generalized tonic-clonic; l = left; m = months; MF = multifocal; N = normal; NA = not applicable; NAV = not available; Par = parietal; SE = status epilepticus; Sz = seizures; TCS = tonic-clonic seizures; y = years; → = change to.
Treatment (sodium channel blockers are highlighted in bold): B6 = vitamin B6; CBZ = carbamazepine; CLB = clobazam; CLZ = clonazepam; LTG = lamotrigine; LEV = levetiracetam; OXC = oxcarbazepine; PB = phenobarbital; PHT = phenytoin; PP = pyridoxal phosphate; ST = sulthiame; TPM = topiramate; VPA = valproate; ZNS = zonisamide.
Clinical characteristics and treatment response of the previously unpublished patients: B(F)NIS
| Patient | Mutation/ inheritance | Age at seizure onset | Epilepsy synd- rome | Initial seizure type | Other seizure types | EEG | MRI | Cognition onset/ follow-up | Neurol- ogical features | Additi- onal features | Age at last follow-up | Seizure outcome (offset: age) | Treatment effects | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Seizure- free | Seizure reduction | No effect | ||||||||||||||
| 1 | A202V/maternal | 1 d | BFNS | F, GTC | NAV | N | NA/N | N | 4 y 6 m | Sz free (2 m) | PB, PHT, TPM, CBZ | VPA, LEV | ||||
| 2 | G828V/de novo | 2 d | BNS | F, C, AU (cluster) | TCS | MF spikes | N | NA/N | N | 14 m | Sz free (2 m) | PHT→OXC | PB, PP, TPM, LEV | |||
| 3 | Q1531K/de novo | 3 d | BNS | S, C, GTC | GTC | NAV | NA | NA/N | N | 14 m | Sz free (5 d) | PB | ||||
| 4 | S863F/de novo | 5 d | BNS→ other | GTC | GTCS, SE, FD | NA→ MF spikes, ESES-like | N | NA/N | N | ADS | 11 y | Sz free (3 m → 10 y) | OXC→ST | LTG, LEV | ||
| 5 | D343G/de novo | 6 d | BNS | GTC (cluster) | F | N | N | NA/N | N | 18 m | Sz free (6 m) | OXC | LEV, PB, PP, CLB | |||
| 6 | F207S/de novo | 9 d | BNS | C | TCS | Spikes par l | N | NA/N | N | 18 m | Sz free (6 m) | PHT | TPM | PB, CLZ, OXC | ||
| 7 | V261M/de novo | 9 d | BNS | GTC | N | N | NA/N | N | 20 m | Sz free (3 m) | CLB | PHT, LEV | B6, VPA | |||
| 8 | R36G/maternal | 16 m | BFIS | F (cluster) | None | NA | N/N | N | 3 y | Sz free (2 y) | ||||||
| 9 | R36G/maternal | 23 m | BFIS | F | C | Slowing | NA | N/N | N | 4 y 5 m | Sz free (NAV) | ZNS | ||||
| Patient | Mutation/ inheritance | Age at seizure onset | Epilepsy synd- rome | Initial seizure type | Other seizure types | EEG | MRI | Cognition onset/ follow-up | Neurol- ogical features | Additi- onal features | Age at last follow-up | Seizure outcome (offset: age) | Treatment effects | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Seizure- free | Seizure reduction | No effect | ||||||||||||||
| 1 | A202V/maternal | 1 d | BFNS | F, GTC | NAV | N | NA/N | N | 4 y 6 m | Sz free (2 m) | PB, PHT, TPM, CBZ | VPA, LEV | ||||
| 2 | G828V/de novo | 2 d | BNS | F, C, AU (cluster) | TCS | MF spikes | N | NA/N | N | 14 m | Sz free (2 m) | PHT→OXC | PB, PP, TPM, LEV | |||
| 3 | Q1531K/de novo | 3 d | BNS | S, C, GTC | GTC | NAV | NA | NA/N | N | 14 m | Sz free (5 d) | PB | ||||
| 4 | S863F/de novo | 5 d | BNS→ other | GTC | GTCS, SE, FD | NA→ MF spikes, ESES-like | N | NA/N | N | ADS | 11 y | Sz free (3 m → 10 y) | OXC→ST | LTG, LEV | ||
| 5 | D343G/de novo | 6 d | BNS | GTC (cluster) | F | N | N | NA/N | N | 18 m | Sz free (6 m) | OXC | LEV, PB, PP, CLB | |||
| 6 | F207S/de novo | 9 d | BNS | C | TCS | Spikes par l | N | NA/N | N | 18 m | Sz free (6 m) | PHT | TPM | PB, CLZ, OXC | ||
| 7 | V261M/de novo | 9 d | BNS | GTC | N | N | NA/N | N | 20 m | Sz free (3 m) | CLB | PHT, LEV | B6, VPA | |||
| 8 | R36G/maternal | 16 m | BFIS | F (cluster) | None | NA | N/N | N | 3 y | Sz free (2 y) | ||||||
| 9 | R36G/maternal | 23 m | BFIS | F | C | Slowing | NA | N/N | N | 4 y 5 m | Sz free (NAV) | ZNS | ||||
ADS = attention deficit disorder; AU = autonomic seizures; BFIS = benign familial infantile seizures; BNS = benign neonatal seizures; BFNS = benign familial neonatal seizures; C = clonic; F = focal; FD = focal dyscognitive; GTC = generalized tonic-clonic; l = left; m = months; MF = multifocal; N = normal; NA = not applicable; NAV = not available; Par = parietal; SE = status epilepticus; Sz = seizures; TCS = tonic-clonic seizures; y = years; → = change to.
Treatment (sodium channel blockers are highlighted in bold): B6 = vitamin B6; CBZ = carbamazepine; CLB = clobazam; CLZ = clonazepam; LTG = lamotrigine; LEV = levetiracetam; OXC = oxcarbazepine; PB = phenobarbital; PHT = phenytoin; PP = pyridoxal phosphate; ST = sulthiame; TPM = topiramate; VPA = valproate; ZNS = zonisamide.
Clinical characteristics and treatment response of the previously unpublished patients: encephalopathy with early onset epilepsy
| Patient | Mutation/ inheritance | Age at seizure onset | Epilepsy syndrome | Initial seizure type | Other seizure types | EEG | MRI | Cognition onset / follow-up | Neurological features | Additional features | Age at last follow-up | Seizure Outcome (offset: age) | Treatment effects | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Seizure- free | Seizure reduction | No effect | Wor- sening | |||||||||||||
| 10 | V423L/de novo | 1 d | OS | M, AP | T | SB→MF spikes | NA | NA/SD | Hypotonia | MC | 34 m (deceased) | Intractable | BR | B6, PP, MDZ, LEV, PHT, RGB, KD, CBZ, LCM | ||
| 11 | E999K/de novo | 1 d | OS→other | T | M, TCS | SB→beta→f spikes | N | NA/SD | Dystonia | Oculogyric crises | 5 y | Sz free (1 m, relapses with low PHT levels) | PHT | PB, B6, PP, VGB, TPM, LEV | ||
| 12 | Q1811E/de novo | 1 d | OS→other | T | GTCS, S, F | SB→Spikes l | N | NA/MD | Hypotonia, unsteady gait | ASD | 8 y | Sz free (4 y) | LTG + VPA + LEV | PHT, TPM | VGB | |
| 13 | M1548V/de novo | 1 d | OS→WS | T | GTC | SB→HA→slowing | N | NA/SD | Hypotonia | ASD | 18 m | Intractable | TPM | PB, PHT, B6, PP, LEV, VGB, CBZ | ||
| 14 | I237N/de novo | 1 d | other | F | F (variable onset) | MF spikes, slowing | HM | NA/SD | Hypotonia | Poor eye contact | 3 y 9 m | Intractable | VPA, CBZ | VGB, LEV, TPM, LTG, B6, PHT, PB, CLZ | ||
| 15 | V887A/de novo | 1 d | OS→WS | M | S | SB→HA | N | NA/SD | Hypotonia | 15 m | Sz free (6 m, relapse with low PHT levels) | PHT | TPM | PB,ST,CS | ||
| 16 | G882R/de novo | 1 d | EIMFS | unilateral TC r/l | T | MF spikes, ictal pattern r / l | N | NA/SD | Hypotonia | MC | 10 m | Intractable | PHT, LCM, ZNS | LEV, CS, B6, PP | ||
| 17 | I1640S/de novo | 1 d | Other | T | F, TC | Spikes r | NA | NA/MD | NAV | 9 y | Sz free (7 y) | LCM | LTG, TPM | B6, PB, VPA, LEV | ||
| 18 | K908E/de novo | 1 d | Other | M | S | Gen+ MF spikes, slowing | At | NA/SD | Nystagmus, hypotonia, dystonia | MC | 8 y | Sz free (7 y 6 m) | LEV, ZNS | |||
| 19 | R1882Q/unknown | 1 d | Other | NA | F →; C | MF + bil spikes, poly spikes | Subtle CD | NA/SD | N | MC | 10 y 6 m | Intractable | CBZ | PB, LEV, OXC, TPM, VPA, CLB, LTG, PHT | ||
| 20 | V1627M/de novo | 2 d | EIMFS | T r/l migrating | F, AP | MF spikes→N | N | NA/MID | Hypotonia | 14 m | Sz free (2 m) | VGB | LEV, B6, PP, TPM, PB,VPA | |||
| 21 | R856Q/de novo | 2 d | OS | T | T | SB→SW | Sinus thrombosis, ischaemia CC | NA/NA | NAV | Connatal sinus thrombosis | 3 m (deceased) | Intractable | MDZ | LEV, PB, PHT, B6, VGB, TPM | ||
| 22 | A1500T/de novo | 2 d | OS→ other | F, T | C | SB→spikes l te→slowing | HM, At | NA/SD | Spasticity | Poor eye contact, MC | 13 y | Sz free (3 m), relapse (12 m), sz free(13 m), relapse (5 y), sz free (10 y) | TPM (3 m), ACTH (13 m) | LTG, VPA | ||
| 23 | M1545V/de novo | 2 d | EIMFS | T, C r/l migrating | F, TCS | SB→MF spikes→N | N | NA/MID | Hypotonia | 12 m | Sz free (3 m, relapses with low PHT levels) | PHT→CBZ | LEV | PB, B6, PP, VPA | ||
| 24 | E430A/de novo | 2 d | Other | F | MF spikes | N | NA/SD | Hypotonia | Regression, ASD | 22 m | Sz free (12m) | PHT | TPM, CLB | |||
| 25 | S1536R/de novo | 2 d | Other | T | AP, SE, TCS | MF spikes, occ slowing | N | NA/MD | Hypotonia | 2 y 8 m | Sz free (6 m, relapses with low PHT levels) | PHT | TPM, PB, LEV, B6 | |||
| 26 | F1597L/de novo | 3 d | EIMFS | T r/l migrating | T→S | SB→spikes r/l te, slowing | At, calcifications | NA/SD | Dystonia | Dysautonomia, irritability | 3 y | Intractable | PHT, CBZ, LTG, BR | B6, PB, LEV, VGB, KD, ST, VPA, LCM, ZNS | ||
| 27 | V424L/de novo | 3 d | Other | F | TCS | SB→MF biocc, slowing | HM | NA/SD | Hypotonia | Poor eye contact | 4 y | Sz free (15 m) | CBZ | CS | PB, VPA | |
| 28 | I891T/de novo | 3 d | Other | T | MF spikes, bisynchrony | N | NA/MD | Coordination difficulties | ADS | 3 y | Sz free (3 m) | PHT | B6, PP, CBZ, CLZ, MDZ, TPM, VPA, ACTH | |||
| 29 | E999K/de novo | 3 d | Other | T r/l | SB→gen SW | At, HM | NA/SD | Dystonia | ASD, nystagmus | 22 y | Sz free (3 y) | CBZ | PB | |||
| 30 | G882E/de novo | 4 d | EIMFS | F, AU, HC r/l M, C migrating | T, AA, | MF spikes, slowing | At, HM | NA/SD | Dystonia | Dysautonomia | 8 y | Intractable | PHT, LCM | VPA, LEV, FBM, GBP, CLB, CLZ, MSX, TPM, PB, KD, CS, LI, ME | ||
| 31 | R1319Q/unknown | 5 d | Other | M | S | MF spikes, slowing | HM | NA/SD | Hypotonia, limb hypertonia | 14 m | Sz free (13 m) | ACTH | TPM, others | |||
| 32 | R1629H/de novo | 6 d | other | S | GTCS, FM | SW fr-ce→MF spikes | N | NA/SD | Hypotonia | 8 y | Sz free (4 m) | PHT | CLZ, MDZ, VPA, LEV, VGB, B6 | |||
| 33 | V423L/de novo | 6 d | OS | F | TCS, T r/l | SB→MF spikes, slowing | T2H | NA/SD | Spasticity | MC | 4 y | Intractable | OXC | PB, LEV, VGB, CS, ZNS, RUF, VPA, TPM, LTG | ||
| 34 | A263V/de novo | 3 w | other | S | T, GTC | Atypical HA→MF spikes, slowing | At, HM | N/SD | Spasticity | MC, scoliosis | 13 y (deceased) | Intractable | ACTH,NZP, CS,MDZ, CLB, LTG, VPA, CLZ,TPM | PB, PHT, TPM, VNS | ||
| 35 | F1651C mosaic/de novo | 6 w | other | GTCS | C, SE | MF spikes | N | N/MID | Hypotonia | 9 m | Sz free (3 m, relapses with low PHT levels) | PHT | PB, OXC, TPM, LEV, B6, PP | |||
| 36 | R1319Q/de novo | 2 m | other | GTC | FC | SW, slowing | HM | N/MD | N | ASD | 3 y 10 m | Sz free (8 m) | VGB | LEV | ||
| 37 | F895S/de novo | 2 m | other | S, M | C, GTC | SB→MF spikes | N | SD/SD | Hypotonia | Optic atrophy | 4 y 8 m | Intractable | VGB, PB, TPM, VPA, CLB | |||
| Patient | Mutation/ inheritance | Age at seizure onset | Epilepsy syndrome | Initial seizure type | Other seizure types | EEG | MRI | Cognition onset / follow-up | Neurological features | Additional features | Age at last follow-up | Seizure Outcome (offset: age) | Treatment effects | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Seizure- free | Seizure reduction | No effect | Wor- sening | |||||||||||||
| 10 | V423L/de novo | 1 d | OS | M, AP | T | SB→MF spikes | NA | NA/SD | Hypotonia | MC | 34 m (deceased) | Intractable | BR | B6, PP, MDZ, LEV, PHT, RGB, KD, CBZ, LCM | ||
| 11 | E999K/de novo | 1 d | OS→other | T | M, TCS | SB→beta→f spikes | N | NA/SD | Dystonia | Oculogyric crises | 5 y | Sz free (1 m, relapses with low PHT levels) | PHT | PB, B6, PP, VGB, TPM, LEV | ||
