





Abstract
The intrinsic complexity of the microbiota combined with technical variability render shotgun metagenomics challenging to analyze for routine clinical or research applications. In silico data generation offers a controlled environment allowing for example to benchmark bioinformatics tools, to optimize study design, statistical power, or to validate targeted applications. Here, we propose assembly_finder and the Metagenomic Sequence Simulator (MeSS), two easy-to-use Bioconda packages, as part of a benchmarking toolkit to download genomes and simulate shotgun metagenomics samples, respectively. Outperforming existing tools in speed while requiring less memory, MeSS reproducibly generates accurate complex communities based on a list of taxonomic ranks and their abundance.
All code is released under MIT License and is available on https://github.com/metagenlab/MeSS and https://github.com/metagenlab/assembly_finder.
1 Introduction
Shotgun metagenomics has been widely applied for large-scale investigations of complex human, animal, and environmental microbiomes (Huttenhower et al. 2012, Oh et al. 2014, Quince et al. 2017). While species composition and functions correlated with various diseases (Zhernakova et al. 2016) or environmental conditions (Techtmann and Hazen 2016), detecting statistically significant biological effect sizes between the microbiome and outcomes is challenging (Quince et al. 2017) and must be backed by adequately powered studies. Similarly, the translation of metagenomics to routine clinical diagnosis requires extensive testing to validate each application and assess its limits (Scherz et al. 2022). However, validating shotgun metagenomics by sequencing real samples is expensive and burdensome (Afshinnekoo et al. 2017). Thus, data emulation is a cost- and time-effective approach for researchers and clinical microbiologists for example to assess workflow performance, study design, and power, or to benchmark bioinformatic tools (Gourlé et al. 2019).
Tools for microbiome simulation employ different strategies, including some using real data. For example, MB-GAN uses a generative adversarial network (Rong et al. 2021) and learns from existing microbiome abundances to simulate realistic abundances. Other tools such as SEQ2MGS (Van Camp and Porollo 2022) rely on real sequencing reads and mixes them together to generate artificial metagenomes. While using existing high-throughput sequencing data generates realistic benchmarking datasets, the data can contain errors, biases and uncertainties that can pose challenges for data interpretation (Pfeifer 2017). In addition, the impact of computational methods on results is huge as different tools provide divergent results in genomic analyses like variant calling (Barbitoff et al. 2022). Thus, a controlled environment, without experimental biases, is needed to correctly benchmark computational methods performance (Milhaven and Pfeifer 2023). Read simulators, which emulate synthetic DNA sequencing reads based on sequencing technologies specific error models, can be used to construct such an environment. Popular short read simulators (Milhaven and Pfeifer 2023) include ART (Huang et al. 2012), InSilicoSeq (Gourlé et al. 2019), Mason (Holtgrewe 2010), and NEAT (Stephens et al. 2016). For long read sequencing, tools like SILICO (Baker et al. 2016), Badread (Wick 2019), and PBSIM3 (Ono et al. 2022) allow both Nanopore and PacBio reads simulation. Most of the read simulators are used to generate whole genome sequencing reads for one genome. Thus, generating sequencing reads for multiple genomes with fine-grain control on abundance profiles for each genome is complicated. To our knowledge, only CAMISIM (Fritz et al. 2019) offers the possibility to generate in silico metagenome sequences of communities for both long and short reads technologies. In fact, CAMISIM simulates metagenomics sequences out of existing 16S profiles or de novo, from a list of genomes (Fritz et al. 2019). CAMISIM offers many customization options like multi-sample time series, but sample generation is slow and only few reference datasets are available on the CAMI challenge website (Meyer et al. 2022).
Here, we propose MeSS and assembly_finder, easy-to-use Bioconda (Grüning et al. 2018) packages that allow scalable and reproducible reference genome download as well as short and long read simulation to mimic shotgun metagenomics sequencing of complex samples.