| 12 | Q1811E/de novo | 1 d | OS→other | T | GTCS, S, F | SB→Spikes l | N | NA/MD | Hypotonia, unsteady gait | ASD | 8 y | Sz free (4 y) | LTG + VPA + LEV | PHT, TPM | VGB | |
| 13 | M1548V/de novo | 1 d | OS→WS | T | GTC | SB→HA→slowing | N | NA/SD | Hypotonia | ASD | 18 m | Intractable | TPM | PB, PHT, B6, PP, LEV, VGB, CBZ | ||
| 14 | I237N/de novo | 1 d | other | F | F (variable onset) | MF spikes, slowing | HM | NA/SD | Hypotonia | Poor eye contact | 3 y 9 m | Intractable | VPA, CBZ | VGB, LEV, TPM, LTG, B6, PHT, PB, CLZ | ||
| 15 | V887A/de novo | 1 d | OS→WS | M | S | SB→HA | N | NA/SD | Hypotonia | 15 m | Sz free (6 m, relapse with low PHT levels) | PHT | TPM | PB,ST,CS | ||
| 16 | G882R/de novo | 1 d | EIMFS | unilateral TC r/l | T | MF spikes, ictal pattern r / l | N | NA/SD | Hypotonia | MC | 10 m | Intractable | PHT, LCM, ZNS | LEV, CS, B6, PP | ||
| 17 | I1640S/de novo | 1 d | Other | T | F, TC | Spikes r | NA | NA/MD | NAV | 9 y | Sz free (7 y) | LCM | LTG, TPM | B6, PB, VPA, LEV | ||
| 18 | K908E/de novo | 1 d | Other | M | S | Gen+ MF spikes, slowing | At | NA/SD | Nystagmus, hypotonia, dystonia | MC | 8 y | Sz free (7 y 6 m) | LEV, ZNS | |||
| 19 | R1882Q/unknown | 1 d | Other | NA | F →; C | MF + bil spikes, poly spikes | Subtle CD | NA/SD | N | MC | 10 y 6 m | Intractable | CBZ | PB, LEV, OXC, TPM, VPA, CLB, LTG, PHT | ||
| 20 | V1627M/de novo | 2 d | EIMFS | T r/l migrating | F, AP | MF spikes→N | N | NA/MID | Hypotonia | 14 m | Sz free (2 m) | VGB | LEV, B6, PP, TPM, PB,VPA | |||
| 21 | R856Q/de novo | 2 d | OS | T | T | SB→SW | Sinus thrombosis, ischaemia CC | NA/NA | NAV | Connatal sinus thrombosis | 3 m (deceased) | Intractable | MDZ | LEV, PB, PHT, B6, VGB, TPM | ||
| 22 | A1500T/de novo | 2 d | OS→ other | F, T | C | SB→spikes l te→slowing | HM, At | NA/SD | Spasticity | Poor eye contact, MC | 13 y | Sz free (3 m), relapse (12 m), sz free(13 m), relapse (5 y), sz free (10 y) | TPM (3 m), ACTH (13 m) | LTG, VPA | ||
| 23 | M1545V/de novo | 2 d | EIMFS | T, C r/l migrating | F, TCS | SB→MF spikes→N | N | NA/MID | Hypotonia | 12 m | Sz free (3 m, relapses with low PHT levels) | PHT→CBZ | LEV | PB, B6, PP, VPA | ||
| 24 | E430A/de novo | 2 d | Other | F | MF spikes | N | NA/SD | Hypotonia | Regression, ASD | 22 m | Sz free (12m) | PHT | TPM, CLB | |||
| 25 | S1536R/de novo | 2 d | Other | T | AP, SE, TCS | MF spikes, occ slowing | N | NA/MD | Hypotonia | 2 y 8 m | Sz free (6 m, relapses with low PHT levels) | PHT | TPM, PB, LEV, B6 | |||
| 26 | F1597L/de novo | 3 d | EIMFS | T r/l migrating | T→S | SB→spikes r/l te, slowing | At, calcifications | NA/SD | Dystonia | Dysautonomia, irritability | 3 y | Intractable | PHT, CBZ, LTG, BR | B6, PB, LEV, VGB, KD, ST, VPA, LCM, ZNS | ||
| 27 | V424L/de novo | 3 d | Other | F | TCS | SB→MF biocc, slowing | HM | NA/SD | Hypotonia | Poor eye contact | 4 y | Sz free (15 m) | CBZ | CS | PB, VPA | |
| 28 | I891T/de novo | 3 d | Other | T | MF spikes, bisynchrony | N | NA/MD | Coordination difficulties | ADS | 3 y | Sz free (3 m) | PHT | B6, PP, CBZ, CLZ, MDZ, TPM, VPA, ACTH | |||
| 29 | E999K/de novo | 3 d | Other | T r/l | SB→gen SW | At, HM | NA/SD | Dystonia | ASD, nystagmus | 22 y | Sz free (3 y) | CBZ | PB | |||
| 30 | G882E/de novo | 4 d | EIMFS | F, AU, HC r/l M, C migrating | T, AA, | MF spikes, slowing | At, HM | NA/SD | Dystonia | Dysautonomia | 8 y | Intractable | PHT, LCM | VPA, LEV, FBM, GBP, CLB, CLZ, MSX, TPM, PB, KD, CS, LI, ME | ||
| 31 | R1319Q/unknown | 5 d | Other | M | S | MF spikes, slowing | HM | NA/SD | Hypotonia, limb hypertonia | 14 m | Sz free (13 m) | ACTH | TPM, others | |||
| 32 | R1629H/de novo | 6 d | other | S | GTCS, FM | SW fr-ce→MF spikes | N | NA/SD | Hypotonia | 8 y | Sz free (4 m) | PHT | CLZ, MDZ, VPA, LEV, VGB, B6 | |||
| 33 | V423L/de novo | 6 d | OS | F | TCS, T r/l | SB→MF spikes, slowing | T2H | NA/SD | Spasticity | MC | 4 y | Intractable | OXC | PB, LEV, VGB, CS, ZNS, RUF, VPA, TPM, LTG | ||
| 34 | A263V/de novo | 3 w | other | S | T, GTC | Atypical HA→MF spikes, slowing | At, HM | N/SD | Spasticity | MC, scoliosis | 13 y (deceased) | Intractable | ACTH,NZP, CS,MDZ, CLB, LTG, VPA, CLZ,TPM | PB, PHT, TPM, VNS | ||
| 35 | F1651C mosaic/de novo | 6 w | other | GTCS | C, SE | MF spikes | N | N/MID | Hypotonia | 9 m | Sz free (3 m, relapses with low PHT levels) | PHT | PB, OXC, TPM, LEV, B6, PP | |||
| 36 | R1319Q/de novo | 2 m | other | GTC | FC | SW, slowing | HM | N/MD | N | ASD | 3 y 10 m | Sz free (8 m) | VGB | LEV | ||
| 37 | F895S/de novo | 2 m | other | S, M | C, GTC | SB→MF spikes | N | SD/SD | Hypotonia | Optic atrophy | 4 y 8 m | Intractable | VGB, PB, TPM, VPA, CLB | |||
AA = atypical absences; A = atonic; AB = absences; ADS = attention deficit disorder; ASD = autism spectrum disorder; AU = autonomic seizures; AP = apnoeic seizures; At = atrophy; Bifr = bifrontal; Bil = bilateral; C = clonic; CC = corpus callosum; CD = cortical dysplasia; Ce = central; DA = drop attacks; ED = epileptiform discharges; F = focal; FD = focal dyscognitive; FC = febrile convulsion; fr = frontal; Gen = generalized; GTC = generalized tonic-clonic; HA = hypsarrhythmia; HC = hemiclonic; HM = hypomyelination; IS = infantile spasms; L = left; m = months; MID = mild intellectual disability; MD = moderate intellectual disability; M = myoclonic; MC = microcephaly; MF = multifocal; N = normal; NA = not applicable; NAV = not available; NCSE = non-convulsive status epilepticus; Occ = occipital; OS = Ohtahara syndrome; Par = parietal; R = right; S = spasms; SE = status epilepticus; SB = suppression burst; SD = severe intellectual disability; SW = spike and waves; Sz = seizures; T = tonic; T2H = T2-hyperintensities; TCS = tonic-clonic seizures; Te = temporal; w = week; y = years; → = change to.
Treatment (sodium channel blockers are highlighted in bold): AZA = acetazolamide; B6 = vitamin B6; BR = bromide; CBZ = carbamazepine; CLB = clobazam; CLZ = clonazepam; CS = corticosteroids; ESM = ethosuximide; FBM = felbamate; GBP = gabapentin; IVIG = intravenous immunoglobulins; KD = ketogenic diet; LCM = lacosamide; LI = lidocaine; LTG = lamotrigine; LEV = levetiracetam; MDZ = Midazolam; ME = metilexine; MSX = mesuximide; OXC = oxcarbazepine; PB = phenobarbital; PHT = phenytoin; PP = pyridoxal phosphate; RGB = retigabine; RUF = rufinamide; ST = sulthiame; STP = stiripentol; TPM = topiramate; VGB = vigabatrin; VNS = vagal nerve stimulation; VPA = valproate; ZNS = zonisamide.
Clinical characteristics and treatment response of the previously unpublished patients: encephalopathy with early onset epilepsy
| Patient | Mutation/ inheritance | Age at seizure onset | Epilepsy syndrome | Initial seizure type | Other seizure types | EEG | MRI | Cognition onset / follow-up | Neurological features | Additional features | Age at last follow-up | Seizure Outcome (offset: age) | Treatment effects | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Seizure- free | Seizure reduction | No effect | Wor- sening | |||||||||||||
| 10 | V423L/de novo | 1 d | OS | M, AP | T | SB→MF spikes | NA | NA/SD | Hypotonia | MC | 34 m (deceased) | Intractable | BR | B6, PP, MDZ, LEV, PHT, RGB, KD, CBZ, LCM | ||
| 11 | E999K/de novo | 1 d | OS→other | T | M, TCS | SB→beta→f spikes | N | NA/SD | Dystonia | Oculogyric crises | 5 y | Sz free (1 m, relapses with low PHT levels) | PHT | PB, B6, PP, VGB, TPM, LEV | ||
| 12 | Q1811E/de novo | 1 d | OS→other | T | GTCS, S, F | SB→Spikes l | N | NA/MD | Hypotonia, unsteady gait | ASD | 8 y | Sz free (4 y) | LTG + VPA + LEV | PHT, TPM | VGB | |
| 13 | M1548V/de novo | 1 d | OS→WS | T | GTC | SB→HA→slowing | N | NA/SD | Hypotonia | ASD | 18 m | Intractable | TPM | PB, PHT, B6, PP, LEV, VGB, CBZ | ||
| 14 | I237N/de novo | 1 d | other | F | F (variable onset) | MF spikes, slowing | HM | NA/SD | Hypotonia | Poor eye contact | 3 y 9 m | Intractable | VPA, CBZ | VGB, LEV, TPM, LTG, B6, PHT, PB, CLZ | ||
| 15 | V887A/de novo | 1 d | OS→WS | M | S | SB→HA | N | NA/SD | Hypotonia | 15 m | Sz free (6 m, relapse with low PHT levels) | PHT | TPM | PB,ST,CS | ||
| 16 | G882R/de novo | 1 d | EIMFS | unilateral TC r/l | T | MF spikes, ictal pattern r / l | N | NA/SD | Hypotonia | MC | 10 m | Intractable | PHT, LCM, ZNS | LEV, CS, B6, PP | ||
| 17 | I1640S/de novo | 1 d | Other | T | F, TC | Spikes r | NA | NA/MD | NAV | 9 y | Sz free (7 y) | LCM | LTG, TPM | B6, PB, VPA, LEV | ||
| 18 | K908E/de novo | 1 d | Other | M | S | Gen+ MF spikes, slowing | At | NA/SD | Nystagmus, hypotonia, dystonia | MC | 8 y | Sz free (7 y 6 m) | LEV, ZNS | |||
| 19 | R1882Q/unknown | 1 d | Other | NA | F →; C | MF + bil spikes, poly spikes | Subtle CD | NA/SD | N | MC | 10 y 6 m | Intractable | CBZ | PB, LEV, OXC, TPM, VPA, CLB, LTG, PHT | ||
| 20 | V1627M/de novo | 2 d | EIMFS | T r/l migrating | F, AP | MF spikes→N | N | NA/MID | Hypotonia | 14 m | Sz free (2 m) | VGB | LEV, B6, PP, TPM, PB,VPA | |||
| 21 | R856Q/de novo | 2 d | OS | T | T | SB→SW | Sinus thrombosis, ischaemia CC | NA/NA | NAV | Connatal sinus thrombosis | 3 m (deceased) | Intractable | MDZ | LEV, PB, PHT, B6, VGB, TPM | ||
| 22 | A1500T/de novo | 2 d | OS→ other | F, T | C | SB→spikes l te→slowing | HM, At | NA/SD | Spasticity | Poor eye contact, MC | 13 y | Sz free (3 m), relapse (12 m), sz free(13 m), relapse (5 y), sz free (10 y) | TPM (3 m), ACTH (13 m) | LTG, VPA | ||
| 23 | M1545V/de novo | 2 d | EIMFS | T, C r/l migrating | F, TCS | SB→MF spikes→N | N | NA/MID | Hypotonia | 12 m | Sz free (3 m, relapses with low PHT levels) | PHT→CBZ | LEV | PB, B6, PP, VPA | ||
| 24 | E430A/de novo | 2 d | Other | F | MF spikes | N | NA/SD | Hypotonia | Regression, ASD | 22 m | Sz free (12m) | PHT | TPM, CLB | |||
| 25 | S1536R/de novo | 2 d | Other | T | AP, SE, TCS | MF spikes, occ slowing | N | NA/MD | Hypotonia | 2 y 8 m | Sz free (6 m, relapses with low PHT levels) | PHT | TPM, PB, LEV, B6 | |||
| 26 | F1597L/de novo | 3 d | EIMFS | T r/l migrating | T→S | SB→spikes r/l te, slowing | At, calcifications | NA/SD | Dystonia | Dysautonomia, irritability | 3 y | Intractable | PHT, CBZ, LTG, BR | B6, PB, LEV, VGB, KD, ST, VPA, LCM, ZNS | ||
| 27 | V424L/de novo | 3 d | Other | F | TCS | SB→MF biocc, slowing | HM | NA/SD | Hypotonia | Poor eye contact | 4 y | Sz free (15 m) | CBZ | CS | PB, VPA | |
| 28 | I891T/de novo | 3 d | Other | T | MF spikes, bisynchrony | N | NA/MD | Coordination difficulties | ADS | 3 y | Sz free (3 m) | PHT | B6, PP, CBZ, CLZ, MDZ, TPM, VPA, ACTH | |||
| 29 | E999K/de novo | 3 d | Other | T r/l | SB→gen SW | At, HM | NA/SD | Dystonia | ASD, nystagmus | 22 y | Sz free (3 y) | CBZ | PB | |||
| 30 | G882E/de novo | 4 d | EIMFS | F, AU, HC r/l M, C migrating | T, AA, | MF spikes, slowing | At, HM | NA/SD | Dystonia | Dysautonomia | 8 y | Intractable | PHT, LCM | VPA, LEV, FBM, GBP, CLB, CLZ, MSX, TPM, PB, KD, CS, LI, ME | ||
| 31 | R1319Q/unknown | 5 d | Other | M | S | MF spikes, slowing | HM | NA/SD | Hypotonia, limb hypertonia | 14 m | Sz free (13 m) | ACTH | TPM, others | |||
| 32 | R1629H/de novo | 6 d | other | S | GTCS, FM | SW fr-ce→MF spikes | N | NA/SD | Hypotonia | 8 y | Sz free (4 m) | PHT | CLZ, MDZ, VPA, LEV, VGB, B6 | |||
| 33 | V423L/de novo | 6 d | OS | F | TCS, T r/l | SB→MF spikes, slowing | T2H | NA/SD | Spasticity | MC | 4 y | Intractable | OXC | PB, LEV, VGB, CS, ZNS, RUF, VPA, TPM, LTG | ||
| 34 | A263V/de novo | 3 w | other | S | T, GTC | Atypical HA→MF spikes, slowing | At, HM | N/SD | Spasticity | MC, scoliosis | 13 y (deceased) | Intractable | ACTH,NZP, CS,MDZ, CLB, LTG, VPA, CLZ,TPM | PB, PHT, TPM, VNS | ||
| 35 | F1651C mosaic/de novo | 6 w | other | GTCS | C, SE | MF spikes | N | N/MID | Hypotonia | 9 m | Sz free (3 m, relapses with low PHT levels) | PHT | PB, OXC, TPM, LEV, B6, PP | |||
| 36 | R1319Q/de novo | 2 m | other | GTC | FC | SW, slowing | HM | N/MD | N | ASD | 3 y 10 m | Sz free (8 m) | VGB | LEV | ||
| 37 | F895S/de novo | 2 m | other | S, M | C, GTC | SB→MF spikes | N | SD/SD | Hypotonia | Optic atrophy | 4 y 8 m | Intractable | VGB, PB, TPM, VPA, CLB | |||
| Patient | Mutation/ inheritance | Age at seizure onset | Epilepsy syndrome | Initial seizure type | Other seizure types | EEG | MRI | Cognition onset / follow-up | Neurological features | Additional features | Age at last follow-up | Seizure Outcome (offset: age) | Treatment effects | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Seizure- free | Seizure reduction | No effect | Wor- sening | |||||||||||||
| 10 | V423L/de novo | 1 d | OS | M, AP | T | SB→MF spikes | NA | NA/SD | Hypotonia | MC | 34 m (deceased) | Intractable | BR | B6, PP, MDZ, LEV, PHT, RGB, KD, CBZ, LCM | ||
| 11 | E999K/de novo | 1 d | OS→other | T | M, TCS | SB→beta→f spikes | N | NA/SD | Dystonia | Oculogyric crises | 5 y | Sz free (1 m, relapses with low PHT levels) | PHT | PB, B6, PP, VGB, TPM, LEV | ||
| 12 | Q1811E/de novo | 1 d | OS→other | T | GTCS, S, F | SB→Spikes l | N | NA/MD | Hypotonia, unsteady gait | ASD | 8 y | Sz free (4 y) | LTG + VPA + LEV | PHT, TPM | VGB | |
| 13 | M1548V/de novo | 1 d | OS→WS | T | GTC | SB→HA→slowing | N | NA/SD | Hypotonia | ASD | 18 m | Intractable | TPM | PB, PHT, B6, PP, LEV, VGB, CBZ | ||
| 14 | I237N/de novo | 1 d | other | F | F (variable onset) | MF spikes, slowing | HM | NA/SD | Hypotonia | Poor eye contact | 3 y 9 m | Intractable | VPA, CBZ | VGB, LEV, TPM, LTG, B6, PHT, PB, CLZ | ||
| 15 | V887A/de novo | 1 d | OS→WS | M | S | SB→HA | N | NA/SD | Hypotonia | 15 m | Sz free (6 m, relapse with low PHT levels) | PHT | TPM | PB,ST,CS | ||
| 16 | G882R/de novo | 1 d | EIMFS | unilateral TC r/l | T | MF spikes, ictal pattern r / l | N | NA/SD | Hypotonia | MC | 10 m | Intractable | PHT, LCM, ZNS | LEV, CS, B6, PP | ||
| 17 | I1640S/de novo | 1 d | Other | T | F, TC | Spikes r | NA | NA/MD | NAV | 9 y | Sz free (7 y) | LCM | LTG, TPM | B6, PB, VPA, LEV | ||
| 18 | K908E/de novo | 1 d | Other | M | S | Gen+ MF spikes, slowing | At | NA/SD | Nystagmus, hypotonia, dystonia | MC | 8 y | Sz free (7 y 6 m) | LEV, ZNS | |||
| 19 | R1882Q/unknown | 1 d | Other | NA | F →; C | MF + bil spikes, poly spikes | Subtle CD | NA/SD | N | MC | 10 y 6 m | Intractable | CBZ | PB, LEV, OXC, TPM, VPA, CLB, LTG, PHT | ||
| 20 | V1627M/de novo | 2 d | EIMFS | T r/l migrating | F, AP | MF spikes→N | N | NA/MID | Hypotonia | 14 m | Sz free (2 m) | VGB | LEV, B6, PP, TPM, PB,VPA | |||
| 21 | R856Q/de novo | 2 d | OS | T | T | SB→SW | Sinus thrombosis, ischaemia CC | NA/NA | NAV | Connatal sinus thrombosis | 3 m (deceased) | Intractable | MDZ | LEV, PB, PHT, B6, VGB, TPM | ||
| 22 | A1500T/de novo | 2 d | OS→ other | F, T | C | SB→spikes l te→slowing | HM, At | NA/SD | Spasticity | Poor eye contact, MC | 13 y | Sz free (3 m), relapse (12 m), sz free(13 m), relapse (5 y), sz free (10 y) | TPM (3 m), ACTH (13 m) | LTG, VPA | ||
| 23 | M1545V/de novo | 2 d | EIMFS | T, C r/l migrating | F, TCS | SB→MF spikes→N | N | NA/MID | Hypotonia | 12 m | Sz free (3 m, relapses with low PHT levels) | PHT→CBZ | LEV | PB, B6, PP, VPA | ||
| 24 | E430A/de novo | 2 d | Other | F | MF spikes | N | NA/SD | Hypotonia | Regression, ASD | 22 m | Sz free (12m) | PHT | TPM, CLB | |||
| 25 | S1536R/de novo | 2 d | Other | T | AP, SE, TCS | MF spikes, occ slowing | N | NA/MD | Hypotonia | 2 y 8 m | Sz free (6 m, relapses with low PHT levels) | PHT | TPM, PB, LEV, B6 | |||
| 26 | F1597L/de novo | 3 d | EIMFS | T r/l migrating | T→S | SB→spikes r/l te, slowing | At, calcifications | NA/SD | Dystonia | Dysautonomia, irritability | 3 y | Intractable | PHT, CBZ, LTG, BR | B6, PB, LEV, VGB, KD, ST, VPA, LCM, ZNS | ||
| 27 | V424L/de novo | 3 d | Other | F | TCS | SB→MF biocc, slowing | HM | NA/SD | Hypotonia | Poor eye contact | 4 y | Sz free (15 m) | CBZ | CS | PB, VPA | |
| 28 | I891T/de novo | 3 d | Other | T | MF spikes, bisynchrony | N | NA/MD | Coordination difficulties | ADS | 3 y | Sz free (3 m) | PHT | B6, PP, CBZ, CLZ, MDZ, TPM, VPA, ACTH | |||
| 29 | E999K/de novo | 3 d | Other | T r/l | SB→gen SW | At, HM | NA/SD | Dystonia | ASD, nystagmus | 22 y | Sz free (3 y) | CBZ | PB | |||
| 30 | G882E/de novo | 4 d | EIMFS | F, AU, HC r/l M, C migrating | T, AA, | MF spikes, slowing | At, HM | NA/SD | Dystonia | Dysautonomia | 8 y | Intractable | PHT, LCM | VPA, LEV, FBM, GBP, CLB, CLZ, MSX, TPM, PB, KD, CS, LI, ME | ||
| 31 | R1319Q/unknown | 5 d | Other | M | S | MF spikes, slowing | HM | NA/SD | Hypotonia, limb hypertonia | 14 m | Sz free (13 m) | ACTH | TPM, others | |||
| 32 | R1629H/de novo | 6 d | other | S | GTCS, FM | SW fr-ce→MF spikes | N | NA/SD | Hypotonia | 8 y | Sz free (4 m) | PHT | CLZ, MDZ, VPA, LEV, VGB, B6 | |||
| 33 | V423L/de novo | 6 d | OS | F | TCS, T r/l | SB→MF spikes, slowing | T2H | NA/SD | Spasticity | MC | 4 y | Intractable | OXC | PB, LEV, VGB, CS, ZNS, RUF, VPA, TPM, LTG | ||
| 34 | A263V/de novo | 3 w | other | S | T, GTC | Atypical HA→MF spikes, slowing | At, HM | N/SD | Spasticity | MC, scoliosis | 13 y (deceased) | Intractable | ACTH,NZP, CS,MDZ, CLB, LTG, VPA, CLZ,TPM | PB, PHT, TPM, VNS | ||
| 35 | F1651C mosaic/de novo | 6 w | other | GTCS | C, SE | MF spikes | N | N/MID | Hypotonia | 9 m | Sz free (3 m, relapses with low PHT levels) | PHT | PB, OXC, TPM, LEV, B6, PP | |||
| 36 | R1319Q/de novo | 2 m | other | GTC | FC | SW, slowing | HM | N/MD | N | ASD | 3 y 10 m | Sz free (8 m) | VGB | LEV | ||
| 37 | F895S/de novo | 2 m | other | S, M | C, GTC | SB→MF spikes | N | SD/SD | Hypotonia | Optic atrophy | 4 y 8 m | Intractable | VGB, PB, TPM, VPA, CLB | |||
AA = atypical absences; A = atonic; AB = absences; ADS = attention deficit disorder; ASD = autism spectrum disorder; AU = autonomic seizures; AP = apnoeic seizures; At = atrophy; Bifr = bifrontal; Bil = bilateral; C = clonic; CC = corpus callosum; CD = cortical dysplasia; Ce = central; DA = drop attacks; ED = epileptiform discharges; F = focal; FD = focal dyscognitive; FC = febrile convulsion; fr = frontal; Gen = generalized; GTC = generalized tonic-clonic; HA = hypsarrhythmia; HC = hemiclonic; HM = hypomyelination; IS = infantile spasms; L = left; m = months; MID = mild intellectual disability; MD = moderate intellectual disability; M = myoclonic; MC = microcephaly; MF = multifocal; N = normal; NA = not applicable; NAV = not available; NCSE = non-convulsive status epilepticus; Occ = occipital; OS = Ohtahara syndrome; Par = parietal; R = right; S = spasms; SE = status epilepticus; SB = suppression burst; SD = severe intellectual disability; SW = spike and waves; Sz = seizures; T = tonic; T2H = T2-hyperintensities; TCS = tonic-clonic seizures; Te = temporal; w = week; y = years; → = change to.
Treatment (sodium channel blockers are highlighted in bold): AZA = acetazolamide; B6 = vitamin B6; BR = bromide; CBZ = carbamazepine; CLB = clobazam; CLZ = clonazepam; CS = corticosteroids; ESM = ethosuximide; FBM = felbamate; GBP = gabapentin; IVIG = intravenous immunoglobulins; KD = ketogenic diet; LCM = lacosamide; LI = lidocaine; LTG = lamotrigine; LEV = levetiracetam; MDZ = Midazolam; ME = metilexine; MSX = mesuximide; OXC = oxcarbazepine; PB = phenobarbital; PHT = phenytoin; PP = pyridoxal phosphate; RGB = retigabine; RUF = rufinamide; ST = sulthiame; STP = stiripentol; TPM = topiramate; VGB = vigabatrin; VNS = vagal nerve stimulation; VPA = valproate; ZNS = zonisamide.
Clinical characteristics and treatment response of the previously unpublished patients: encephalopathy with late onset epilepsy
| Patient | Mutation/ inheritance | Age at seizure onset | Epilepsy syndrome | Initial seizure type | Other seizure types | EEG | MRI | Cognition onset /follow-up | Neurological features | Additional features | Age at last follow-up | Seizure outcome (offset: age) | Treatment effects | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Seizure- free | Sz reduction | No effect | Wors- ening | |||||||||||||
| 38 | G899S/de novo | 3 m | Other | HC | GTC, FD, AA | Bifr SW | N | NA/SD | N | ASD | 7 y | Intractable | TPM, LEV, RUF, CLZ, CLB | PB, B6, VPA, LCM, ZNS, KD | OXC | |
| 39 | T227I/de novo | 3 m | WS | T, AP | S | HA→post spikes, slowing | N | SD/SD | Hypotonia | MC | 2 y | Intractable | LEV, ZNS | B6, VGB, CS, ACTH, TPM, KD, PB, OXC | ||
| 40 | A733T/de novo | 3 m | Other | HC r/l | AA, DA, FD | MF spikes, slowing | N | MD/MD | N | 12 y | Intractable | LEV | VPA, ESM, LTG, OXC | |||
| 41 | R1882P/de novo | 4 m | Other | T, C r | MF spikes, SW- status te→ESES-like | N | N/SD | N | Regression, ASD, aggression | 6 y | Sz free (8 m) | LEV | PB, VPA, ST, KD, LTG | |||
| 42 | L1665F/de novo | 4 m | Other | TS, SE | GTC (series) | N | N | N/SD | N | Stereotyped behaviour | 6 y 3 m | Sz free (7 m) | CBZ | VPA | PB | |
| 43 | L1342P/de novo | 6 m | WS | S | HA→MF spikes, slowing | At | SD/SD | Axial hypotonia, limb spasticity | Apnoeas, no eye contact | 16 y | Intractable | VGB, CS | ||||
| 44 | L881P/de novo | 6 m | WS→LGS | S | T, TC, AA | HA→MF spikes | At, HM, cerebellar at | SD/SD | Spasticity, hemiparesis | Small stature, early puberty | 18 y | Intractable | TPM, MDZ, CLB | VPA, LTG, PB, LEV, VGB, KD | RUF | |
| 45 | I1281F/de novo | 7 m | WS→LGS | S | T, TC | HA→MF spikes, slowing, diffuse SW | At | NA/SD | 16 y | Intractable | VNS, VPA, CLZ, RUF, TPM, LTG | |||||
| 46 | R853Q/de novo | 8 m | WS→LGS | S | AA, T, F (4 y) | HA→for bil spikes, SW, Slowing | T2H, at | N/SD | Dystonia | Preterm 28 w, MC, regression | 6 y 6 m | Intractable | ACTH | VPA, CLB, TPM | LTG, FBM, LEV | CBZ |