2 Features
MeSS streamlines metagenomic sample generation by automating genome download, community design and read simulation in one command line. MeSS takes as input a table of genomes with their abundances, to produce shuffled, anonymous and compressed reads, and optionally their alignment to their reference sequence. Since reads and alignment files can occupy large volumes (Numanagić et al. 2016), they are compressed by default to minimize storage footprint (Sun et al. 2021). In addition, MeSS outputs taxonomic profiles, in biobox format, containing exact expected sequence and taxonomic abundances (Sun et al. 2021). To ensure scalability and efficient resource usage, users can set the number of CPUs and memory required by the pipeline and use built-in scheduler profiles like SLURM (Yoo et al. 2003) to run on high-performance computing clusters. Lastly, MeSS provides an easy subcommand to create metagenome templates for healthy individuals covering four human body sites. These templates can be used to create scenario-based simulations like spiking in a respiratory pathogen to mimic an infection of respiratory airways.
Read simulation involves running command line tools that require different software dependencies and file format processing, which makes it tedious for inexperienced users to run manually. To address this, MeSS uses Snakemake, a workflow engine that automates steps ensuring scalability and reproducibility while facilitating software dependency deployment (Köster and Rahmann 2012). Despite these advantages, running a Snakemake pipeline requires extensive setup, from copying the workflow directory, configuration file modification to inputting the absolute path to a Snakefile, which can be overwhelming for new users (Roach et al. 2022). To improve user experience, MeSS uses Snaketool (Roach et al. 2022) to conveniently launch Snakemake from the command line without the previously mentioned hassles.
2.1 Assembly download with the Bioconda package assembly_finder
To simulate metagenomic sequences of microbial communities, genome assemblies of representative taxa can be downloaded from public databases such as NCBI RefSeq (O’Leary et al. 2016). While the assembly summary table in NCBI’s ftp contains all links to access the data, their download via ftp is slow and often results in connection timeouts. Therefore, we developed assembly_finder (Fig. 1A), a wrapper around the NCBI datasets command line (O’Leary et al. 2024) to retrieve large numbers of genome sequences. Starting from lists of scientific names, taxonomic identifiers (taxids) (Federhen 2012), or genome accessions, assembly_finder combines NCBI datasets commands, for a fast and easy download of genome sequences and metadata.
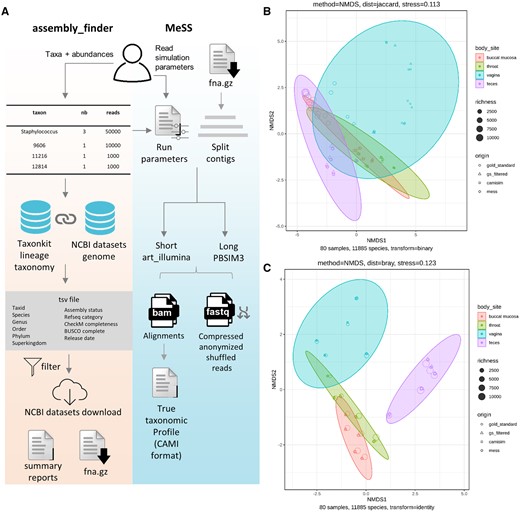
MeSS and assembly_finder workflows and assessment of simulated communities. (A) Main steps of assembly_finder and MeSS workflows. (B) Analysis of real and simulated datasets with NMDS ordination of beta diversity using Jaccard. (C) Bray–Curtis. Circles correspond to real samples, triangles to real samples filtered at 200 reads per taxa, squares and diamonds to CAMISIM and MeSS simulated samples, respectively. Shape size reflects the number of species present in the sample. Each ellipse groups a different body site: buccal mucosa (red), throat (green), vagina (blue), and feces (purple).