| 47 | A1652P/de novo | 9 m | WS | S | atypical HA→ slowing | HM | SD/SD | Hypotonia | ASD | 3 y | Intractable | VGB, LEV, NZP | VPA, TPM, CS | PHT | ||
| 48 | R1319W/de novo | 10 m | WS→LGS | T, S | TCS, A, AA | HA→spikes occ →spikes cue-par r | N | SD/SD | Hypotonia | Diabetes, ASD | 10 y | Sz free (7 y) | CBZ, TPM, ESM, ZNS, VPA, FBM, LEV, B6, KD, CS, LTG | |||
| 49 | E1211K / de novo | 11 m | WS→other | S | FC, SE, M | HA→MF spikes, slowing | N | SD/SD | Hypotonia | 4 y | Intractable | LEV, LTG, PB, KD | ST, VGB, CS | |||
| 50 | R853Q/de novo | 13 m | WS | S | T, AU, M | HA→MF spikes, ESES-like | N | MD/SD | Hypotonia, choreo-athetosis | 8 y | Intractable | VGB, PB, CLB | ST, TPM, LEV, ZNS, ESM, LTG, PHT, OXC, CS, KD,AZA | |||
| 51 | H930Q/de novo | 15 m | MAE | TCS | A, MAS, T, AA | slowing occ | N | N/MD | N | ASD | 6 y | Sz free (18 m) | LEV + TPM | VPA, OXC, CLB | ||
| 52 | P1622S/de novo | 2 y | MAE | MA, DA | T, AA, M | Spike-slow-waves te-par bil | N | MD/MD | Ataxia | ASD, MC | 3 y | Intractable | TPM | LEV, ESM, B6, PB, CLB, VGB, PP, KD | OXC, LTG | |
| 53 | F612S/de novo | 2 y 6 m | Other | TCS | M | fr spikes r/l, fr delta | N | MD/MD | N | 3 y | Intractable | CLB, LEV, RUF | ||||
| 54 | c.605 + 1G > T/ de novo | 2 y 6 m | Other | TCS | T, A | midline spikes ce, slowing→ bitemp spikes | N | N/SD | Hypotonia | ASD, regression | 7 y | Intractable | LEV, CLB, KD | ZNS, LTG, FBM, TPM, RUF, B6, IVIG, VPA, PB, CS | ||
| 55 | V1528Cfs*7/de novo | 3 y | LGS | TCS | T, SE | Spike-slow-waves, Beta (sleep) | N | SD/SD | Spasticity | ASD, MC | 9 y | Sz free (6 y) | VPA + LTG | CLZ, ESM | ||
| 56 | R853Q/unknown | 3 y | Other | TS | S | HA | CC hypoplasia | SD/SD | Pyramidal signs, hand dystonia | Arachnodactyly | 25 y | Intractable | VPA, ACTH | |||
| 57 | C1170Vfs*15/de novo | 3 y | Other | TCS | M, AB | gen SW, l fr SW, high voltage theta | HiS | MD/SD | Hypertonic limbs | ASD | 17 y | Intractable | VPA, LTG, Br, LEV, CLB | CBZ, OXC, TPM, STP, ST, KD | ||
| 58 | G1223R/de novo | 3 y | Other | F, M | TCS, M, A, AA, NCSE | gen SW, PSW, slowing | N | N/MD | Ataxia | Regression | 19 y | Intractable | VPA, CLZ, CLB, LTG, LEV | PHT, PB, CBZ, TPM, VNS, CS | ||
| 59 | R1235*/de novo | 3 y 4 m | Other | FC | GTC, DA | gen SW, PSW | HM | MD/MD | N | ASD | 14 y | Intractable | LEV, ZNS | VPA, CLB, LTG | ||
| 60 | A1773V/de novo | 3 y 6 m | Other | F | Sharp waves r fr (sleep), slowing | N | MD/SD | N | ASD | 4 y | Sz free (4 y) | LEV | ||||
| 61 | K1933M/de novo | 4 y | Other | F, TCS | AA | MF spikes, bil SW→ESES-like | N | SD/SD | Ataxia, parkinsonian gait | ASD, hyperkinetic and aggressive behaviour | 17 y | Intractable | ST, VPA, TPM, CLB, CS | LTG, LCM | CBZ | |
| 62 | c.698‐1G>T, splice site/de novo | 4 y 6 m | Other | F | f spikes→MF, ESES-like→N | N | N/MID | N | 10 y | Sz free (9 y) | TPM | others | ||||
| 63 | W1716*/de novo | 4 y 7 m | Other | Febrile T (cluster) | AA | Gen SW | N | MD/MD | N | Psychosis (17 y) | 25 y | Sz free (9 y) | VPA | PB | CBZ | |
| 64 | N503Kfs*19/de novo | 6 y | Other | TCS (cluster) | Bil sharp waves | N | SD/SD | ASD | 20 y | Sz free (6 y) | VPA | |||||
| 65 | W281*/de novo | 7y | Other | F | sec. Gen. TCS | MF spikes→ESES- like | N | MD/MD | Clumsiness | 13 y | Sz free (10 y) | ST | ||||
| 66 | S1656F/de novo | 8 y 11 m | LGS | GTC | AA | Bifr SW, slowing | N | MD/MD | Hypotonia, crouched gait | Agitation | 12 y | Intractable | LLTG, VPA | TTPM | ||
| Patient | Mutation/ inheritance | Age at seizure onset | Epilepsy syndrome | Initial seizure type | Other seizure types | EEG | MRI | Cognition onset /follow-up | Neurological features | Additional features | Age at last follow-up | Seizure outcome (offset: age) | Treatment effects | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Seizure- free | Sz reduction | No effect | Wors- ening | |||||||||||||
| 38 | G899S/de novo | 3 m | Other | HC | GTC, FD, AA | Bifr SW | N | NA/SD | N | ASD | 7 y | Intractable | TPM, LEV, RUF, CLZ, CLB | PB, B6, VPA, LCM, ZNS, KD | OXC | |
| 39 | T227I/de novo | 3 m | WS | T, AP | S | HA→post spikes, slowing | N | SD/SD | Hypotonia | MC | 2 y | Intractable | LEV, ZNS | B6, VGB, CS, ACTH, TPM, KD, PB, OXC | ||
| 40 | A733T/de novo | 3 m | Other | HC r/l | AA, DA, FD | MF spikes, slowing | N | MD/MD | N | 12 y | Intractable | LEV | VPA, ESM, LTG, OXC | |||
| 41 | R1882P/de novo | 4 m | Other | T, C r | MF spikes, SW- status te→ESES-like | N | N/SD | N | Regression, ASD, aggression | 6 y | Sz free (8 m) | LEV | PB, VPA, ST, KD, LTG | |||
| 42 | L1665F/de novo | 4 m | Other | TS, SE | GTC (series) | N | N | N/SD | N | Stereotyped behaviour | 6 y 3 m | Sz free (7 m) | CBZ | VPA | PB | |
| 43 | L1342P/de novo | 6 m | WS | S | HA→MF spikes, slowing | At | SD/SD | Axial hypotonia, limb spasticity | Apnoeas, no eye contact | 16 y | Intractable | VGB, CS | ||||
| 44 | L881P/de novo | 6 m | WS→LGS | S | T, TC, AA | HA→MF spikes | At, HM, cerebellar at | SD/SD | Spasticity, hemiparesis | Small stature, early puberty | 18 y | Intractable | TPM, MDZ, CLB | VPA, LTG, PB, LEV, VGB, KD | RUF | |
| 45 | I1281F/de novo | 7 m | WS→LGS | S | T, TC | HA→MF spikes, slowing, diffuse SW | At | NA/SD | 16 y | Intractable | VNS, VPA, CLZ, RUF, TPM, LTG | |||||
| 46 | R853Q/de novo | 8 m | WS→LGS | S | AA, T, F (4 y) | HA→for bil spikes, SW, Slowing | T2H, at | N/SD | Dystonia | Preterm 28 w, MC, regression | 6 y 6 m | Intractable | ACTH | VPA, CLB, TPM | LTG, FBM, LEV | CBZ |
| 47 | A1652P/de novo | 9 m | WS | S | atypical HA→ slowing | HM | SD/SD | Hypotonia | ASD | 3 y | Intractable | VGB, LEV, NZP | VPA, TPM, CS | PHT | ||
| 48 | R1319W/de novo | 10 m | WS→LGS | T, S | TCS, A, AA | HA→spikes occ →spikes cue-par r | N | SD/SD | Hypotonia | Diabetes, ASD | 10 y | Sz free (7 y) | CBZ, TPM, ESM, ZNS, VPA, FBM, LEV, B6, KD, CS, LTG | |||
| 49 | E1211K / de novo | 11 m | WS→other | S | FC, SE, M | HA→MF spikes, slowing | N | SD/SD | Hypotonia | 4 y | Intractable | LEV, LTG, PB, KD | ST, VGB, CS | |||
| 50 | R853Q/de novo | 13 m | WS | S | T, AU, M | HA→MF spikes, ESES-like | N | MD/SD | Hypotonia, choreo-athetosis | 8 y | Intractable | VGB, PB, CLB | ST, TPM, LEV, ZNS, ESM, LTG, PHT, OXC, CS, KD,AZA | |||
| 51 | H930Q/de novo | 15 m | MAE | TCS | A, MAS, T, AA | slowing occ | N | N/MD | N | ASD | 6 y | Sz free (18 m) | LEV + TPM | VPA, OXC, CLB | ||
| 52 | P1622S/de novo | 2 y | MAE | MA, DA | T, AA, M | Spike-slow-waves te-par bil | N | MD/MD | Ataxia | ASD, MC | 3 y | Intractable | TPM | LEV, ESM, B6, PB, CLB, VGB, PP, KD | OXC, LTG | |
| 53 | F612S/de novo | 2 y 6 m | Other | TCS | M | fr spikes r/l, fr delta | N | MD/MD | N | 3 y | Intractable | CLB, LEV, RUF | ||||
| 54 | c.605 + 1G > T/ de novo | 2 y 6 m | Other | TCS | T, A | midline spikes ce, slowing→ bitemp spikes | N | N/SD | Hypotonia | ASD, regression | 7 y | Intractable | LEV, CLB, KD | ZNS, LTG, FBM, TPM, RUF, B6, IVIG, VPA, PB, CS | ||
| 55 | V1528Cfs*7/de novo | 3 y | LGS | TCS | T, SE | Spike-slow-waves, Beta (sleep) | N | SD/SD | Spasticity | ASD, MC | 9 y | Sz free (6 y) | VPA + LTG | CLZ, ESM | ||
| 56 | R853Q/unknown | 3 y | Other | TS | S | HA | CC hypoplasia | SD/SD | Pyramidal signs, hand dystonia | Arachnodactyly | 25 y | Intractable | VPA, ACTH | |||
| 57 | C1170Vfs*15/de novo | 3 y | Other | TCS | M, AB | gen SW, l fr SW, high voltage theta | HiS | MD/SD | Hypertonic limbs | ASD | 17 y | Intractable | VPA, LTG, Br, LEV, CLB | CBZ, OXC, TPM, STP, ST, KD | ||
| 58 | G1223R/de novo | 3 y | Other | F, M | TCS, M, A, AA, NCSE | gen SW, PSW, slowing | N | N/MD | Ataxia | Regression | 19 y | Intractable | VPA, CLZ, CLB, LTG, LEV | PHT, PB, CBZ, TPM, VNS, CS | ||
| 59 | R1235*/de novo | 3 y 4 m | Other | FC | GTC, DA | gen SW, PSW | HM | MD/MD | N | ASD | 14 y | Intractable | LEV, ZNS | VPA, CLB, LTG | ||
| 60 | A1773V/de novo | 3 y 6 m | Other | F | Sharp waves r fr (sleep), slowing | N | MD/SD | N | ASD | 4 y | Sz free (4 y) | LEV | ||||
| 61 | K1933M/de novo | 4 y | Other | F, TCS | AA | MF spikes, bil SW→ESES-like | N | SD/SD | Ataxia, parkinsonian gait | ASD, hyperkinetic and aggressive behaviour | 17 y | Intractable | ST, VPA, TPM, CLB, CS | LTG, LCM | CBZ | |
| 62 | c.698‐1G>T, splice site/de novo | 4 y 6 m | Other | F | f spikes→MF, ESES-like→N | N | N/MID | N | 10 y | Sz free (9 y) | TPM | others | ||||
| 63 | W1716*/de novo | 4 y 7 m | Other | Febrile T (cluster) | AA | Gen SW | N | MD/MD | N | Psychosis (17 y) | 25 y | Sz free (9 y) | VPA | PB | CBZ | |
| 64 | N503Kfs*19/de novo | 6 y | Other | TCS (cluster) | Bil sharp waves | N | SD/SD | ASD | 20 y | Sz free (6 y) | VPA | |||||
| 65 | W281*/de novo | 7y | Other | F | sec. Gen. TCS | MF spikes→ESES- like | N | MD/MD | Clumsiness | 13 y | Sz free (10 y) | ST | ||||
| 66 | S1656F/de novo | 8 y 11 m | LGS | GTC | AA | Bifr SW, slowing | N | MD/MD | Hypotonia, crouched gait | Agitation | 12 y | Intractable | LLTG, VPA | TTPM | ||
AA = atypical absences; A = atonic; AB = absences; ADS = attention deficit disorder; ASD = autism spectrum disorder; AU = autonomic seizures; AP = apneic seizures; At = atrophy; Bifr = bifrontal; Bil = bilateral; C = clonic; CC = corpus callosum; Ce = central; DA = drop attacks; ED = epileptiform discharges; F = focal; FD = focal dyscognitive; FC = febrile convulsion; fr = frontal; Gen = generalized; GTC = generalized tonic-clonic; HA = hypsarrhythmia; HC = hemiclonic; HiS = hippocampal sclerosis; HM = hypomyelination; IS = infantile spasms; L = left; LGS = Lennox-Gastaut syndrome; MAS = myoclonic-atonic seizures; = MAE = myoclonic-atonic epilepsy; m = months; MID = mild intellectual disability; MD = moderate intellectual disability; M = myoclonic; MC = microcephaly; MF = multifocal; N = normal; NA = not applicable; NAV = not available; NCSE = non-convulsive status epilepticus; Occ = occipital; Par = parietal; R = right; S = spasms; SE = status epilepticus; SD = severe intellectual disability; SW = spike and waves; Sz = seizures; T = tonic; T2H = T2-hyperintensities; TCS = tonic-clonic seizures; Te = temporal; w = week; WS = West syndrome; y = years;→ = change to.
Treatment (sodium channel blockers are highlighted in bold): AZA = acetazolamide; B6 = vitamin B6; BR = bromide; CBZ = carbamazepine; CLB = clobazam; CLZ = clonazepam; CS = corticosteroids; ESM = ethosuximide; FBM = felbamate; GBP = gabapentin; IVIG = intravenous immunoglobulins; KD = ketogenic diet; LCM = lacosamide; LTG = lamotrigine; LEV = levetiracetam; MDZ = midazolam; MSX = mesuximide; OXC = oxcarbazepine; PB = phenobarbital; PHT = phenytoin; PP = pyridoxal phosphate; RGB = retigabine; RUF = rufinamide; ST = sulthiame; STP = stiripentol; TPM = topiramate; VGB = vigabatrin; VNS = vagal nerve stimulation; VPA = valproate; ZNS = zonisamide.
Clinical characteristics and treatment response of the previously unpublished patients: encephalopathy with late onset epilepsy
| Patient | Mutation/ inheritance | Age at seizure onset | Epilepsy syndrome | Initial seizure type | Other seizure types | EEG | MRI | Cognition onset /follow-up | Neurological features | Additional features | Age at last follow-up | Seizure outcome (offset: age) | Treatment effects | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Seizure- free | Sz reduction | No effect | Wors- ening | |||||||||||||
| 38 | G899S/de novo | 3 m | Other | HC | GTC, FD, AA | Bifr SW | N | NA/SD | N | ASD | 7 y | Intractable | TPM, LEV, RUF, CLZ, CLB | PB, B6, VPA, LCM, ZNS, KD | OXC | |
| 39 | T227I/de novo | 3 m | WS | T, AP | S | HA→post spikes, slowing | N | SD/SD | Hypotonia | MC | 2 y | Intractable | LEV, ZNS | B6, VGB, CS, ACTH, TPM, KD, PB, OXC | ||
| 40 | A733T/de novo | 3 m | Other | HC r/l | AA, DA, FD | MF spikes, slowing | N | MD/MD | N | 12 y | Intractable | LEV | VPA, ESM, LTG, OXC | |||
| 41 | R1882P/de novo | 4 m | Other | T, C r | MF spikes, SW- status te→ESES-like | N | N/SD | N | Regression, ASD, aggression | 6 y | Sz free (8 m) | LEV | PB, VPA, ST, KD, LTG | |||
| 42 | L1665F/de novo | 4 m | Other | TS, SE | GTC (series) | N | N | N/SD | N | Stereotyped behaviour | 6 y 3 m | Sz free (7 m) | CBZ | VPA | PB | |
| 43 | L1342P/de novo | 6 m | WS | S | HA→MF spikes, slowing | At | SD/SD | Axial hypotonia, limb spasticity | Apnoeas, no eye contact | 16 y | Intractable | VGB, CS | ||||
| 44 | L881P/de novo | 6 m | WS→LGS | S | T, TC, AA | HA→MF spikes | At, HM, cerebellar at | SD/SD | Spasticity, hemiparesis | Small stature, early puberty | 18 y | Intractable | TPM, MDZ, CLB | VPA, LTG, PB, LEV, VGB, KD | RUF | |
| 45 | I1281F/de novo | 7 m | WS→LGS | S | T, TC | HA→MF spikes, slowing, diffuse SW | At | NA/SD | 16 y | Intractable | VNS, VPA, CLZ, RUF, TPM, LTG | |||||
| 46 | R853Q/de novo | 8 m | WS→LGS | S | AA, T, F (4 y) | HA→for bil spikes, SW, Slowing | T2H, at | N/SD | Dystonia | Preterm 28 w, MC, regression | 6 y 6 m | Intractable | ACTH | VPA, CLB, TPM | LTG, FBM, LEV | CBZ |
| 47 | A1652P/de novo | 9 m | WS | S | atypical HA→ slowing | HM | SD/SD | Hypotonia | ASD | 3 y | Intractable | VGB, LEV, NZP | VPA, TPM, CS | PHT | ||
| 48 | R1319W/de novo | 10 m | WS→LGS | T, S | TCS, A, AA | HA→spikes occ →spikes cue-par r | N | SD/SD | Hypotonia | Diabetes, ASD | 10 y | Sz free (7 y) | CBZ, TPM, ESM, ZNS, VPA, FBM, LEV, B6, KD, CS, LTG | |||
| 49 | E1211K / de novo | 11 m | WS→other | S | FC, SE, M | HA→MF spikes, slowing | N | SD/SD | Hypotonia | 4 y | Intractable | LEV, LTG, PB, KD | ST, VGB, CS | |||
| 50 | R853Q/de novo | 13 m | WS | S | T, AU, M | HA→MF spikes, ESES-like | N | MD/SD | Hypotonia, choreo-athetosis | 8 y | Intractable | VGB, PB, CLB | ST, TPM, LEV, ZNS, ESM, LTG, PHT, OXC, CS, KD,AZA | |||
| 51 | H930Q/de novo | 15 m | MAE | TCS | A, MAS, T, AA | slowing occ | N | N/MD | N | ASD | 6 y | Sz free (18 m) | LEV + TPM | VPA, OXC, CLB | ||
| 52 | P1622S/de novo | 2 y | MAE | MA, DA | T, AA, M | Spike-slow-waves te-par bil | N | MD/MD | Ataxia | ASD, MC | 3 y | Intractable | TPM | LEV, ESM, B6, PB, CLB, VGB, PP, KD | OXC, LTG | |
| 53 | F612S/de novo | 2 y 6 m | Other | TCS | M | fr spikes r/l, fr delta | N | MD/MD | N | 3 y | Intractable | CLB, LEV, RUF | ||||
| 54 | c.605 + 1G > T/ de novo | 2 y 6 m | Other | TCS | T, A | midline spikes ce, slowing→ bitemp spikes | N | N/SD | Hypotonia | ASD, regression | 7 y | Intractable | LEV, CLB, KD | ZNS, LTG, FBM, TPM, RUF, B6, IVIG, VPA, PB, CS | ||
| 55 | V1528Cfs*7/de novo | 3 y | LGS | TCS | T, SE | Spike-slow-waves, Beta (sleep) | N | SD/SD | Spasticity | ASD, MC | 9 y | Sz free (6 y) | VPA + LTG | CLZ, ESM | ||
| 56 | R853Q/unknown | 3 y | Other | TS | S | HA | CC hypoplasia | SD/SD | Pyramidal signs, hand dystonia | Arachnodactyly | 25 y | Intractable | VPA, ACTH | |||
| 57 | C1170Vfs*15/de novo | 3 y | Other | TCS | M, AB | gen SW, l fr SW, high voltage theta | HiS | MD/SD | Hypertonic limbs | ASD | 17 y | Intractable | VPA, LTG, Br, LEV, CLB | CBZ, OXC, TPM, STP, ST, KD | ||
| 58 | G1223R/de novo | 3 y | Other | F, M | TCS, M, A, AA, NCSE | gen SW, PSW, slowing | N | N/MD | Ataxia | Regression | 19 y | Intractable | VPA, CLZ, CLB, LTG, LEV | PHT, PB, CBZ, TPM, VNS, CS | ||
| 59 | R1235*/de novo | 3 y 4 m | Other | FC | GTC, DA | gen SW, PSW | HM | MD/MD | N | ASD | 14 y | Intractable | LEV, ZNS | VPA, CLB, LTG | ||
| 60 | A1773V/de novo | 3 y 6 m | Other | F | Sharp waves r fr (sleep), slowing | N | MD/SD | N | ASD | 4 y | Sz free (4 y) | LEV | ||||
| 61 | K1933M/de novo | 4 y | Other | F, TCS | AA | MF spikes, bil SW→ESES-like | N | SD/SD | Ataxia, parkinsonian gait | ASD, hyperkinetic and aggressive behaviour | 17 y | Intractable | ST, VPA, TPM, CLB, CS | LTG, LCM | CBZ | |
| 62 | c.698‐1G>T, splice site/de novo | 4 y 6 m | Other | F | f spikes→MF, ESES-like→N | N | N/MID | N | 10 y | Sz free (9 y) | TPM | others | ||||
| 63 | W1716*/de novo | 4 y 7 m | Other | Febrile T (cluster) | AA | Gen SW | N | MD/MD | N | Psychosis (17 y) | 25 y | Sz free (9 y) | VPA | PB | CBZ | |
| 64 | N503Kfs*19/de novo | 6 y | Other | TCS (cluster) | Bil sharp waves | N | SD/SD | ASD | 20 y | Sz free (6 y) | VPA | |||||
| 65 | W281*/de novo | 7y | Other | F | sec. Gen. TCS | MF spikes→ESES- like | N | MD/MD | Clumsiness | 13 y | Sz free (10 y) | ST | ||||
| 66 | S1656F/de novo | 8 y 11 m | LGS | GTC | AA | Bifr SW, slowing | N | MD/MD | Hypotonia, crouched gait | Agitation | 12 y | Intractable | LLTG, VPA | TTPM | ||
| Patient | Mutation/ inheritance | Age at seizure onset | Epilepsy syndrome | Initial seizure type | Other seizure types | EEG | MRI | Cognition onset /follow-up | Neurological features | Additional features | Age at last follow-up | Seizure outcome (offset: age) | Treatment effects | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Seizure- free | Sz reduction | No effect | Wors- ening | |||||||||||||
| 38 | G899S/de novo | 3 m | Other | HC | GTC, FD, AA | Bifr SW | N | NA/SD | N | ASD | 7 y | Intractable | TPM, LEV, RUF, CLZ, CLB | PB, B6, VPA, LCM, ZNS, KD | OXC | |
| 39 | T227I/de novo | 3 m | WS | T, AP | S | HA→post spikes, slowing | N | SD/SD | Hypotonia | MC | 2 y | Intractable | LEV, ZNS | B6, VGB, CS, ACTH, TPM, KD, PB, OXC | ||
| 40 | A733T/de novo | 3 m | Other | HC r/l | AA, DA, FD | MF spikes, slowing | N | MD/MD | N | 12 y | Intractable | LEV | VPA, ESM, LTG, OXC | |||
| 41 | R1882P/de novo | 4 m | Other | T, C r | MF spikes, SW- status te→ESES-like | N | N/SD | N | Regression, ASD, aggression | 6 y | Sz free (8 m) | LEV | PB, VPA, ST, KD, LTG | |||
| 42 | L1665F/de novo | 4 m | Other | TS, SE | GTC (series) | N | N | N/SD | N | Stereotyped behaviour | 6 y 3 m | Sz free (7 m) | CBZ | VPA | PB | |
| 43 | L1342P/de novo | 6 m | WS | S | HA→MF spikes, slowing | At | SD/SD | Axial hypotonia, limb spasticity | Apnoeas, no eye contact | 16 y | Intractable | VGB, CS | ||||
| 44 | L881P/de novo | 6 m | WS→LGS | S | T, TC, AA | HA→MF spikes | At, HM, cerebellar at | SD/SD | Spasticity, hemiparesis | Small stature, early puberty | 18 y | Intractable | TPM, MDZ, CLB | VPA, LTG, PB, LEV, VGB, KD | RUF | |
| 45 | I1281F/de novo | 7 m | WS→LGS | S | T, TC | HA→MF spikes, slowing, diffuse SW | At | NA/SD | 16 y | Intractable | VNS, VPA, CLZ, RUF, TPM, LTG | |||||
| 46 | R853Q/de novo | 8 m | WS→LGS | S | AA, T, F (4 y) | HA→for bil spikes, SW, Slowing | T2H, at | N/SD | Dystonia | Preterm 28 w, MC, regression | 6 y 6 m | Intractable | ACTH | VPA, CLB, TPM | LTG, FBM, LEV | CBZ |
| 47 | A1652P/de novo | 9 m | WS | S | atypical HA→ slowing | HM | SD/SD | Hypotonia | ASD | 3 y | Intractable | VGB, LEV, NZP | VPA, TPM, CS | PHT | ||
| 48 | R1319W/de novo | 10 m | WS→LGS | T, S | TCS, A, AA | HA→spikes occ →spikes cue-par r | N | SD/SD | Hypotonia | Diabetes, ASD | 10 y | Sz free (7 y) | CBZ, TPM, ESM, ZNS, VPA, FBM, LEV, B6, KD, CS, LTG | |||
| 49 | E1211K / de novo | 11 m | WS→other | S | FC, SE, M | HA→MF spikes, slowing | N | SD/SD | Hypotonia | 4 y | Intractable | LEV, LTG, PB, KD | ST, VGB, CS | |||
| 50 | R853Q/de novo | 13 m | WS | S | T, AU, M | HA→MF spikes, ESES-like | N | MD/SD | Hypotonia, choreo-athetosis | 8 y | Intractable | VGB, PB, CLB | ST, TPM, LEV, ZNS, ESM, LTG, PHT, OXC, CS, KD,AZA | |||
| 51 | H930Q/de novo | 15 m | MAE | TCS | A, MAS, T, AA | slowing occ | N | N/MD | N | ASD | 6 y | Sz free (18 m) | LEV + TPM | VPA, OXC, CLB | ||
| 52 | P1622S/de novo | 2 y | MAE | MA, DA | T, AA, M | Spike-slow-waves te-par bil | N | MD/MD | Ataxia | ASD, MC | 3 y | Intractable | TPM | LEV, ESM, B6, PB, CLB, VGB, PP, KD | OXC, LTG | |
| 53 | F612S/de novo | 2 y 6 m | Other | TCS | M | fr spikes r/l, fr delta | N | MD/MD | N | 3 y | Intractable | CLB, LEV, RUF | ||||
| 54 | c.605 + 1G > T/ de novo | 2 y 6 m | Other | TCS | T, A | midline spikes ce, slowing→ bitemp spikes | N | N/SD | Hypotonia | ASD, regression | 7 y | Intractable | LEV, CLB, KD | ZNS, LTG, FBM, TPM, RUF, B6, IVIG, VPA, PB, CS | ||
| 55 | V1528Cfs*7/de novo | 3 y | LGS | TCS | T, SE | Spike-slow-waves, Beta (sleep) | N | SD/SD | Spasticity | ASD, MC | 9 y | Sz free (6 y) | VPA + LTG | CLZ, ESM | ||
| 56 | R853Q/unknown | 3 y | Other | TS | S | HA | CC hypoplasia | SD/SD | Pyramidal signs, hand dystonia | Arachnodactyly | 25 y | Intractable | VPA, ACTH | |||
| 57 | C1170Vfs*15/de novo | 3 y | Other | TCS | M, AB | gen SW, l fr SW, high voltage theta | HiS | MD/SD | Hypertonic limbs | ASD | 17 y | Intractable | VPA, LTG, Br, LEV, CLB | CBZ, OXC, TPM, STP, ST, KD | ||
| 58 | G1223R/de novo | 3 y | Other | F, M | TCS, M, A, AA, NCSE | gen SW, PSW, slowing | N | N/MD | Ataxia | Regression | 19 y | Intractable | VPA, CLZ, CLB, LTG, LEV | PHT, PB, CBZ, TPM, VNS, CS | ||
| 59 | R1235*/de novo | 3 y 4 m | Other | FC | GTC, DA | gen SW, PSW | HM | MD/MD | N | ASD | 14 y | Intractable | LEV, ZNS | VPA, CLB, LTG | ||
| 60 | A1773V/de novo | 3 y 6 m | Other | F | Sharp waves r fr (sleep), slowing | N | MD/SD | N | ASD | 4 y | Sz free (4 y) | LEV | ||||
| 61 | K1933M/de novo | 4 y | Other | F, TCS | AA | MF spikes, bil SW→ESES-like | N | SD/SD | Ataxia, parkinsonian gait | ASD, hyperkinetic and aggressive behaviour | 17 y | Intractable | ST, VPA, TPM, CLB, CS | LTG, LCM | CBZ | |
| 62 | c.698‐1G>T, splice site/de novo | 4 y 6 m | Other | F | f spikes→MF, ESES-like→N | N | N/MID | N | 10 y | Sz free (9 y) | TPM | others | ||||
| 63 | W1716*/de novo | 4 y 7 m | Other | Febrile T (cluster) | AA | Gen SW | N | MD/MD | N | Psychosis (17 y) | 25 y | Sz free (9 y) | VPA | PB | CBZ | |
| 64 | N503Kfs*19/de novo | 6 y | Other | TCS (cluster) | Bil sharp waves | N | SD/SD | ASD | 20 y | Sz free (6 y) | VPA | |||||
| 65 | W281*/de novo | 7y | Other | F | sec. Gen. TCS | MF spikes→ESES- like | N | MD/MD | Clumsiness | 13 y | Sz free (10 y) | ST | ||||
| 66 | S1656F/de novo | 8 y 11 m | LGS | GTC | AA | Bifr SW, slowing | N | MD/MD | Hypotonia, crouched gait | Agitation | 12 y | Intractable | LLTG, VPA | TTPM | ||
AA = atypical absences; A = atonic; AB = absences; ADS = attention deficit disorder; ASD = autism spectrum disorder; AU = autonomic seizures; AP = apneic seizures; At = atrophy; Bifr = bifrontal; Bil = bilateral; C = clonic; CC = corpus callosum; Ce = central; DA = drop attacks; ED = epileptiform discharges; F = focal; FD = focal dyscognitive; FC = febrile convulsion; fr = frontal; Gen = generalized; GTC = generalized tonic-clonic; HA = hypsarrhythmia; HC = hemiclonic; HiS = hippocampal sclerosis; HM = hypomyelination; IS = infantile spasms; L = left; LGS = Lennox-Gastaut syndrome; MAS = myoclonic-atonic seizures; = MAE = myoclonic-atonic epilepsy; m = months; MID = mild intellectual disability; MD = moderate intellectual disability; M = myoclonic; MC = microcephaly; MF = multifocal; N = normal; NA = not applicable; NAV = not available; NCSE = non-convulsive status epilepticus; Occ = occipital; Par = parietal; R = right; S = spasms; SE = status epilepticus; SD = severe intellectual disability; SW = spike and waves; Sz = seizures; T = tonic; T2H = T2-hyperintensities; TCS = tonic-clonic seizures; Te = temporal; w = week; WS = West syndrome; y = years;→ = change to.
Treatment (sodium channel blockers are highlighted in bold): AZA = acetazolamide; B6 = vitamin B6; BR = bromide; CBZ = carbamazepine; CLB = clobazam; CLZ = clonazepam; CS = corticosteroids; ESM = ethosuximide; FBM = felbamate; GBP = gabapentin; IVIG = intravenous immunoglobulins; KD = ketogenic diet; LCM = lacosamide; LTG = lamotrigine; LEV = levetiracetam; MDZ = midazolam; MSX = mesuximide; OXC = oxcarbazepine; PB = phenobarbital; PHT = phenytoin; PP = pyridoxal phosphate; RGB = retigabine; RUF = rufinamide; ST = sulthiame; STP = stiripentol; TPM = topiramate; VGB = vigabatrin; VNS = vagal nerve stimulation; VPA = valproate; ZNS = zonisamide.
Clinical characteristics of the previously unpublished patients: intellectual disability and/or autism without epilepsy
| Patient | Mutation/ inheritance | EEG | MRI | Cognition onset/ follow-up | Neurological features | Additional features | Age at follow-up |
|---|---|---|---|---|---|---|---|
| 67 | K1387Sfs*4/de novo | NA | NA | MD | N | ASD | 5 y 4 m |
| 68 | R1435*/de novo | N | T2H | SD | N | ASD, early puberty | 7 y 8 m |
| 69 | T1711Lfs*8/de novo | Slowing | N | SD | Hypotonia | Rett-like, ASD | 9 y 8 m |
| 70 | G1744E/de novo | N | N | MD | N | ASD | 4 y |
| 71 | c.386+2T > C/de novo | NA | NA | MD | NAV | Episodic ataxia, ASD | 13 y |
| Patient | Mutation/ inheritance | EEG | MRI | Cognition onset/ follow-up | Neurological features | Additional features | Age at follow-up |
|---|---|---|---|---|---|---|---|
| 67 | K1387Sfs*4/de novo | NA | NA | MD | N | ASD | 5 y 4 m |
| 68 | R1435*/de novo | N | T2H | SD | N | ASD, early puberty | 7 y 8 m |
| 69 | T1711Lfs*8/de novo | Slowing | N | SD | Hypotonia | Rett-like, ASD | 9 y 8 m |
| 70 | G1744E/de novo | N | N | MD | N | ASD | 4 y |
| 71 | c.386+2T > C/de novo | NA | NA | MD | NAV | Episodic ataxia, ASD | 13 y |
ASD = autism spectrum disorder; m = months; MD = moderate intellectual disability; N = normal; NA = not applicable; NAV = not available; SD = severe intellectual disability; T2H = T2-hyperintensities; y = years.