For each query, assembly metadata like assembly status, assembly level, and taxonomy identifiers are retrieved using the datasets summary genome command. For all unique taxonomy identifiers a complete lineage taxonomy, from kingdom to species is retrieved using taxonkit (Shen and Ren 2021). Next, genome assemblies are ranked according to metrics present in the assembly metadata table, which include RefSeq category (reference, or representative), assembly status (complete, chromosome, scaffold, contig), BUSCO (Manni et al. 2021a,b) and checkM (Parks et al. 2015) completeness scores. BUSCO and checkM completeness estimate genome quality based on expected gene content (Manni et al. 2021a,b), and thus, genomes with high BUSCO and checkM scores rank higher in the summary table. Optionally, assembly_finder can rank assemblies at any given taxonomic level, for example to download the 10 best-ranking genomes for each bacterial genus.
2.2 Sample community design
For each metagenomic sample, MeSS accepts a list of local genomes or a list of taxa to be downloaded with assembly_finder. By default, MeSS supports manually assigning quantities to each taxon or genome. For example, sequence counts (bases or reads), sequence abundances, or taxonomic abundances can be set and are converted to coverage depths by normalizing to genome sizes. Alternatively, MeSS can draw abundances from an even (same taxon with equal proportions) or a log-normal distribution, frequently used to model microbial communities (Curtis et al. 2002, Unterseher et al. 2011).
2.3 Read simulation
To choose read simulators to include in MeSS, we established the following selection criteria. First, the simulator should generate fastq files. Second, it must generate a “ground truth” SAM/BAM file to know the genomic location of reads, which is crucial for benchmarking and tool comparison (Milhaven and Pfeifer 2023). Third, it needs low computational costs and must be installable via bioconda. As both ART and PBSIM3 fit the criteria and ART performed well in benchmarks (Milhaven and Pfeifer 2023), they were included in MeSS for short and long read simulation, respectively. ART simulates an equal amount of reads per contig (Milhaven and Pfeifer 2023) and PBSIM3 generates reads per contig. Thus, to have a uniform read sampling across the genome for ART, and compatibility with PBSIM3, multi contig fasta files are split into separate contig files. Simulated reads per contig are then concatenated into one paired or single fastq file per sample. Simulated reads contain information like fasta header name, and genome position from which the read was sampled. To hide this information in the anonymized reads, read headers are replaced by their index position in the fastq file using seqkit (Shen et al. 2016). Moreover, to avoid downstream biases in the analysis, reads are shuffled with seqkit as well. Optionally, MeSS generates as output alignment files which are converted to the BAM format using SAMtools (Li et al. 2009) and Bioconvert (Caro et al. 2023). Average coverage and read counts are extracted from BAMs using “samtools coverage” (Li et al. 2009), which is then used to create a taxonomic profile file following the biobox format (Belmann et al. 2015) using “taxonkit profile2cami” (Shen and Ren 2021). As highlighted by Sun et al., metagenomics profiles can contain either relative sequence or taxonomic abundances, and neglecting the difference between these two metrics can bias benchmarking results (Sun et al. 2021). To avoid this, MeSS provides profiles with both sequence (read percentages) and taxonomic (coverage depth percentages) abundances.
Technical replicates from the same metagenome can be generated by setting the number of replicates and the standard deviation of genome quantity value (reads, bases, coverage depths, and sequence or taxonomic abundances) between replicates. By default, the standard deviation between replicates is zero, and the difference is the sequence genomic origin set by the seed. Overall, the pipeline outputs compressed fastq files, a true taxonomic abundance profile in CAMI format, as well as BAM files to verify reads alignments to their reference genome.