Clinical characteristics of the previously unpublished patients: intellectual disability and/or autism without epilepsy
| Patient | Mutation/ inheritance | EEG | MRI | Cognition onset/ follow-up | Neurological features | Additional features | Age at follow-up |
|---|---|---|---|---|---|---|---|
| 67 | K1387Sfs*4/de novo | NA | NA | MD | N | ASD | 5 y 4 m |
| 68 | R1435*/de novo | N | T2H | SD | N | ASD, early puberty | 7 y 8 m |
| 69 | T1711Lfs*8/de novo | Slowing | N | SD | Hypotonia | Rett-like, ASD | 9 y 8 m |
| 70 | G1744E/de novo | N | N | MD | N | ASD | 4 y |
| 71 | c.386+2T > C/de novo | NA | NA | MD | NAV | Episodic ataxia, ASD | 13 y |
| Patient | Mutation/ inheritance | EEG | MRI | Cognition onset/ follow-up | Neurological features | Additional features | Age at follow-up |
|---|---|---|---|---|---|---|---|
| 67 | K1387Sfs*4/de novo | NA | NA | MD | N | ASD | 5 y 4 m |
| 68 | R1435*/de novo | N | T2H | SD | N | ASD, early puberty | 7 y 8 m |
| 69 | T1711Lfs*8/de novo | Slowing | N | SD | Hypotonia | Rett-like, ASD | 9 y 8 m |
| 70 | G1744E/de novo | N | N | MD | N | ASD | 4 y |
| 71 | c.386+2T > C/de novo | NA | NA | MD | NAV | Episodic ataxia, ASD | 13 y |
ASD = autism spectrum disorder; m = months; MD = moderate intellectual disability; N = normal; NA = not applicable; NAV = not available; SD = severe intellectual disability; T2H = T2-hyperintensities; y = years.
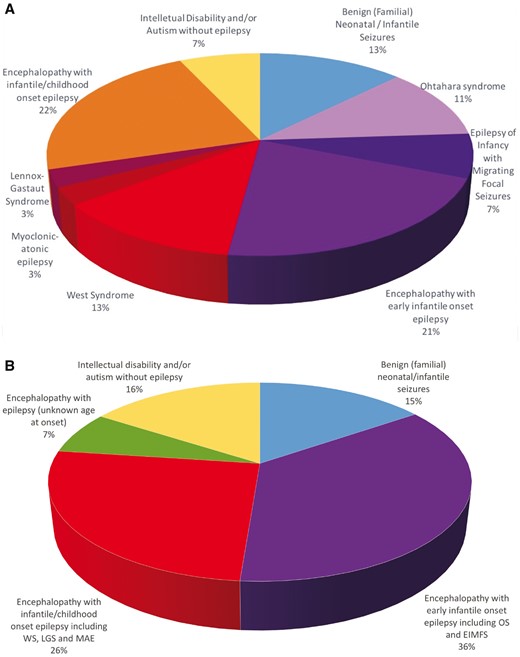
Distribution of patients according to phenotype and age at epilepsy onset. (A) Epilepsy phenotypes in the previously unpublished cohort (n = 66). ‘Patients with Lennox-Gastaut syndrome’ refers to patients with Lennox-Gastaut syndrome not evolving from West syndrome. (B) Phenotypes in the overall cohort (n = 201). LGS = Lennox-Gastaut syndrome; MAE = myoclonic-atonic epilepsy; OS = Ohtahara syndrome; WS = West syndrome.
Phenotypic features
Benign (familial) neonatal/infantile seizures
We identified nine unpublished patients (Table 1) and 24 probands from the literature with B(F)NIS due to a SCN2A mutation, as well as 109 mutation-positive family members (Berkovic et al., 2004; Striano et al., 2006; Herlenius et al., 2007; Heron et al., 2010; Liao et al., 2010b; Lemke et al., 2012; Lauxmann et al., 2013; Zara et al., 2013; Grinton et al., 2015; Johannesen et al., 2016; Schwarz et al., 2016). The mutations occurred de novo in 6/33 of the probands. Age at seizure onset ranged from the first day of life to 23 months. Approximately half of the children had seizure onset within the first month of life. Seizure types were predominantly focal clonic, tonic, and generalized tonic-clonic, frequently occurring in clusters. Interictal EEG showed mostly focal or multifocal spikes, but was normal in some cases. All children became seizure-free at a median age of 5 months (range 5 days to 2 years), and remained seizure-free with normal cognitive development until last follow-up at a median age of 5.5 years (range 7 months to 34 years, data available from 28 cases). A single proband developed a second epilepsy phenotype during later childhood with marked activation of multifocal spikes during sleep and partial cognitive deterioration, resembling electrical status epilepticus during slow sleep (ESES), and two had isolated seizures until the age of 2 and 14 years, respectively. Five children with two recurrent mutations (A263V and R1882G) exhibited episodic ataxia later in life (Liao et al., 2010a; Johannesen et al., 2016; Schwarz et al., 2016).
Encephalopathy with early infantile epilepsy
Twenty-eight new patients (Table 2) and 43 previously published ones (Ogiwara et al., 2009; Dhamija et al., 2013; Nakamura et al., 2013; Touma et al., 2013; Baasch et al., 2014; Martin et al., 2014; Matalon et al., 2014; Zerem et al., 2014; Fukasawa et al., 2015; Howell et al., 2015; Allen et al., 2016; Trump et al., 2016) had epilepsy onset within the first 3 months of life. Thirty-one had an identifiable epilepsy syndrome, i.e. Ohtahara syndrome (18 cases) or EIMFS (13 cases). The remaining 40 patients had unclassifiable epilepsies. The predominant seizure types in these were focal, tonic, and tonic-clonic seizures or spasms. Initial EEGs showed a suppression burst pattern in 25 cases (18 with Ohtahara syndrome, two with EIMFS, and five with unclassifiable epilepsies), and multifocal spikes in the majority of the remaining cases. Regardless of the epileptic syndrome, all patients fulfilled the criteria of encephalopathy as they had intellectual disability, being severe in 54/71 cases. Six children had autism spectrum disorder. Additional features included muscular hypotonia (n = 32), microcephaly (n = 15), marked dystonic or choreatic movement disorders (n = 8), spasticity (n = 3), or dysautonomia (n = 5). Seven patients in this subgroup were deceased at time of follow-up, and causes of death included severe infections and status epilepticus.
Encephalopathy with infantile/childhood epilepsy
This group included 29 unpublished (Table 3) and 29 previously published patients (Haug et al., 2001; Sugawara et al., 2001; Kamiya et al., 2004; Ogiwara et al., 2009; Shi et al., 2009; Kobayashi et al., 2012; Allen et al., 2013; Nakamura et al., 2013; Sundaram et al., 2013; Hackenberg et al., 2014; D’Gama et al., 2015; Howell et al., 2015; Mercimek-Mahmutoglu et al., 2015; Samanta and Ramakrishnaiah, 2015; Wong et al., 2015; Dimassi et al., 2016; Horvath et al., 2016). Sixteen presented with West syndrome or infantile spasms, which evolved into Lennox-Gastaut syndrome in 5/16 patients. Two cases were diagnosed as Dravet syndrome, two as Lennox-Gastaut syndrome and two as myoclonic-atonic epilepsy. The majority of the remaining patients with seizure onset after 3 months of age had unclassifiable epilepsies mainly with generalized seizure types including generalized tonic-clonic (15/36, occurring in clusters in four), absence (n = 12) and myoclonic seizures (n = 8). EEG showed mainly generalized spikes and waves or multifocal spikes. Interestingly, four patients with unclassifiable epilepsies (Patients 41, 61, 62 and 65) and one patient (Patient 50) with West syndrome showed an ESES-like marked activation of spikes and waves during sleep, prompting AED treatment to reduce spike-wave activity in order to prevent ESES-induced neuropsychological sequelae. Cognition before seizure onset varied from normal to severely delayed, and was reported abnormal during follow-up in all cases, with severe cognitive impairment in 35/58. Autism spectrum disorder was found in 18 children. Additional features included muscular hypotonia (n = 16) and marked choreatic or dyskinetic movement disorder in six patients.
Encephalopathy with unspecified onset of epilepsy
In 10 of the previously published patients (Need et al., 2012; Wang et al., 2012; Carvill et al., 2013; Saitoh et al., 2015; Li et al., 2016), data were limited, and age at seizure onset was not available. One case was classified as Lennox-Gastaut syndrome, the others as encephalopathies with epilepsy that were not further characterized.
Intellectual disability and/or autism without epilepsy
This subgroup consisted of patients with a verified SCN2A mutation, but no signs of epileptic seizures. Five unpublished (Table 4) and 27 previously published cases fulfilled these criteria (Weiss et al., 2003; Rauch et al., 2012; Sanders et al., 2012; Jiang et al., 2013; Tavassoli et al., 2014; Codina-Sola et al., 2015; D’Gama et al., 2015; Carroll et al., 2016; Li et al., 2016). The unpublished cases, aged 4 to 13 years, exhibited autism with moderate to severe intellectual disability. From the previously published individuals, 14 had autism, nine had intellectual disability without autism, and four had schizophrenia.
Estimated frequency of SCN2A mutations in the Danish population
Via the electronic population databases of National Statistics at the Statens Serum Institute (Denmark), we calculated the birth cohort from 2006–14, giving 550 261 births. In the same period at least seven Danish children with an SCN2A mutation causing the reported phenotypes were born, making a total minimum frequency of approximately 1/78 608 births.
Seizure outcome and treatment effects
Antiepileptic treatment effects on seizures were analysed in all unpublished patients with epilepsy for which sufficiently detailed clinical information was available (n = 66, Tables 1–4). Besides classical AEDs, corticosteroids or adrenocorticotropic hormones (ACTH) were tried in 19 children, ketogenic diet in 13, vagal nerve stimulation in three, and immunoglobulins in one.
Benign (familial) neonatal/infantile seizures
All children became seizure-free at a median age of 3 months (range: 5 days to 2 years). All but one remained seizure-free until last follow-up at a median age of 20 months (range: 14 months to 11 years) (n = 9, Table 1). Patient 4 developed an ESES-like picture at the age of 8 years, and became seizure-free again at the age of 10 years. Seizures stopped spontaneously in three patients, and with AED treatment in six. The sequence of events strongly suggested that seizure freedom was reached by treatment and not by natural history in those cases. Phenytoin was effective in two patients, oxcarbazepine in two, zonisamide in one and clobazam in one. Initial AED treatment failed in seven cases, and six received more than two AEDs (mean 4.3) before the seizures stopped.
Encephalopathy with early infantile epilepsy with onset younger than 3 months
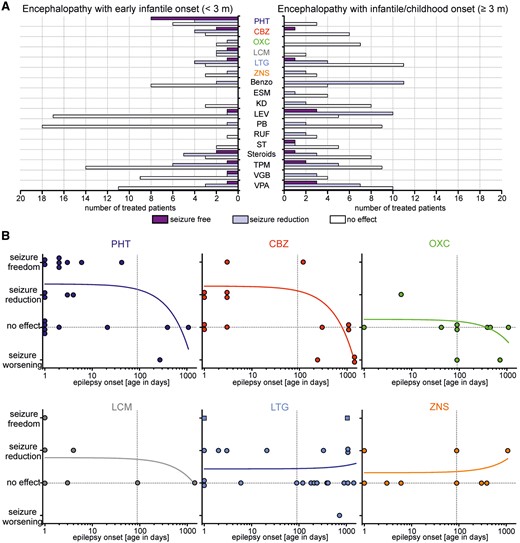
Treatment effects on epileptic seizures in patients with SCN2A-related encephalopathies. (A) Number of treated patients and their seizure outcome according to the judgement of the treating physician (purple = seizure free; light purple = seizure reduction; open bar = no effect) that have been treated with different AEDs. Only effects of AEDs that have been used in more than three patients are shown. (B) Effects of treatments with specific SCBs on seizures as a function of the age of onset of the epilepsy in days. Each dot represents one treatment period with the respective SCB of one patient. Squares in the lamotrigine graph represent a combination of lamotrigine with additional AEDs. Patient 23 has been excluded for carbamazepine, as the seizure freedom correlated with phenytoin treatment. Lines represent linear regressions highlighting the dependence of treatment effects with SCBs on age of onset of the epilepsy. Only for phenytoin the slope was significantly different from 0 (phenytoin: P = 0.03; carbamazepine: P = 0.06; oxcarbazepine: P = 0.43; lacosamide: P = 0.50; lamotrigine: P = 0.66; zonisamide: P = 0.52). benzo = benzodiazepines; CBZ = carbamazepine; ESM = ethosuximide; KD = ketogenic diet; LCM = lacosamide; LTG = lamotrigine; LEV = levetiracetam; OXC = oxcarbazepine; PB = phenobarbital; PHT = phenytoin; RUF = rufinamide; ST = sulthiame; TPM = topiramate; VGB = vigabatrin; VPA = valproate; ZNS = zonisamide.
Encephalopathy with infantile/childhood epilepsy with onset at 3 months or older
Ten children became seizure-free during follow-up (median observation period after seizure freedom: 3.5 years, range 1 month to 16 years) (n = 29, Table 3). Eight of nine patients with West syndrome were resistant to treatment, including steroids or ACTH in six. One child (Patient 46) responded to ACTH, but later developed drug-resistant Lennox-Gastaut syndrome. Among the patients with other epilepsy phenotypes, different AEDs were effective in single cases each, including levetiracetam (n = 2) and valproate (n = 2). Seizure reduction occurred most frequently with levetiracetam (n = 9), benzodiazepines (n = 9), and valproate (n = 7). Ineffective AEDs included lamotrigine (n = 10), valproate (n = 10), phenobarbital (n = 9), and topiramate (n = 9). Drug-induced aggravation of seizures occurred in seven children with carbamazepine (n = 3), oxcarbazepine (n = 2), phenytoin, lamotrigine and rufinamide, and remitted after discontinuation of the respective AED (see also Fig. 2B and Table 3). Atypical absences were the predominant seizure types in these. In a boy with myoclonic-atonic epilepsy (Patient 52), the frequency of drop attacks and tonic seizures increased markedly after introduction of oxcarbazepine, and lamotrigine provoked episodes of status epilepticus.
Response to sodium channel blocker versus non-sodium channel blocker treatment
We tested the significance of effects of SCBs versus non-SCBs on seizure outcome of patients with encephalopathy and epilepsy with onset <3 months and ≥3 months by applying a χ2 test (Supplementary Table 3). Treatment with phenytoin and carbamazepine or with all SCBs considered together showed a significantly better response for patients with onset of epilepsy <3 months than for those with onset ≥3 months (P < 0.01 and P < 0.001, respectively). In contrast, patients with epilepsy onset ≥3 months responded significantly better to non-SCBs (P < 0.001).
Impact of the genetic diagnosis on treatment decisions
In most patients, genetic diagnosis was only made late during follow-up and had no impact on treatment decisions, because patients were already seizure-free or many AEDs had been tried before. In some cases, however, suspicion or confirmation of a mutation in SCN2A led to specific treatment trials with SCBs: three children (Patients 15, 23 and 25) with severe early onset epilepsies and one (Patient 2) with de novo BNIS became seizure-free with phenytoin, and three children with severe early onset EIMFS (Patients 26 and 30) or Ohtahara syndrome (Patient 33) showed partial responses to some SCBs, whereas other types of AEDs had failed before. In contrast, three children with late onset epilepsies (Patients 39, 40 and 41) showed no effect on SCB trials.
Genetic findings
Mutations were missense in all children with B(F)NIS or encephalopathies with epilepsy onset <3 months. The majority of the missense mutations (both inherited and de novo) affects highly evolutionarily conserved amino acids and no obvious correlation between the position of the mutation and the severity of the associated phenotype was observed. In the subgroup of infantile/childhood epilepsies, mutations were missense in 45, early stop codons in six, frameshift in five (both predicting truncated proteins), and altered splice-sites in two. All non-missense mutations occurred in children with seizure onset beyond the first year of life. In children with intellectual disability or autism without epilepsy, mutations were missense (n = 8), frameshift (n = 7), nonsense (n = 8), splices-site (n = 5) or in-frame deletions (n = 2). Twenty-eight recurrent mutations were observed (Table 5), and some of them were associated with specific phenotypes.
Recurrent mutations
| Mutation | Frequency % (n cases) | Phenotypes |
|---|---|---|
| M136I | 1 (2) | EE, EIMFS |
| V213D | 1 (2) | EOEE |
| V261M | 1 (2) | BNS |
| A263V/T | 3.5 (7) | BNS, EE, OS |
| V423L | 1 (2) | OS |
| V430Q/G/A | 1.5 (3) | BFIS, OS |
| N503K fs | 1 (2) | ID |
| R853Q | 4.5 (9) | WS, EE, LGS |
| R856L/Q | 1 (2) | OS,EIMFS |
| G882R/E | 1 (2) | EIMFS |
| K905N | 1 (2) | EE |
| F928C | 1 (2) | EIMFS, EE |
| R937C | 1 (2) | ID |
| C595* | 1 (2) | ASD |
| E999V/K | 3 (6) | EIEE, OS, EOEE |
| G1013* | 1 (2) | ASD |
| I1021Y* | 1 (2) | LGS, EE |
| E1211K | 1 (2) | WS |
| V1282F | 1 (2) | Schizophrenia |
| R1319Q/W | 3 (6) | BFIS, WS, EE |
| V1326V/D | 1 (2) | EIMFS, OS |
| S1336Y | 1.5 (3) | OS |
| L1342P | 2.5 (5) | EOEE, WS |
| R1435* | 1 (2) | ASD |
| Q1531K | 1 (2) | BFNS |
| T1623N | 1 (2) | OS, EE |
| R1629L/H | 1 (2) | EE |
| R1882G/Q/L | 5 (10) | BIS, OS, EE |
| Mutation | Frequency % (n cases) | Phenotypes |
|---|---|---|
| M136I | 1 (2) | EE, EIMFS |
| V213D | 1 (2) | EOEE |
| V261M | 1 (2) | BNS |
| A263V/T | 3.5 (7) | BNS, EE, OS |
| V423L | 1 (2) | OS |
| V430Q/G/A | 1.5 (3) | BFIS, OS |
| N503K fs | 1 (2) | ID |
| R853Q | 4.5 (9) | WS, EE, LGS |
| R856L/Q | 1 (2) | OS,EIMFS |
| G882R/E | 1 (2) | EIMFS |
| K905N | 1 (2) | EE |
| F928C | 1 (2) | EIMFS, EE |
| R937C | 1 (2) | ID |
| C595* | 1 (2) | ASD |
| E999V/K | 3 (6) | EIEE, OS, EOEE |
| G1013* | 1 (2) | ASD |
| I1021Y* | 1 (2) | LGS, EE |
| E1211K | 1 (2) | WS |
| V1282F | 1 (2) | Schizophrenia |
| R1319Q/W | 3 (6) | BFIS, WS, EE |
| V1326V/D | 1 (2) | EIMFS, OS |
| S1336Y | 1.5 (3) | OS |
| L1342P | 2.5 (5) | EOEE, WS |
| R1435* | 1 (2) | ASD |
| Q1531K | 1 (2) | BFNS |
| T1623N | 1 (2) | OS, EE |
| R1629L/H | 1 (2) | EE |
| R1882G/Q/L | 5 (10) | BIS, OS, EE |
ASD = autistism spectrum disorder (without seizures); BFIS = benign familial infantile seizures; BIS = benign infantile seizures; BNS = benign neonatal seizures; EE = epileptic encephalopathy; EIEE = early infantile epileptic encephalopathy; EIMFS = epilepsy of infancy with migrating focal seizures; EOEE = early onset epileptic encephalopathy; ID = intellectual disability (without seizures); LGS = Lennox-Gastaut syndrome; OS = Ohtahara syndrome; WS = West syndrome.
Recurrent mutations
| Mutation | Frequency % (n cases) | Phenotypes |
|---|---|---|
| M136I | 1 (2) | EE, EIMFS |
| V213D | 1 (2) | EOEE |
| V261M | 1 (2) | BNS |
| A263V/T | 3.5 (7) | BNS, EE, OS |
| V423L | 1 (2) | OS |
| V430Q/G/A | 1.5 (3) | BFIS, OS |
| N503K fs | 1 (2) | ID |
| R853Q | 4.5 (9) | WS, EE, LGS |
| R856L/Q | 1 (2) | OS,EIMFS |
| G882R/E | 1 (2) | EIMFS |
| K905N | 1 (2) | EE |
| F928C | 1 (2) | EIMFS, EE |
| R937C | 1 (2) | ID |
| C595* | 1 (2) | ASD |
| E999V/K | 3 (6) | EIEE, OS, EOEE |
| G1013* | 1 (2) | ASD |
| I1021Y* | 1 (2) | LGS, EE |
| E1211K | 1 (2) | WS |
| V1282F | 1 (2) | Schizophrenia |
| R1319Q/W | 3 (6) | BFIS, WS, EE |
| V1326V/D | 1 (2) | EIMFS, OS |
| S1336Y | 1.5 (3) | OS |
| L1342P | 2.5 (5) | EOEE, WS |
| R1435* | 1 (2) | ASD |
| Q1531K | 1 (2) | BFNS |
| T1623N | 1 (2) | OS, EE |
| R1629L/H | 1 (2) | EE |
| R1882G/Q/L | 5 (10) | BIS, OS, EE |
| Mutation | Frequency % (n cases) | Phenotypes |
|---|---|---|
| M136I | 1 (2) | EE, EIMFS |
| V213D | 1 (2) | EOEE |
| V261M | 1 (2) | BNS |
| A263V/T | 3.5 (7) | BNS, EE, OS |
| V423L | 1 (2) | OS |
| V430Q/G/A | 1.5 (3) | BFIS, OS |
| N503K fs | 1 (2) | ID |
| R853Q | 4.5 (9) | WS, EE, LGS |
| R856L/Q | 1 (2) | OS,EIMFS |
| G882R/E | 1 (2) | EIMFS |
| K905N | 1 (2) | EE |
| F928C | 1 (2) | EIMFS, EE |
| R937C | 1 (2) | ID |
| C595* | 1 (2) | ASD |
| E999V/K | 3 (6) | EIEE, OS, EOEE |
| G1013* | 1 (2) | ASD |
| I1021Y* | 1 (2) | LGS, EE |
| E1211K | 1 (2) | WS |
| V1282F | 1 (2) | Schizophrenia |
| R1319Q/W | 3 (6) | BFIS, WS, EE |
| V1326V/D | 1 (2) | EIMFS, OS |
| S1336Y | 1.5 (3) | OS |
| L1342P | 2.5 (5) | EOEE, WS |
| R1435* | 1 (2) | ASD |
| Q1531K | 1 (2) | BFNS |
| T1623N | 1 (2) | OS, EE |
| R1629L/H | 1 (2) | EE |
| R1882G/Q/L | 5 (10) | BIS, OS, EE |
ASD = autistism spectrum disorder (without seizures); BFIS = benign familial infantile seizures; BIS = benign infantile seizures; BNS = benign neonatal seizures; EE = epileptic encephalopathy; EIEE = early infantile epileptic encephalopathy; EIMFS = epilepsy of infancy with migrating focal seizures; EOEE = early onset epileptic encephalopathy; ID = intellectual disability (without seizures); LGS = Lennox-Gastaut syndrome; OS = Ohtahara syndrome; WS = West syndrome.
The A263V variant (de novo) has previously been described in patients with benign neonatal/infantile seizures and childhood onset episodic ataxia (Liao et al., 2010a; Johannesen et al., 2016; Schwarz et al., 2016). However, a twin pair with the same mutation was identified to have Ohtahara syndrome, one of them being deceased, and the other having severe intellectual disability (Touma et al., 2013). Furthermore, we detected a new case with this mutation (Patient 34, de novo), a male who suffered from severe encephalopathy with early infantile epilepsy. He died at the age of 13 years due to status epilepticus.
The R853Q variant was found in nine independent patients, six of whom were previously published and three unpublished (Patients 46, 50 and 56) (Allen et al., 2013; Nakamura et al., 2013; Samanta and Ramakrishnaiah, 2015; Li et al., 2016). Six of these patients presented with West syndrome at 11–13 months of age, all were severely intellectually disabled, and most had intractable seizures.
The R1319Q variant was previously reported in three different families with BFIS (Berkovic et al., 2004), while we found it twice (Patients 31 and 36) occurring de novo in patients with encephalopathy with early infantile epilepsy. In line with the previously reported cases, however, the two patients became seizure-free.
Ten mutations were found at amino acid position 1882, with variable substitutions. R1882G resulted in benign infantile seizures with late onset episodic ataxia in two cases, and was shown to cause a gain-of-function (Schwarz et al., 2016). In contrast, R1882Q (Patient 19) (Carvill et al., 2013; Howell et al., 2015; Trump et al., 2016), R1882L (Baasch et al., 2014) and R1882P (Patient 41) resulted in severe phenotypes with intellectual disability.
Functional studies
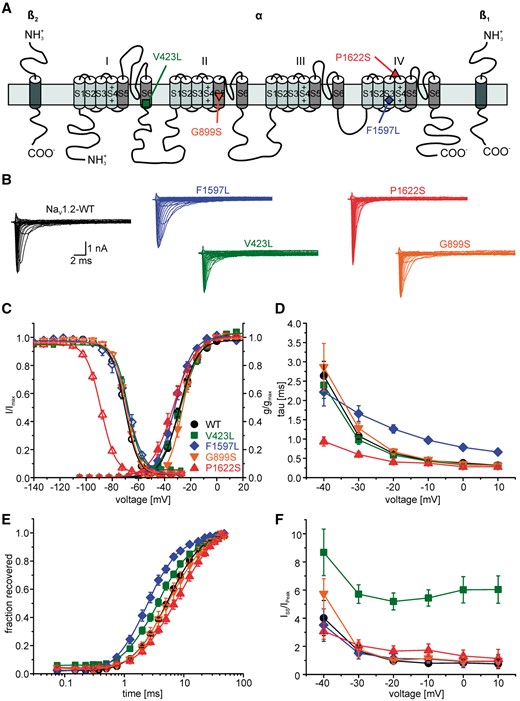
Functional studies reveal pronounced gain-of-function changes for the V423L and F1597L mutations and loss-of-function changes for the P1622S and G899S mutations. (A) Schematic of the human Nav1.2 α-subunit together with β1 and β2 subunits showing the locations of the four functionally studied mutations (V423L green square; F1597L blue diamond; G899S orange inverted triangle; P1622S red triangle). (B) Representative current traces of whole-cell Na+ currents recorded from tsA201 cells transfected with either Nav1.2 wild-type or mutant channels. (C) Voltage dependence of steady state Na+ channel activation and inactivation revealing a significant depolarizing shift of the activation curve for G899S (loss-of-function) as well as a significant hyperpolarizing shift of the inactivation curve for P1622S (loss-of-function) in comparison with the wild-type. Lines represent fits of Boltzmann functions. (D) Voltage dependence of the fast inactivation time constant for wild-type and mutant channels revealing a slowing of fast inactivation for F1597L and an acceleration for P1622S. (E) The time course of recovery from fast inactivation determined at −100 mV showed significant changes between wild-type and mutant channels. F1597L mutant channels showed a significantly faster recovery (gain-of-function), whereas P1622S mutant channels showed a significant slowing of the recovery from fast inactivation compared to wild-type channels (loss-of-function). Lines represent fits of exponential functions yielding the time constant τrec. (F) Voltage dependence of the persistent sodium current showing a large increase for V423L compared to the wild-type. Current amplitudes recorded at the end of a 70-ms depolarization were normalized to the peak current amplitude (steady state current/initial peak current). Shown are means ± SEM.
We found gain-of-function effects for the mutations V423L and F1597L, whereas the mutations G899S and P1622S showed loss-of-function effects (Fig. 3 and Supplementary Table 2). The analysis of the V423L mutation revealed a change in slope of activation (Fig. 3C and Supplementary Table 2) as well as a doubling of the window current and a dramatic increase in the persistent sodium current compared with the wild-type (Fig. 3B, C and F). For F1597L mutant channels, we observed a hyperpolarizing shift of the activation curve (Fig. 3C), fast inactivation time constants were significantly larger for mutant channels (Fig. 3D) and recovery from fast inactivation was accelerated (Fig. 3E and Supplementary Table 2).
In contrast, electrophysiological analysis of the mutations G899S and P1622S revealed profound loss-of-function changes. The most prominent change for P1622S mutant channels was a significant hyperpolarizing shift of the fast inactivation curve (Fig. 3C). The effect of the G899S mutation on channel kinetics was not as pronounced as for P1622S, consisting of a depolarizing shift and a slope change of steady-state activation (Fig. 3C). Both mutations thus predict a decrease of channel availability and membrane excitability in neurons expressing mutant Nav1.2 channels, an effect that would be further enhanced by SCBs. Functional consequences of known SCN2A variants that have been reported in the literature are listed in Supplementary Table 1.
Discussion
Our study of 71 new patients with SCN2A mutations and additional 130 previously reported cases—including a physiological characterization of some mutations and existing functional data from the literature—reveal evidence that there are two distinct groups among symptomatic SCN2A mutation carriers with epilepsy. The first group is represented by (i) early infantile epilepsy with onset before 3 months of age; (ii) missense mutations with gain-of-function effects of different severity with correlation to the severity of the clinical phenotype; and (iii) a relatively good response to SCBs. The second group is characterized by (i) a later epilepsy onset, i.e. after 3 months of age; (ii) frequent loss-of-function mutations (truncations and splice site mutations, but also missense mutations with loss-of-function effects); and (iii) a relatively poor response to SCBs. The phenotypic spectrum in the early onset group comprises B(F)NIS, Ohtahara syndrome, EIMFS and unclassified encephalopathies, whereas the late onset cases include West syndrome, Lennox-Gastaut syndrome, myoclonic-atonic epilepsy, and focal epilepsies with an ESES-like picture. A third, smaller group is represented by (i) intellectual disability and/or autism without epilepsy; and (ii) frequent loss-of-function mutations (truncations).
In the benign end of the spectrum, both familial and de novo mutations are found. These patients are characterized by a normal cognitive development and self-limited epilepsy with cessation of seizures mostly during the first year of life. Seizure semiology shows considerable variation, the typical ‘clustering’ is not always present, and family history may be negative due to the presence of de novo mutations. Seizures may be initially difficult to treat: in our cohort of unpublished patients, seizures were initially drug-resistant in 6/9 children (as discussed further below). However, EEG might help to rule out a severe epileptic phenotype, showing typically a normal background activity with or without multifocal spikes, but never a suppression burst pattern. After cessation of neonatal/infantile seizures, five children developed episodic ataxia and pain (Liao et al., 2010a; Johannesen et al., 2016; Schwarz et al., 2016).
The group with encephalopathy and epilepsy is the largest among the SCN2A carriers; 69% of the cohort falls into this category (Fig. 1). Half of those patients had a seizure onset in the early infantile period (<3 months). In the late onset group, seizure onset was usually before the age of 4 years (only five patients had a later seizure debut).
Fifty-four of the 139 children had an identifiable epileptic syndrome (32%). In the early infantile group, Ohtahara syndrome (n = 18) and EIMFS (n = 13) constitute the most important specific phenotypes. Whereas Ohtahara syndrome was the first syndrome to be reported in SCN2A-related encephalopathies, EIMFS was only recently recognized (Howell et al., 2015). In EIMFS, KCNT1 mutations have been reported as the most common underlying genetic cause so far (Barcia et al., 2012; Ohba et al., 2015). In KCNT1-related EIMFS, the prognosis seems to be uniformly poor, and affected children show severe disability with ongoing seizures during follow-up (Barcia et al., 2012; Ohba et al., 2015). In our SCN2A cohort, 2/5 patients with EIMFS became seizure-free at the age of 2 and 12 months with vigabatrin and phenytoin, respectively, and showed mild intellectual disability. Thus, prognosis seems to be more favourable in SCN2A-related EIMFS compared to those caused by KCNT1 mutations.