3 Benchmarks
3.1 Comparison of simulated and real samples
To evaluate MeSS ability to generate realistic metagenomes, 20 samples from four different body sites (buccal mucosa, throat, gut, and vagina) were retrieved from the Human Microbiome Project (Huttenhower et al. 2012). Reads were classified with Kraken2 (Wood et al. 2019) using a standard Kraken2 index from June 2024 obtained from Index zone. Sequence abundances were estimated at the species level using Bracken (Lu et al. 2017). Across all 20 samples, 1498 unique taxa, to which at least 200 reads were attributed at species level, formed the real expected community. Species taxids were inputted to assembly_finder to download one reference genome per taxid. The downloaded genomes were used for read simulation using MeSS and CAMISIM, the current state-of-the-art metagenomic sample simulator (Fritz et al. 2019). To ensure a fair comparison, we ran MeSS and CAMISIM with the same parameters to simulate 100 bp paired reads following the empHiseq2k error profile using 16 CPUs within a Nextflow (Di Tommaso et al. 2017) pipeline available here (https://github.com/metagenlab/benchmark-MeSS-CAMISIM). To demonstrate that simulated samples have the same community structure as real samples, we evaluated alpha and beta diversity indexes. As shown in Supplementary Fig. S1, all alpha diversity indices evaluating richness and/or abundance from simulated and real datasets strongly correlate (0.96 Spearman’s R squared, on average, across all indexes). This demonstrates that MeSS simulates mock communities with similar evenness and richness compared to expected communities.
To assess whether species composition and abundance in simulated samples are comparable to the real communities, pairwise Jaccard and Bray–Curtis distances were computed and visualized by NMDS ordination plots (Fig. 1B). Real samples cluster closely with their simulated counterparts, and group by body sites, with some expected similarities between throat and mouth metagenomes. A PERMANOVA with Bray–Curtis distance showed that centroids and dispersion differ significantly among body sites (F = 16.05, P = 0.001), but not among simulated and real communities (F = 0.08, P = 1.000), confirming that the species and their relative abundances are conserved between real and simulated sample metagenomes. The code to reproduce the figures is available at https://github.com/metagenlab/MeSS-figures.
3.2 Resource usage
CPU and RAM usage of MeSS was compared with CAMISIM de novo mode using 2000 bacterial reference genomes. Sixteen subsamples were simulated with an increasing number of genomes, first ranging from 1 to 10, and then doubling up to 640, and a coverage depth of 1x. As shown in Supplementary Fig. S2, MeSS exhibits a lower linear time complexity when compared to CAMISIM. Time complexity is defined as the process time needed to simulate metagenomes as a function of the number of genomes. Overall, MeSS is around 18x faster than CAMISIM and uses less RAM (Supplementary Fig. S3). The code to benchmark resources usage is available at https://github.com/metagenlab/benchmark-MeSS-CAMISIM.
4 Conclusion
assembly_finder together with MeSS can be used as a scalable, reproducible, and easy-to-use toolkit for benchmarking metagenomic software. In fact, with its simple command line, one can gather genomes and mix them into communities to mimic shotgun metagenomics sequencing of complex communities. It offers fast assembly download and parallel read simulation with an extensive list of parameters such as number of replicates, choice of sequencing technology, sequencing error profile, and read pairing. MeSS can generate specific communities, either inferred from taxonomic profiling of real metagenomic samples, or communities with random lognormal or even distributions. Moreover, we bundled metagenome templates for healthy individuals, which can be useful to create scenario-based simulations for example by spiking in microorganisms to benchmark tools for pathogen detection. MeSS offers improved efficiency to available computational resources, outperforming CAMISIM. Both MeSS and assembly_finder source codes are available at https://github.com/metagenlab/MeSS and https://github.com/metagenlab/assembly_finder.
Author contributions
Farid Chaabane (Conceptualization [equal], Formal analysis [lead], Methodology [equal], Software [lead], Writing—original draft [lead]), Trestan Pillonel (Conceptualization [equal], Methodology [equal], Software [supporting], Supervision [supporting], Writing—review & editing [supporting]), and Claire Bertelli (Conceptualization [equal], Funding acquisition [lead], Methodology [supporting], Project administration [lead], Resources [lead], Supervision [lead], Writing—review & editing [lead])
Supplementary data
Supplementary data are available at Bioinformatics online.
Conflict of interest: None declared.
Funding
The initial software development was financed by internal funds and the maintenance and further enhancement of the tool is supported as a part of NCCR Microbiomes, a National Centre of Competence in Research, funded by the Swiss National Science Foundation [225148].


{kind=link}