Epilepsies with onset beyond the early infantile period are increasingly recognized in SCN2A-related disorders. In particular, West syndrome has recently emerged as the most important specific phenotype, accounting for 16 patients so far. Of note, one recurring mutation (R853Q), which has been found in nine cases so far, is frequently associated with West syndrome (six cases), and should therefore be considered in the diagnostic work-up of children with West syndrome.
The intellectual disability/autism group without epilepsy is most likely an underestimate of the actual number of cases, since it is not common practice in all countries/hospitals to perform genetic testing in patients with intellectual disability and/or autism without seizures. This group of patients represents 16% of the cohort.
The relative frequency of the disease groups as represented in Fig. 1A for the newly identified cases with epilepsy and in Fig. 1B for all cases probably contains biases, as (i) benign neonatal-infantile epilepsies were the first entity in which SCN2A mutations were detected suggesting a relative over-representation of those and may be also the severe early onset cases in the literature; and (ii) since cases without epilepsy are probably underdiagnosed.
Genotype
Recurrent mutations are seen both in the benign and severe end of the spectrum (Table 5). However, even with the same mutation, a quite large phenotypic variance is observed. A remarkable phenotype was found for R853Q mutation carriers in whom 6/9 were affected by West syndrome. Furthermore, three of the patients with an A263V mutation showed a BNIS phenotype with late onset episodic ataxia, while three others with the same mutation had more severe phenotypes. Thus, both the mutation itself and other genetic or environmental factors contribute to the individual phenotype.
Interestingly, we found only missense mutations in the early onset epilepsies, whereas truncations, splice-site and nonsense mutations were solely seen in the epilepsies with a later onset and in the group of cases without epilepsy. This observation, along with the functional effects described in the results section and discussed below, could have implications for treatment in these two major groups of SCN2A patients. Supplementary Table 1 provides an overview of all known SCN2A mutations, the associated clinical phenotypes and known functional consequences.
Prevalence
We estimated the frequency of SCN2A mutations causing the reported phenotypes in the Danish population to be 1/78 608. This number will most likely be an underestimate. There is not a strong tradition for a systematic genetic screening of patients with isolated autism or intellectual disability, thus there might be a recruitment bias towards patients with epilepsy.
Seizure outcome and treatment effects
In B(F)NIS families, seizures are reported to be controlled by AEDs (Berkovic et al., 2004; Striano et al., 2006; Herlenius et al., 2007). In the unpublished cohort, however, many patients were resistant to various AEDs, including phenobarbital, topiramate, levetiracetam and valproate, whereas SCBs (especially oxcarbazepine and phenytoin), were completely or partially effective in 6/9 cases, suggesting a specific effect of SCBs in this subgroup. However, and as known from previous studies, three patients became seizure-free without any AEDs, which is part of the natural history of these patients. Thus, the effect of SCBs in these patients should be interpreted with some caution, although we observed a clear correlation between introduction of the drugs and seizure freedom.
Apart from B(F)NIS, we found a different pattern of seizure outcome and AED effects in our cohort according to the age at seizure onset. Of all patients with encephalopathy and epilepsy onset <3 months, 17/28 (61%) became seizure-free, 10 of them during SCB treatment, mainly with phenytoin (n = 8), whereas other standard AEDs (e.g. phenobarbital, levetiracetam) were largely ineffective. Interestingly, in five children recurrent declines of phenytoin plasma levels resulted in prompt seizure relapses that were reversible after adjusting the phenytoin dosage. These observations underline the beneficial effect of phenytoin on seizure activity in these patients.
To date, there are only few reports on treatment response in SCN2A-related epilepsies. In the series of Nakamura et al. (2013) on 15 children with early onset seizures, epilepsies were described as intractable in 12 of 15 cases. Phenytoin was tried in five children with seizure onset between 1 day and 6 weeks of age, and showed partial effects in four of them. Zonisamide showed some effects in 4/6 children. Two children became seizure-free with lamotrigine at the age of 6 months and 6 years, respectively. Howell et al. (2015) reported improvement of seizure control in 11/15 patients with early seizure onset, phenytoin was reported to be partially effective in seven children with neonatal seizure onset. Sawaishi et al. (2002) described a striking effect of lidocaine, a prototypic sodium channel blocker, in a patient with Ohtahara syndrome due to a SCN2A mutation (Sawaishi et al., 2002; Ogiwara et al., 2009).
Taken together, patients with early onset epilepsies were difficult to treat but responded relatively well to SCBs, in particular to phenytoin in appropriate dosages. The high dosages needed to control seizures completely might not have been reached in many patients who did not become seizure-free.
In contrast, only 10/29 (34%) children with infantile/childhood epilepsy became seizure-free, and seizures mostly did not respond to SCBs. In the nine children with West syndrome, seizures and hypsarrhythmia in the EEG were mostly resistant to standard treatment including steroids and ketogenic diet. Nakamura et al. (2013) reported on eight children with West syndrome in their series. All were intractable, suggesting that SCN2A-related West syndrome is particularly difficult to treat. However, some case reports describe treatment responses to ACTH (Nakamura et al., 2013), topiramate (Ogiwara et al., 2009; Sundaram et al., 2013; Samanta and Ramakrishnaiah, 2015) and transient effects of prednisolone (Matalon et al., 2014).
Seizure worsening associated with SCB treatment was observed in seven children of our cohort, all of whom had seizure onset beyond the age of 3 months. Hackenberg et al. (2014) reported an increase of seizure frequency related to carbamazepine treatment in a child with seizure onset at 3 months of age. Howell et al. (2015) reported the appearance of myoclonus with vigabatrin and lamotrigine in one child. SCBs are known to aggravate seizure activity in epileptic syndromes that are caused by loss-of-function mutations in Nav1.1 channels, e.g. Dravet syndrome, putatively because they further reduce sodium channel activity in inhibitory neurons (Brunklaus et al., 2014) expressing Nav1.1 as the major sodium channel (Catterall, 2014). As discussed below, a similar mechanism may apply for loss-of-function mutations in Nav1.2 channels.
The statistical χ2 test confirmed our impression of differential treatment effects of SCBs and non-SCBs in early versus late onset encephalopathies with epilepsy. The late onset cases responded significantly better to non-SCBs than the early onset ones, which may indicate that the epilepsy in the early onset cases is more difficult to treat. This observation even strengthens the finding of a significantly better response to SCBs of the early onset compared to the late onset cases, and suggests that the higher seizure freedom rate of the early onset group (61%, compared to only 34% of the late onset group) is the likely consequence of a specific pharmaco-response to gain-of-function SCNA2 mutations (see discussion below on functional effects).
In summary, SCBs seem to have positive effects on seizures in early infantile onset epilepsies, but are not effective or can even worsen seizure activity in epilepsies with onset at 3 months of age or later. However, these conclusions have to be taken with care, as the natural history of these conditions is unknown, our observations are purely retrospective and the exact duration from AED introduction to seizure freedom was not clear in all cases.
Functional studies and their pharmacological and neurophysiological interpretation
Out of the diversity of SCN2A mutations that have been identified until now, only a small number have been studied functionally. SCN2A mutations can lead to either augmented or reduced Nav1.2 activity (Kamiya et al., 2004; Scalmani et al., 2006; Xu et al., 2007; Ogiwara et al., 2009; Liao et al., 2010a, b; Lossin et al., 2012; Rauch et al., 2012; Lauxmann et al., 2013; Sundaram et al., 2013; Codina-Sola et al., 2015; Schwarz et al., 2016). Here, we functionally analysed four additional mutations, two of which were identified in patients suffering from encephalopathy with early infantile onset epilepsy. For these mutations (V423L and F1597L), a clear gain-of-function effect was found (Fig. 3). V423L particularly showed a tremendous increase in persistent sodium current, probably explaining the extremely severe phenotype with highly drug-resistant Ohtahara syndrome in both children affected by this mutation. We hypothesize that this persistent current was too large to be sufficiently reduced by SCBs in clinically relevant dosages. F1597L caused an accelerated recovery from fast inactivation, and the patient showed an EIMFS phenotype with a prompt response to phenytoin treatment. In contrast, we have shown clear loss-of-function effects for two mutations found in children with infantile/childhood epilepsy (G899S and P1622S). The phenotype consisted of tonic-clonic seizures and absences in one and myoclonic-atonic epilepsy in the other child, which was clearly different from the other two mutations. This difference was equally seen with regard to the treatment response, which showed seizure aggravation upon SCB treatment in both cases.
In previous studies, several disease-causing mutations in SCN2A have been functionally analysed. In children with severe early onset epilepsies, gain-of-function mutations were described (Ogiwara et al., 2009; this study, see above). Missense mutations from patients with B(F)NIS have been characterized to cause a gain-of-function (Scalmani et al., 2006; Xu et al., 2007; Liao et al., 2010a, b; Lauxmann et al., 2013; Schwarz et al., 2016) with one of these mutations (A263V) also causing severe epilepsy in some cases (see above). Few studies also indicated some biophysical features indicating a loss-of-function (Scalmani et al., 2006; Misra et al., 2008); however, studies in neurons by Scalmani et al. (2006) suggested a net gain-of-function with increased excitability for two such cases so that the main effect seems to be a gain-of-function (R223Q and R1319Q), matching the effective SCB treatment of patients carrying the R1319Q mutation. These distinct effects of the same mutation analysed in neuronal versus non-neuronal cells could be caused by post-translational modifications, phosphorylation, trafficking and protein–protein interactions (such as with β-subunits, which we also used in our study) of the channels which can be quite different in neuronal cells and heterologous expression systems (for review see Shao et al., 2009). It has been shown for mutations in Nav1.1 that—besides the β-subunits as the closest interacting partners of α-subunits—ankyrin, calmodulin or other endogenous proteins can also have essential roles for intracellular trafficking and functional expression (Rusconi et al., 2007; Cestele et al., 2013). Such effects may also apply for the mutations we studied here in tsA201 cells. Furthermore, modifier genes might affect the clinical severity and the variability of phenotypes seen in epilepsy patients with gain-of-function SCN2A mutations (Bergren et al., 2005; Hawkins and Kearney, 2016).
In contrast to the observed gain-of-function effects, missense loss-of-function and nonsense mutations have been identified in patients with later onset epilepsies, although two mutations also showed a hyperpolarizing shift of the activation curve as a gain-of-function feature (Kamiya et al., 2004; Ogiwara et al., 2009; Lossin et al., 2012). Finally, autism spectrum disorder without seizures was also associated with loss-of-function mutations (Kamiya et al., 2004; Rauch et al., 2012; Sanders et al., 2012; Carvill et al., 2013; Codina-Sola et al., 2015; D’Gama et al., 2015; Howell et al., 2015; Carroll et al., 2016; Horvath et al., 2016; Li et al., 2016; Trump et al., 2016) (Supplementary Table 1).
These findings strengthen the hypothesis that there is a link between gain-of-function SCN2A mutations, early onset epilepsy, and effectiveness of SCBs on the one hand, and loss-of-function mutations, later onset epilepsy, and ineffectiveness of SCBs on the other hand. Gain-of-function versus loss-of-function mutations affecting the neuronal excitability of different neurons during specific developmental stages might explain the variation in seizure onset and the response to SCBs in early infantile syndromes. Early in development, the Nav1.2 channel is highly expressed at nodes of Ranvier and axon initial segments and is partially replaced during development by the Nav1.6 channel (Kaplan et al., 2001; Liao et al., 2010b), which could be confirmed in adult human hippocampal brain slices (Liao et al., 2010b). Nav1.2 channels are therefore considered to contribute to action potential generation and propagation and influence the axonal firing frequency during early development. Hence, mutations causing gain-of-function effects can alter the characteristic firing patterns of Nav1.2-expressing neurons and cause hyperexcitability, which can be dampened by SCB treatment and thus improve seizure outcome. In contrast, loss-of-function mutations cannot drive Nav1.2-expressing cells into hyperexcitability during early development, and therefore patients may not exhibit early onset seizures. However, loss-of-function mutations seem to lead to severe epileptic phenotypes later in development. In the more mature brain, unmyelinated axons express the Nav1.2 channel, such as mossy fibres projecting from the hippocampal dentate gyrus into the CA3 region (Kaplan et al., 2001; Liao et al., 2010b). The most prevalent targets of mossy fibres apart from CA3 pyramidal cells are GABAergic parvalbumin-positive interneurons. Mossy fibres contact inhibitory basket cells ∼50 times more frequently than pyramidal cells mediating a powerful feedforward inhibition (Acsady et al., 1998). Therefore, a reduced excitability of dentate granule cells due to Nav1.2 loss-of-function mutations can result in CA3 hyperexcitability, which can spread to subsequent hippocampal regions and further. A treatment with SCBs can therefore act in a similar way as hypothesized for loss-of-function Nav1.1 mutations. Here, SCBs are predicted to further reduce the activity of inhibitory neurons expressing Nav1.1 as major sodium channel. Similarly, SCBs could further reduce the activity of Nav1.2-expressing dentate granule cells, which in turn would activate inhibitory neurons less effectively. As mossy fibres are not the only unmyelinated fibres within the brain, other unmyelinated inhibitory neurons expressing Nav1.2 channels could also contribute to neuronal hyperexcitability. Only parvalbumin-positive inhibitory neurons within the cortical layers have been shown to be myelinated (Micheva et al., 2016). Unmyelinated inhibitory neurons expressing Nav1.2 channels are controlling the activity of excitatory neurons and provide feedforward inhibition (Kepecs and Fishell, 2014). In early development, a reduced activity of these inhibitory neurons due to Nav1.2 loss-of-function mutations could be of little or no consequence, as excitatory Nav1.2-expressing neurons within the cortex also show reduced excitability. As mentioned above, the Nav1.2 channel is partially replaced by the Nav1.6 channel at nodes of Ranvier and axon initial segments during development, but is still expressed in unmyelinated fibres, such as those from inhibitory neurons. Additionally, the neonatal splice variant of Nav1.2 has been shown to have a seizure protective role during early development (Gazina et al., 2015). A reduced excitability of unmyelinated cortical inhibitory neurons later in development could therefore lead to hyperexcitable cortical networks. A disruption of the excitatory/inhibitory balance caused by SCN2A loss-of-function mutations can therefore cause seizures and additionally underlie neuropsychiatric diseases and autism, reflected by a higher prevalence in patients with loss-of-function mutations and late onset phenotypes in our cohort.
In summary, our study reflects the large spectrum of SCN2A-related disorders and identifies SCN2A mutations as one of the most common genetic causes of epilepsy. We have established two distinct groups with seizure onset either before or after 3 months of age, which show phenotypic differences, gain-of-function versus loss-of-function Nav1.2 abnormalities and a likely related differential response to treatment with SCBs.
Abbreviations
- ACTH
adrenocorticotropic hormone
- AED
antiepileptic drug
- B(F)NIS
benign (familial) neonatal/infantile seizures
- EIMFS
epilepsy of infancy with migrating focal seizures
- ESES
electrical status epilepticus during slow sleep
- SCB
sodium channel blocker
Acknowledgements
We wish to thank the patients and their families for the participation in this study and Stephan Müller for assistance in performing mutagenesis.
Funding
K.L.H. is employed by and receives a salary from Ambry Genetics. I.H. was supported by intramural funds of the University of Kiel, by a grant from the German Research Foundation (HE5415/3-1) within the EuroEPINOMICS framework of the European Science Foundation, and additional grants of the German Research Foundation (DFG, HE5415/5-1, HE 5415/6-1), German Ministry for Education and Research (01DH12033, MAR 10/012), and grant by the German chapter of the International League Against Epilepsy (DGfE). H.L. was supported by the EU FP7 project EpiPGX (279062), the EuroEPINOMICS framework of the European Science Foundation (DFG grant Le1030/11-2), by the German Ministry for Education and Research (BMBF) rare disease network IonNeurONet (01GM1105A), as well as the German chapter of the International League Against Epilepsy (DGfE) and the foundation “no epilep”. P.d.J. was supported by ESF/Euro-Epinomics (GA136.11.N and FWO/ESF-ECRP). H.S. is a PhD fellow of the Fund for Scientific Research Flanders (1125416N).
Supplementary material
Supplementary material is available at Brain online.
References
Author notes
*,#These authors contributed equally to this work.


{kind=link}
{kind=link}
{kind=link}